

Abstract
Behavioral genetic studies of humans have associated variation in the DTNBP1 gene with schizophrenia and its cognitive deficit phenotypes. The protein encoded by DTNBP1, dysbindin-1, is expressed in forebrain neurons where it interacts with proteins mediating vesicular trafficking and exocytosis. It has been shown that loss of dysbindin-1 results in a decrease in glutamate release in the prefrontal cortex; however the mechanisms underlying this decrease are not fully understood. In order to investigate this question, we evaluated dysbindin-1 null mutant mice, using electrophysiological recordings of prefrontal cortical neurons, imaging studies of vesicles, calcium dynamics and Western blot measures of synaptic proteins and Ca2+ channels.
Dysbindin-1 null mice showed a decrease in the ready releasable pool of synaptic vesicles, decreases in quantal size, decreases in the probability of release and deficits in the rate of endo- and exocytosis compared with wild-type controls. Moreover, the dysbindin-1 null mice show decreases in the [Ca2+]i, expression of L- and N- type Ca2+ channels and several proteins involved in synaptic vesicle trafficking and priming. Our results provide new insights into the mechanisms of action of dysbindin-1.
Keywords: prefrontal cortex, calcium, dysbindin, synapsin, synaptotagmin, synaptic vesicles
Introduction
Schizophrenia is a relatively common neuropsychiatric disorder that often involves debilitating and treatment-refractory cognitive deficits that can significantly limit the psychosocial function of affected persons (Green et al., 2000)). The disorder is highly heritable, and a number of candidate susceptibility genes have emerged recently (Ayalew et al., 2012; Gejman et al., 2010).
Of these putative risk genes, the gene encoding dystrobrevin-binding-protein-1 (i.e., dysbindin-1) -- DTNBP1 is of particular interest. DTNBP1 lies within the chromosome 6p24-22 susceptibility locus (Straub et al., 1995), and multiple associations have been reported between variants of DTNBP1 and schizophrenia (Maher et al., 2010; Talbot et al., 2009). Beyond association between sequence variants and the disorder, a large proportion of schizophrenia patients exhibit lower dysbindin-1C protein in tissue from the PFC (Tang et al, 2009). There are three isoforms of dysbindin: dysbindin 1A, 1B and 1C. Dysbindin 1C is mainly found in postsynaptic sites in human tissue; however it has also been reported in presynaptic sites (Talbot et al., 2011). In mice, dysbindin 1C is the only form of protein found preysnaptically (Talbot et al., 2008). Amongst its functions, dysbindin-1 is involved in the control of presynaptic release of glutamate. Recent studies (Chen et al., 2008; Jentsch et al., 2009) have reported that reduced expression of dysbindin-1 in mice dampened glutamate release in the PFC and hippocampus.
Dysbindin-1 is part of the Biogenesis of Lysosome-related Organelle Complex 1 (BLOC-1 complex) (Starcevic and Dell’Angelica, 2004) which is compromised by 8 proteins (dysbindin, snapin, muted, pallidin, cappuccino and BLOS 1–3). This complex has been related to multiple cellular functions including synaptic vesicle dynamics and stabilization of the t-SNARE complex (Larimore et al., 2011; Mullin et al., 2011; Newell-Litwa et al., 2009, 2010). Interestingly, decreases in dysbindin-1 reduce the level of snapin (Feng et al., 2008) which, in turn, affects its association with SNAP25 and the interactions between SNAP25 and the calcium sensor synaptotagmin-1, thus impairing priming of vesicles. Moreover, changes in dysbindin-1 produce changes in synapsin 1 (Numakawa et al., 2004), which controls the movement of synaptic vesicles from the reserve pool to the ready-releasable pool (RRP) (Cesca et al., 2010), consequently facilitating synaptic vesicle trafficking following high frequency stimulation.
Here, we provide evidence that mice with loss dysbindin-1 expression exhibit a decrease in glutamate release that may be underlie by decreases in the expression of L- and N- type Ca2+ channels, resulting in deficits in [Ca2+]i, abnormalities in synaptic vesicle priming and deficits in the replenishment of the ready releasable pool.
Methods
Animals
Studies were performed on mice carrying a large genomic deletion (exons 6–7; introns 5–7, Li et al., 2003) contained wholly within the DTNBP1 gene. We used mice that had been backcrossed to the C57Bl/6J background (Jackson Laboratories, Bar Harbor, Maine). All animals were genotyped as previously described (Jentsch et al., 2009). All the WT mice were littermates of the dys−/−mice. Male mice were used in the electrophysiological and molecular experiments described here; with the exception of the studies using FM 1–43 (for which the subjects were 20–30 days of age), all subjects were 45–60 days of age at the time of study. All experimental protocols were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee.
Electrophysiology
Brain slices (300 μm) were prepared from 10 dysbindin-1 wild-type (WT) and 13 null mutant (dys−/−) mice. Subjects were anesthetized with isoflurane (Abbott Laboratories). The brain was removed, and coronal slices containing the infralimbic and prelimbic PFC were cut at 300 μm thickness in ice-cold high-sucrose solution containing (in mM): sucrose, 200; KCl, 1.9; Na2HPO4, 1.2; NaHCO3, 33; MgCl2, 6; CaCl2, 0.5; D-Glucose, 10; ascorbic acid, 0.4. Slices were incubated at 33°C for at least 1 h before recordings; the incubation medium contained (in mM): NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 25; MgCl, 4, CaCl, 1, D-Glucose, 10; sucrose, 15; ascorbic acid, 0.4, aerated with 5%CO2/95%O2. After incubation, slices were transferred to a submerged chamber and superfused with oxygenated artificial cerebrospinal fluid (aCSF) (in mM): 125 NaCl, 2.5 KCl, 25 NaHCO3, 2.0 CaCl2, 1.3 MgCl2, 10 D-Glucose and 0.4 ascorbic acid at room temperature. Recordings were made using a Multiclamp 700B amplifier (Axon Instruments, CA), connected to a computer running Windows XP and Axograph X software. All recordings were obtained from pyramidal neurons in layers V or VI of the prelimbic or infralimbic cortex, identified using infrared-differential interference contrast optics and video-microscopy.
Voltage clamp
Picrotoxin (50 μM) was included in the perfusion solution to block GABAA receptors. For voltage-clamp recordings, electrodes (3–7 MΩ resistance in situ) were filled with a solution containing (in mM): 135 CsCl, 10 HEPES, 2 MgCl2, 1 EGTA, 4 NaCl, 2 Na-ATP, 0.3 tris-GTP, 1 QX-314, 10 phosphocreatine; 285 mOsmols. Series resistances (10–20 MΩ), and input resistances were continually monitored throughout the experiment via a −1 mV (100 ms) hyperpolarizing pulse. Pyramidal neurons were clamped at −80 mV. Electric stimulation was delivered via a bipolar concentric electrode positioned in layer II of the PFC. Evoked EPSCs (eEPSCs) were elicited via the stimulation electrode, and the amplitude of the eEPSCs was adjusted to 75% of the maximum amplitude. In order to deplete the RRP, we use a protocol consisting of 20 pulses (1 msec duration) at 40 Hz (delivered 30 times). Miniature EPSCs (mEPSCs) were measured after adding 1 μM TTX to the buffer solution. In order to control for a possible role for endocanabinoid release, depletion experiments were repeated using a minimal stimulation protocol. Briefly, the amplitude of the eEPSCs was adjusted to the minimum amount of current that elicited a constant amplitude eEPSC across 5 trials, then 20 pulses (1 msec duration) at 40 Hz (delivered 30 times) were applied.
Preparation of Synaptosomes
Infralimbic and prelimbic PFC (referred as PFC) tissue from three animals/genotype (unless otherwise indicated) was pooled together to make N=1 in each group. The tissue was homogenized with 10 strokes in a Potter homogenizer holding 5 ml of ice-cold isolation buffer containing 320 nM sucrose, 1 mM Na-EDTA, 10 mM Tris-HEPES (pH 7.4) and a protease inhibitor cocktail (Sigma, catalog # P8340). The homogenates were centrifuged at 600 g for 10 min to obtain a pellet fraction (P1) enriched in cell debris, intact cells and nuclei. The post-nuclear fraction (S1) was collected and centrifuged for 15 min at 9,200 g. The pellet was collected and washed by resuspension in Krebs buffer containing 125 mM NaCl, 5 mM KCl, 0.1 mM MgCl2, 1 mM CaCl2, 10 mM D-Glucose and 10 mM HEPES-NAOH, pH 7.4. After washing, the P2 pellet was resuspended in Krebs buffer, and protein concentration was determined by a Bradford assay (Bio Rad). The pellet was a crude synaptosomal fraction.
Loading of Synaptosomes with FM1-43
Synaptosomes were loaded with dye according to Meffert et al. (1994) with modifications. In brief aliquots of synaptosomes (0.4–0.5 mg of protein/ml) were resuspended in Krebs buffer and loaded with 5 μM of FM1-43 for 10 min at 30ºC, followed by the addition of 40 mM KCl for 1 min. Following loading, synaptosomes were pelleted by brief centrifugation followed by washing, repelleting, and resuspension in Krebs buffer containing 1mM CaCl2. Fluorescence measurements were carried out as described by Meffert et al (1994).
Determination of Synaptosomal [Ca2+]i
Synaptosomal fractions from PFC were incubated with 5 μM Fluo-3 and Fluo-4 and 4 μM Fura-2 (Molecular Probes) for 30 min at 37ºC (Yamaguchi et al., 1998). Samples were centrifuged at 10,000 g for 3 min, and pellets were resuspended with 200 μl of pre-warmed Krebs buffer. Fluo-3, Fluo-4 and Fura-2 loaded synaptosomes were placed in 96-well microplates (0.3 mg per well), and plates inserted in a fluorometer (Fluoroskan Ascent-Thermo Labsystems; Waltham, MA). [Ca2+]i was measured by determining the changes in the ratio (R) of fluorescence at 340 (F1) and 380 (F2) nm of excitation for Fura-2 and for Fluo-3 and Fluo-4 measured at 488 nm of excitation, with an emission cut-off of 510 nm. Synaptosomal [Ca2+]i was calculated according to the formula [Ca2+]i = Kd. B(R Rmin)/(Rmax R), using 0.4% Triton X-100 and 7.5 mM EG pH 8.0 to calculate Rmax and Rmin values, respectively.
FM 1-43 staining and destaining
PFC slices (250 μm) from 7 WT and 9 dys −/− mice were labeled with FM1-43 (8 μM; Invitrogen) for 5 min in aCSF containing CNQX (10 μM). They were transferred to CNQX + FM1-43 in high [K+]0 (45 mM) and 2 mM Ca2+ for 15 min to stimulate uptake of FM1-43 via endocytosis of vesicles. All labeling and washing protocols were performed in the presence of CNQX to prevent synaptically-driven action potentials from accelerating dye release. After loading, slices were washed for 10 min prior to imaging. After taking basal images, slices were washed for 15 min in dye-free aCSF containing 100 μM sulforhodamin-101 and scanned. Depolarization-dependent de-staining was obtained by application of 90 mM K+. All FM1-43 experiments were performed using a Zeiss LSM 510 confocal laser-scanning microscope with a 40 X objective. Images were captured after FM1-43 uptake, after application of the sulforhodamine (S-Rhd) quenching and again after 15 min of high K+ depolarization. The filtering strategy used an emission filter with a narrow band pass at 540 ± 20 nm, a range of wavelengths over which FM1-43 emits but S-Rhd does not. Each image was 1024 × 1024 pixels. To monitor exocytosis, the brightness of single cells (containing clusters of synaptic vesicles) was quantified during all destaining or at specific time intervals.
Immunoblotting
Isolated synaptosomes were lysed in 0.1 M phosphate buffer (pH 7.2) containing 0.1% sodium dodecyl sulfate, 1% IGEPal, 1% protease, and 1% phosphatase inhibitor cocktails (Sigma), and centrifuged at 14,000g for 10 min at 4°C. Aliquots of supernatant (40–50 μg of protein) were separated on NuPAGE 4–12% Bis-Tris gels and 3–8% Tris-Acetate gels (Invitrogen Inc., Carlsbad, CA) and transferred onto nitrocellulose membranes, immunoblotted overnight at 4°C with either rabbit polyclonal antibodies to SNAP-25 (1:1,000, Abcam cat. # 53723), snapin (1:1,000 Abcam cat. # 102710), syntaxin-1a pS14 (1:500 Abcam Cat. # 663574), synapsin 1 (1:15,000, Epitomics, Inc cat. # 3077), synapsin 2 (1:5,000 Abcam cat. # 13258), Cacna1b (1:400, Alomone Labs, Cat. # ACE-003), Cacna1c (1:300, Alamone Labs (cat # ACE-002) and mouse monoclonal antibodies to synaptotagmin-I (1:1,000, Abcam cat.# 77314), syntaxin 1a (1:2,000 Abcam cat # 78539), actin (1:2,000) and AP3 (Developmental Studies Hybridoma Bank from Univ. of Iowa, 1:1,000). Horseradish peroxidase-conjugated secondary antibodies at a concentration of 1:5,000 (Abcam) were applied, and detection performed using chemiluminescence (Pierce Biotechnology Inc., Rockford, IL). The immunoblotting experiments were performed four times/antibody and were quantitatively analyzed using software (Imaging Station, Carestream Health, Inc, Rochester, NY).
Statistical Analysis
Protein analysis
Groups were compared using analysis of variance plus Student-Newman-Keuls post hoc test. Data shown are mean ± SEM. Numbers of animals were 4 per group. Significance was set as p<0.05. When only two groups were compared Student-t-test were used (p< 0.05).
Electrophysiology
All figures present data as mean ± SEM. The cumulative probability was calculated from the average of 6 neurons in the WT mice and 5 neurons in dys−/− mice. Nq was calculated by fitting the amplitude of the last 6 eEPSCs with a linear regression extrapolated to zero time. Probability of release (Pr) was calculated from the amplitude of the first eEPSC/Nq. N =releasable quanta, q= quantal size.
Results
Loss of dysbindin-1 expression causes decreased release probability and numbers of vesicles
In order to investigate the origins of the lower glutamate release reported in dys −/− (Chen et al., 2008; Jentsch et al., 2009), we used high frequency stimulation (Dobrunz and Stevens, 1997; Schneggenburger et al., 1999; Taschenberger et al., 2002) to assess the number of ready releasable quanta (Nq). The method is based on the premise that high frequency stimulation-induced depression depends upon depletion of the ready releasable pool (RRP) of vesicles and that this can be estimated by calculating the cumulative amplitude of eEPSCs over time intervals that are shorter than the time required for recovery from depression. Figure 1A shows that pyramidal cells recorded in PFC brain slices of dys −/− mice (n=7 neurons; 5 animals) exhibit smaller amplitude eEPSCs (WT: 74.5 ± 4.9 pA; dys −/−: 35.1 ± 2.9 pA, t-test p< 1.8 × 10−7) and a faster depletion of the RRP, compared to WT mice (n=7 neurons; 6 animals, lineal regression WT: y=28844.7; dys −/− y=1291.9, Fig 1B, t-test p= 6.4 × 10−14). Dys −/− mice exhibit a an apparent decrease of 22 % in the size of the RRP of synaptic vesicles (Nq): 1.4 ± 0.74 pA (n=6 neurons) vs 1.1 ± 0.8 pA (n=6 neurons) for dys−/− and WT mice respectively, (Fig 1C). Moreover, dys −/− mice have a lower probability of release (Pr) (36% lower; WT mice: 0.26 ± 0.06; dys−/− mice 0.16 ± 0.05, Fig 1D (n=7 neurons/genotype)), a phenomenon that depends upon the number of synaptic vesicles in the RRP (Deng et al., 2010). Results for individual cells are depicted in Figures 1C (Nq) and 1D (Pr).
Figure 1.
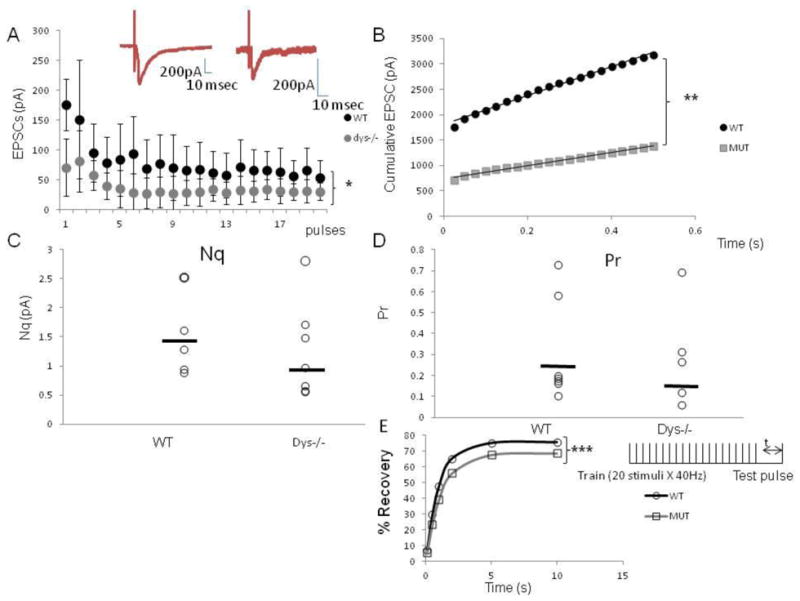
Dy−/− mice exhibit a smaller RRP size and a lower Pr in PFC brain slices as shown with a maximum stimulation protocol. A) Average amplitude of eEPSc for 6 dysbindin-1 WT cells and 5 dys −/− cells in response to 20 stimuli at 40 Hz. The stimulus intensity was adjusted at 75% of the maximum before the train of stimulation was delivered. The amplitude of each eEPSC was measured by resetting the baseline each time at a point within 0.5 ms before the stimulation artifact. The dys−/− mice show signficantly smaller amplitude EPSCs; insert shows representative traces of eEPSCs, (note the difference in scale) B) Cumulative amplitude histogram of eEPSCs. For each group (WT, dys−/−) the eEPSCs amplitudes were fit with a linear regression to estimate the ready releasable pool size (Nq). Dys −/− mice exhibit a significant decrease in average Nq (WT: 2844.7; dys −/−:1291.9). C) Plot showing the individual values of Nq for each recorded neuron, in average dys −/− mice show an apparent decrease by 22% in Nq (WT: 1.4 ± 0.7 pA; Dys −/−: 1.1 ± 0.82 pA), D) Plot showing probability of release. For each cell, Pr was calculated as the ratio of the first eEPScs amplitude divided by its Nq. On average, dys −/− mice have a smaller probability of release (36% lower; WT mice: 0.26 ± 0.06; dys−/− mice 0.16± 005), E) Time course of recovery of the RRP expressed in percentage. The % of recovery was calculated by %R= (Amp test pulse-Average of steady state)/(amp of first EPSCS in train-Amp of steady state) X 100. Dys −/− mice show a slower recovery of the RRP (WT=1.004 s; Dys −/− =1.179s). T-test, * p< 1.8 × 10−7, ** p< 6.4 × 10−14, *** p< 0.001.
The kinetics of the mEPSCs in dys−/− mice were normal: rise time (WT: 2.94±0.1 msec; dys−/−: 3.16 ±0.04 msec), decay time (WT: 5.8 ± 0.9 msec; dys−/−: 6.0 ± 0.84 msec) and area under the curve (WT: 59.8±17.3; dys−/−: 41.1 ± 0.15). We also measured the rate of recovery from vesicle depletion following high-frequency stimulation (Fig 1E). The time course of the recovery after the 40 Hz train was fitted with a single exponential function, resulting in a time constant for WT mice of 1.004-s and for dys−/− mice of 1.179-s (t-test p< 0.001). The results indicate that dys−/− mice have a slower recovery of the RRP.
Using the minimal stimulation protocol (Dobruns and Stevens, 1997, Fig 2), we confirmed the results obtained with the stronger stimulation protocol. Dys −/− mice exhibited smaller amplitude eEPSCs (WT: 54 ± 0.22 pA; dys−/−: 23 ± 0.04 pA, t-test, p< 1.0×10−10, Fig 2A) and a faster depletion of the RRP (linear regression WT y=21922vs dys −/− y=7311.5, t-test p=2.3 × 10 −5 Fig 2B). Dys −/− mice showed an apparent decrease of 58% in Nq (dys−/− mice: 0.5 ± 0.21 pA, Fig 2C; WT mice: 1.2 ± 1.5 pA). Moreover, on average, dys −/− mice have an apparent lower probability of release (16.6% lower; WT mice: 1.2± 0.48; dys−/− mice: 1.0 ± 0.5, Fig 2D). The amplitude of mEPSCs was significantly decreased in dys −/− mice (WT n=10 neurons, 5 mice: 15.8 ± 1.4 pA vs dys−/− n=16 neurons, 7 mice 12.7 ±0.8 pA n=10, F(2,33) =2.2 p=0.04 Fig. 2E), evidence in support of a reduction in quantal size. Moreover, the frequency of mEPSCs was also significantly decreased (WT: 0.6 ± 0.1 Hz, vs dys−/−: 0.3 ± 0.03 Hz, F (2,33)=4.1, p=0.01, Fig 2E), supporting a decrease in release probability
Figure 2.
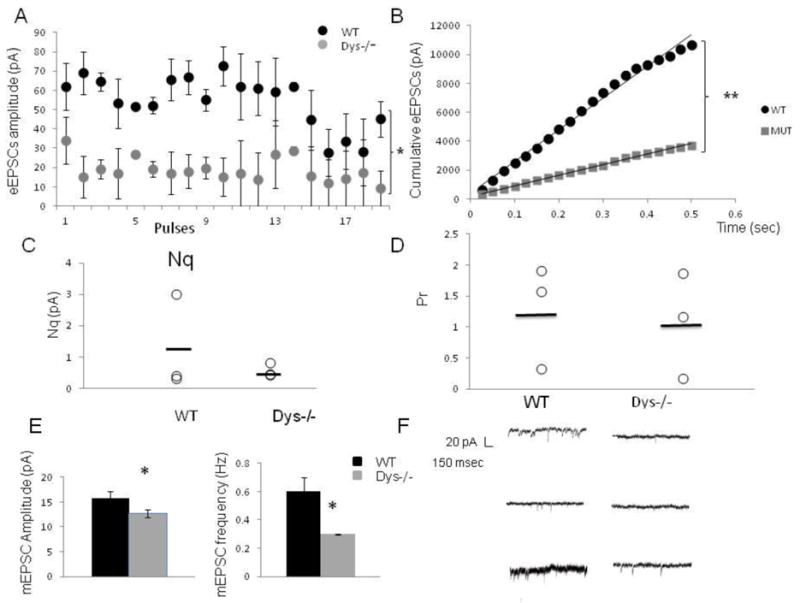
Minimal stimulation protocol. A) Average amplitude of eEPSc for 3 WT and 3 dys −/− cells in response to 20 stimuli at 40 Hz using minimal stimulation. The amplitude of each eEPSC was measured by resetting the baseline each time at a point within 0.5 ms before the stimulation artifact. The stimulus intensity was adjusted at the minimum amount of current that elicited a reliable responses 5/5 times before delivering the train of stimulation. The dys −/− mice show smaller amplitude EPSCs, (WT: 5.4 ± 0.22 pA; dys −/−: 2.30 ± 0.4 pA t-test, *p< 1.0 × 10−10), B) Cumulative amplitude histogram of eEPScs. For each group (WT, dys−/−) the eEPSCs amplitudes were fit with a linear regression to estimate the ready releasable pool size (Nq). Dys −/− mice exhibit a significant decrease in average Nq (WT: 21922; dys −/−:7311.5, p< 2.3 × 10−5) C) Plot showing the individual values of Nq for each recorded neuron, in average dys −/− mice show an apparent decrease of 58 % in Nq (WT: 1.2 ± 1.5 pA; dys −/−: 0.5 ± 0.21 pA. D) Plot showing probability of release. For each cell, Pr was calculated as the ratio of the first eEPScs amplitude divided by its Nq. On average, dys −/−mice have an apparent smaller probability of release (16.6% lower; WT mice: 1.2 ± 0.48; dys −/−mice 1.0 ± 0.5), E) Average amplitude of mEPSCs, dys −/− mice exhibit a significant decrease in the amplitude of mEPSCs (WT:15.8 ± 1.4 pA; dys −/−: 12.7 ± 0.8 pA p=0.04). Also, histogram showing the significant decrease in frequency of mEPSCs (WT: 0.6 ± 0.1 Hz, dys−/−: 0.3 ± 0.03 Hz, p=0.01). F) Representative traces showing the significant decrease in frequency of mEPSCs.
Furthermore, we investigated the size of the RRP using FM1-43 imaging in PFC slices (WT n=15 cells, 7 animals and dys −/− n= 17 cells, 9 animals). FM1-43 dye is much more fluorescent when partitioned in membranes, so dye release from synaptic vesicles can be followed as a decrease in fluorescence (Betz et al., 1992). This protocol is unable to discriminate glutamatergic from GABAergic synapses, but gives insights into the changes in vesicle dynamics after loss of dysbindin-1. To estimate the amount of exocytosis induced by depolarization, the magnitude of the fluorescence decrease after KCl addition was calculated and normalized to that occurring in WT animals. Figure 3A shows the basal loading of FM1-43. Following K+ depolarization, cortical slices labeled with FM1-43 display a nonspecific background staining that partially or completely obscures fluorescence arising from synapses. The addition of S-Rhd markedly reduces the overall fluorescence (Fig. 3B). The fluorescence signal emitted by clusters of synaptic vesicles loaded with FM1-43 reflects rates of synaptic vesicle exo- and endocytosis. Following 15 min of depolarization, we found a decrease of 43% FM1-43 uptake in the PFC of dys −/− mice (p<0.001, Fig. 3C-G), suggesting a reduction in the number of synaptic vesicles. The background level of fluorescence remaining after S-Rhd in WT and dys −/− mice was similar. However, caution have to follow the interpretation of these results as it is possible that changes in dye uptake are occurring in other domains of the neurons and may not necessary represent synaptic changes.
Fig 3.
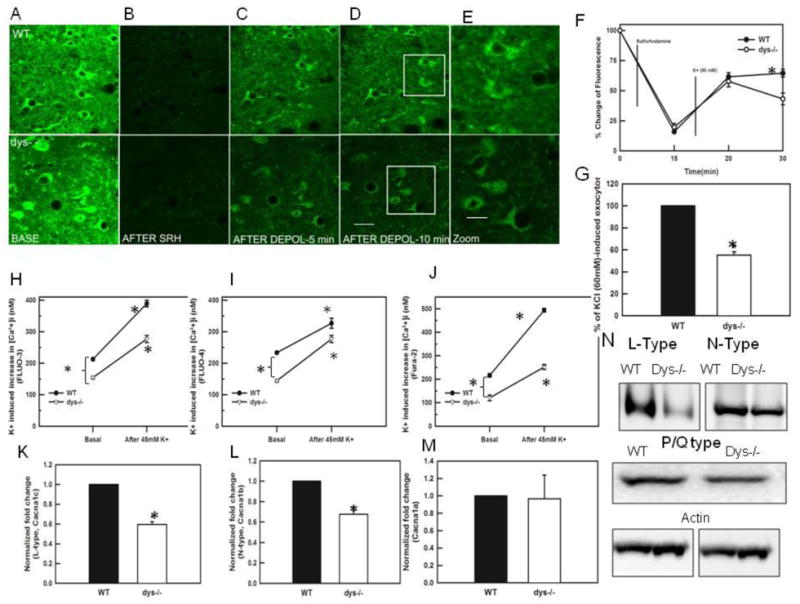
FM1-43 labeling of PFC slices in dysbindin-1 WT and dys −/−mice. A) PFC brain slices 250 μm thick from young animals were loaded with FM1-43 (8 μM) for 15 min, washed 10–15 minutes with perfusion buffer and imaged immediately B) the slices were perfused with sulforhodamine (SRH100 μM) for 15 min. The picture shows the representative quenching of the FM1-43 fluorescence, revealing the underlying synaptic labeling. C-D) Time course showing the loss of FM 1–43 fluorescence during high K+ (90 mM) depolarization. A significant decrease in labeling between WT and dys −/− mice can be observed after 15 minutes of depolarization with high K+ (total time =30 min). Scale bar = 50 μm, E) High magnification (400 X) of the inserts in D, showing the FM1-43 labeling in dysbindin-1 WT and dys −/− mice. (Scale bar = 20 μm). F) Graph showing the pooled data (WT n=15 cells, 7 animals and dys −/− = 17 cells, 9 animals) illustrating changes in FM1-43 fluorescence content. After 15 minutes of depolarization with high K+ there was significant decrease (p<0.001) in the fluorescence in dys −/− PFC. The abscissa axis depicts total times in minutes G) Flourometric quantification of synaptosomal FM1-43. The graph shows that dys −/− mice exhibit a significant decrease (p< 0.001) in % of KCl (60 mM)-induced exocytosis (n=4 animals/genotype) 15 minutes after the depolarization. H) graph showing the change in synaptosomal [Ca2+]i measured with FLUO-3, dys −/− mice show significantly lower basal levels of [Ca2+]i (*p< 0.008), following depolarization with 45 mM K+, the [Ca2+]i levels increased in WT significantly (*p=0.001) as well as in dys −/− mice (*p=0.001), I) Graph showing the change in synaptosomal [Ca2+]i measured with FLUO-4. Dys −/− mice exhibits significant decreases in basal levels (*p<0.001). High K+ stimulation increased significantly the [Ca2+]i levels in dys −/− mice (*p< 0.002) and WT mice (* p <0.006). J) We also assessed [Ca2+]i levels using the radiometric indicator (Fura-2) in the synaptosomal preparation. Dys −/− mice showed significantly lower baseline values (WT: 216.11 ± 7.27; dys −/−: 120.4 ±12.02, *p<0.001, Fig. 3J). Following depolarization with 45 mM K+, dysbindin-1 WT and dys −/− mice show a 228% and 209% increase in Ca2+ levels respectively (WT: 228%, 493.4 ± 7.70 dys −/− 251.8 ± 103, * p< 0.001. K) Using Western Blots to measure the levels of L-type Ca2+ channel protein, we found a significant decrease in dys −/− mice (* p< 0.02), L) Western Blots assessing levels of N-type Ca2+ channels lassos show a significant decrease in in dys −/− mice (*p<0.001). M) Western Blots did not show changes in the levels of P/Q–type Ca2+ channels between WT and dys−/− mice. N) Western blots showing the decreases in N- and L-type Ca2+ channels in dys −/− mice.
In summary, dys −/− mice exhibit decreases in the size of the RRP, decreases in the probability of release, decreases in quantal size, a slower recovery of the RRP and decreases in the rate of synaptic vesicle endocytosis.
Absence of dysbindin-1 expression affects calcium signaling
The result showing slower recovery of the RRP and decreases in probability of release in the dys −/− mice suggested that Ca2+-related mechanisms may be affected in the dys −/− mice. Therefore, we investigated synaptic [Ca2+]i using quantitative Ca2+ imaging with Fluo- 3 and Fluo-4 in a synaptosomal preparation. Using Fluo-3, we found that the basal levels of [Ca2+]i in dys −/− mice are significantly decreased (WT:212.6 ± 4.18 nM; dys−/−: 154.3±3.7 nM, F(3,4)=14.8 p=0.008, Fig 3H). After depolarizing the synaptosomes with 45 mM K+, dysbindin-1 WT and dys −/− mice respectively exhibit 183% and 179% increases in [Ca2+]i (WT baseline: 212.6 ± 4.18 nM; K+: 389.1 ± 10.4nM, p< 0.001 vs dys−/−: 154.3 ±1.7; K+: 276.7 ± 11.5 nM, p= 0.001, Fig 3H). Similar results were found using Fluo-4 at baseline (233.3 ± 1.4 nM vs dys −/−: 144.2 ± 3.79 nM F(3,4)=59.6, p< 0.001) and at 45 mM K+ (326.7± 15.7 for WT vs dys−/−: 276.7 ± 11.5 p=0.002, Fig 3I). In average, we found that dys −/− mice exhibited 27% and 38% decreases in [Ca2+]i respectively when compared with WT mice using either Fluo-3 or Fluo-4. Furthermore, in order to assess [Ca2+]i levels in a more quantitative manner, we used a radiometric indicator (Fura-2) in the synaptosomal preparation. Again, the dys−/− mice showed significantly lower baseline values (44% lower. WT: 216.11 ± 7.27 vs dys −/−: 120.4 ±12.02, F(3,8)= 276.5 p< 0.001, Fig. 3J). Following depolarization with 45 mM K+, dysbindin-1 WT and dys −/− mice show a 228% and 209% increase in Ca2+ levels respectively (WT: 228%, 493.4 ± 7.70 dys −/− 251.8 ± 103, F(3,8)= 276.5 p< 0.001, Fig. 3J). These results suggest that in dys −/− mice the [Ca2+]i levels are significantly decreased.
After finding decreases in [Ca2+]i, we assessed the levels of proteins for P/Q, N- and L-type Ca2+ channels using Western blots. Dysbindin-1 null mice exhibit a significant decrease in N-(F(1,6)=69.6, p<0.001, results from 4 Western blots each containing pooled PFC of 3 animal/genotype) and L-type channels (F(1,6)=216.3, p< 0.001, Fig 3K-L, results from 4 Western blots each containing pooled PFC of 3 animal/genotype), but we did not find significant changes in the protein levels for P/Q- type Ca2+ channels (Fig 3M). Figure 3N shows representative Western Blots for L, N and P/Q-type Ca2+ channels.
In order to control for changes in Ca2+ buffering between WT and dys −/− mice we assessed the protein levels of parvalbumin (Fig. 4A1). It was found that dys −/− mice have significant lower levels of parvalbumin (F (1,6)= 181.1, p<0.010). These results suggest that the reduction in [Ca2+]i found in dys−/− mice is not due to an increase in buffering.
Figure 4.
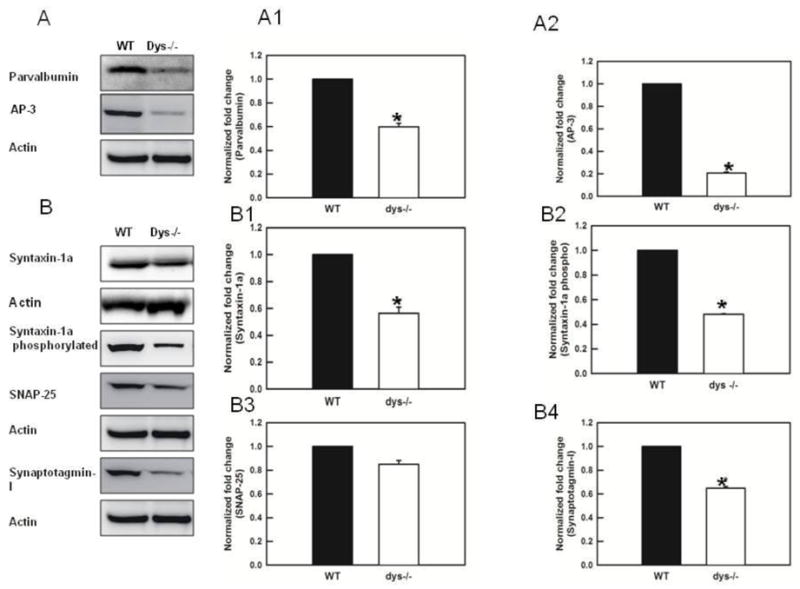
Dysbindin-1 deletion decreases significantly the levels of parvalbumin, AP3, syntaxin 1, syntaxin 1 pS14 and synaptotgamin-1 measured in synaptosomes of the PFC. A) Blots and histogram showing significant decreases in parvalbumin (*p< 0.001) in dys −/− mice. This result indicates that decreases in [Ca2+]i in dys −/− mice are not due to an increase in buffering. A2) Western blots show a signficnat decrease in the levels of AP3 in dys −/− mice (p< 0.01) B) Western blots showing the changes in proteins assessed in dys −/− mice. B1) Quantification of expression levels of syntaxin 1a. Dys −/− mice expressed a significant reduction the levels of syntaxin 1a (p< 0.001), B2) Levels of syntaxin1 pS14 are also significantly decreased in dys −/− mice (p< 0.001). B3) Levels of SNAP-25 were not altered in the dys −/− mice. B4) Levels of synaptotagmin 1 were significantly decreased (p< 0.001) in dys −/− mice.
In summary, our results suggest that dys −/− mice have significant decreases in [Ca2+]i and significantly lower levels of N- and L-type Ca2+ channels, suggesting that decreases in Ca2+ may be involved in the deficits found in Pr and replenishment of the RRP.
Alteration in proteins involved in synaptic vesicle trafficking and priming
Our results in PFC slices show that dys −/− mice have a smaller RRP and slower recovery of the RRP. Therefore, using a synaptosomal preparation we explored the levels of proteins involved in vesicle trafficking from the reserve pool to the RRP (synapsin I and II), the adaptor protein AP3 and the synaptic vesicle priming proteins involved in the RRP syntaxin 1 pS14 and snapin 1.
We found significant decreases in the levels of the adaptor protein AP-3 in the dys −/− mice (20.6 ± 1.6% decrease; F(1,4)=7353.4, p< 0.001, results from 3 Western blots each containing pooled PFC of 3 animal/genotype, Fig 4A2).
Syntaxin 1 interacts with N-type calcium channels (Chapman et al., 1995) and synaptotagmin-1 (Bennett et al., 1992). In the RRP, this complex effectively brings vesicle membranes into close proximity with presynaptic membrane release sites, providing both the temporal and spatial resolution needed for fast neurotransmission (Yoshida et al., 1992). The expression levels of syntaxin 1 were significantly lower in the dys−/− mice (56 ± 6% decrease, F(1,2)=95.7, p< 0.01, Fig 4B1, results from 3 Western blots, each blot contains the pooled PFC of 3 animals/genotype). Moreover, when levels of the syntaxin 1 pS14 were measured it, it was found that the phosphorilated protein was also significantly lower in dys−/− mice (48.1 ± 4.9% decrease, F(1,4)=3243.2, p<0.001, Fig 4B2, results from 3 Western blots each containing pooled PFC of 3 animal/genotype) and the ratio of syntaxin 1 pS14/syntaxin 1 was also reduced (p=0.024). On the other hand, levels of SNAP 25, a protein part of the tSNARE complex that binds syntaxin 1, were not altered (Fig 4B3, results from 3 Western blots each containing pooled PFC of 3 animal/genotype).
The expression levels of synaptotagmin-1 were also significantly different in dys −/− mice (64.9 ± 2.3% decrease, F(1,4)=652.4, p< 0.001, Fig 4B4, 2 blots, each blot contains the pooled PFC of 3 animals/genotype). Synaptotagmin −1 is a protein that plays an important role in priming synaptic vesicles for release (Chapman 2008; Chicka et al., 2008).
The trafficking of synaptic vesicles from the reserve pool to the RRP is mediated by the accumulation of Ca2+ inside presynaptic terminals. This Ca2+ accumulation leads to the phosphorylation of synapsin I and II by CaM kinase II at low frequency stimulation (Benfenati et al., 1990) or by MAPK at both low and high frequency stimulation (Chi et al., 2001, 2003). The phosphorylation of synapsins decreases the affinity of synaptic vesicles for actin (Schiebler et al., 1986), allowing them to move to the docking sites for membrane fusion (Greengard et al., 1993). We found that levels of both synapsin I and II were significantly decreased in dys −/− mice (60.9 ± 10% decrease, F(1,4)=4031.9, p< 0.001; 69.8 ± 8.1% decrease, F(1,8)=68.0, p<0.01 respectively, Fig 5A1-A2 results from 3 Western blots each containing pooled PFC of 3 animal/genotype).
Fig 5.
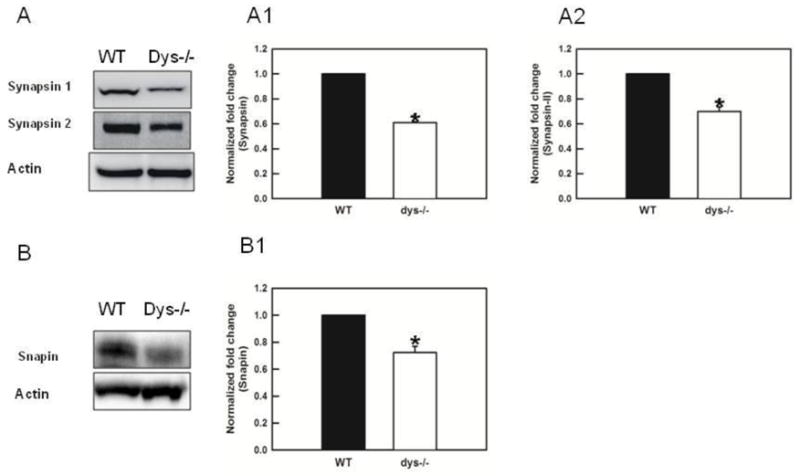
Loss of dysbindin decreases the levels of synapsins and snapin. A) Representative Western blots. A1) Levels of synpasin I are significantly decreased in dys −/− mice (p< 0.001). A2) Levels of synapsin II were also significantly decreased in dys −/− mice (p<0.001). B) Western blots for snapin. B1) Levels of snapin are significantly decreased in dys −/− mice (p< 0.02).
Measures of snapin, a component of the BLOC-1 show a significant decrease in dys −/− mice (72.2 ± 6% decrease, F(1,2)=33.6 p=0.028, Fig 5B1, results from 2 Western blots each containing pooled PFC of 3 animal/genotype). Levels of snapin have been shown to be reduced in dys−/− mice previously by Feng and colleagues (2008, (16)).
In summary, dys −/− mice exhibit significant decreases in several proteins linked to movement of synaptic vesicles to the RRP and in proteins linked to priming of synaptic vesicles. Of note is that the low sample size for some experiments, as well as the relatively small number of mice used in some measures warrant replication of the findings presented here.
Discussion
Levels of the protein dysbindin-1 have been found to be reduced in synaptic tissue of schizophrenia patients, including the PFC (Talbot et al., 2004, 2011; Tang et al., 2009). More importantly, dysbindin-1 has been suggested to play an important role in the negative symptoms and cognitive deficits present in this disorder (Maher et al., 2010; Tang et al., 2009; Talbot et al., 2004). Several authors have demonstrated that decreases in dysbindin-1 reduce glutamate release (Chen et al., 2008; Jentsch et al., 2009; Numakawa et la., 2004), and dysbindin-1 deficient mice exhibit associated deficits in working memory tasks (Jentsch et al., 2009).
Our results indicate the PFC neurons in dys −/− mice have a smaller RRP, smaller quantal size, a small probability of release and a slower recovery of the RRP. These deficits may contribute to the decrease in glutamate release and are consistent with data already published (Chen et al., 2008). Our results also reveal what may be the molecular basis for the deficits in synaptic glutamate release in mice lacking dysbindin-1, namely significant decreases in L- and N-type Ca2+ channels, basal and evoked [Ca2+]i and proteins engaged in synaptic vesicle recruitment to the active zone and in priming of active zone vesicles for release. These results are truly novel. Moreover, our findings of deficits in release-regulating mechanisms in dys −/− mice may point to a common deficiency at other synapses and preliminary results show that dys −/− mice have a significant reduction in frequency of mIPSCs.
Previous experiments by Chen and colleagues (2008) demonstrated that dys−/− mice on the original background (DBA/2J) exhibit a decrease in the number of large density core vesicles and a decrease in the RRP in chromaffin cells. Our experiments in C57B1/6J mice demonstrate that dys −/− mice exhibit decreases in the rate of exo- and endocytosis of synaptic vesicles in the PFC.
An adequate [Ca2+]i is needed for the correct functioning of synaptic vesicle dynamics, so we assessed [Ca2+]i. A difference from the findings of Dickman and Davis (2009) that have shown that dysbindin in the fruit fly is critical for homeostatic modulation of neurotranmission but independent of Ca2+ influx, we found that dys −/− mice have lower presynaptic calcium content. It is possible that the decreases in [Ca2+]i found in dys −/− mice were due to an increase in Ca2+ buffering, thus we assessed levels of parvalbumine (PV). We found that dys −/− mice have a significant reduction in PV. This result together with the report from Carlson and colleagues (2011) showing that dysbindin-deficient mice have a decrease in the levels of PV but not a reduction in PV cells suggest that dys −/− mice do not have increases in Ca2+ buffering.
Synaptic vesicle trafficking and priming of vesicles depend on the interactions of a number of proteins. For synaptic trafficking, synapsin I and II are critical proteins highly dependent on Ca2+ for phosphorylation. Numakawa and colleagues (2004) reported changes in the levels of synapsin I in neuronal cultures following dysbindin-1 knockout or overexpression, and post mortem studies have shown a reduction in synapsin proteins in brains from schizophrenia patients (Vawter et al., 2002, but see Talbot et al., 2004). Our results confirm that in dys −/− mice, there is a significant decrease in the levels of synapsins I and II.
Furthermore, we found significant reductions in the levels of syntaxin1 pS14, and syntaxin1 levels. Syntaxin-1 is a key protein in ion channel regulation (Caterall, 2000) and synaptic exocytosis. Syntaxin 1 phosphorylation can occur by casein kinase 2 (CK2), and phosphorylation of syntaxin enhances its interaction with synaptotagmin (Risinger and Bennett, 1999). We cannot discard the possibility that the decreases in syntaxin 1 pS14 found in dys −/− mice are due to a reduce activity of CK2, as we did not measured it. Interestingly, it has been shown that CK2-mediated phosphorylation of syntaxin 1 is deficient in schizophrenia. This decrease in phosphorylation in turn could affect the binding of syntaxin 1 to its protein partners and result in abnormal neurotransmitter release and synaptic transmission (Castillo et al., 1999; Sheng et al., 1994). Again, the decreases in [Ca2+]i found in dys−/− mice could underlie the decreases in phosphorylation of syntaxin 1 and thereby decrease it interaction with synaptotagmin.
We also found significant decreases in the levels of protein for N- and L-type channels in dys −/− mice, but not P/Q-type, suggesting a mechanism for the decreases in [Ca2+]i. These results are supported by the decreases in levels of syntaxin 1 and synaptotagmin 1, proteins that interact with N-type Ca2+ channels (Chapman, 2008; Sheng et al., 1994; Yoshida et al., 1992). Synaptotagmin 1 is thought to serve as a calcium sensor for neurotransmitter release.
We did not find changes in the levels of SNAP 25, another t-SNARE protein. However the variability in our dys −/− mice was very high, probably obscuring significant differences. Similarly, Chen and colleagues (2008) also found normal level of SNAP-25 in dys−/− mice in the hippocampus.
One common link to all our results is Ca2+ dependence. Therefore, we propose that mice that exhibit loss of dysbindin 1 also show decreases in [Ca2+]i and this results in deficits in trafficking and priming of synaptic vesicles, suggesting a mechanism that mediate the decreases in glutamate release reported in the dys −/− mice (Chen et al., 2008; Jentsch et al., 2009). We found that both genotypes showed similar percentage of increase in [Ca2+]i following depolarization with high K+. This finding may provide support to the idea that the existent Ca2+ mechanisms are intact, but the decrease in the number of Ca2+ channels in the dys −/− mice have a powerful impact in [Ca2+]i.
Dysbindin 1 is part of the BLOC-1 complex and several authors, (Ghiani et la., 2010; Starcevic and Dell’Angelica, 2004) have demonstrated that loss of dysbindin is accompanied by loss of all the BLOC-1 members, therefore, our results could be interpreted as consequence of deficiencies in the BLOC-1 complex rather than only from the loss of dysbindin. Indeed, many of our results are similar to the findings in snapin muted mice (Pan et al., 2009; Tian et al., 2005) and we found a significant decrease in the levels of snapin in the dys −/− mice, suggesting that the role of dysbindin is just to stabilize the BLOC-1 complex. This suggest the possibility that BLOC-1, and not their isolated proteins, is an important player in the cognitive deficits observed in several neuropsychiatric disorders. The role of BLOC-1 in synaptic trafficking could also explain the mechanisms mediating the reduced levels of Ca2+ channels observed in the dysbindin-deficient mice.
Imaging experiments in living sensory neurons show that, within seconds of receptor activation, calcium channels are cleared from the membrane and sequestered in clathrin-coated vesicles. Khanna and colleagues (2007) have proposed that the N-type Ca2+ channels form an endocytotic complex together with SNAP 25, matrix proteins, AP 180 and intersectin. This endocytotic complex, in turns interacts with clathrin proteins that coat the complex in order to begin the endocytosis process. Coat recruitment is mediated by adaptor proteins like AP2 and AP3 (Danglot and Galli, 1998). Several authors (Newell-Litwa et al., 2010; Pan et al., 2009) have shown that the presence of BLOC-1 complex is linked to adaptor proteins. Indeed, interactions of AP3 with BLOC-1 greatly facilitates trafficking of proteins and in cells lacking BLOC-1 or AP3, proteins are miss targeted, thus, it has been proposed that BLOC-1 facilitate AP3 recognition of specific cargos (Newell-Litwa et al., 2009) decreasing rapid degradation of the cargo. As loss of dysbindin elicits loss of BLOC-1 (Ghiani et al., 2010), the normal interactions between BLOC-1 and AP3 are disrupted. We propose that in dysbindin deficient animals the N-type Ca2+ channel endocytotic complex is not coated due to the loss of BLOC-1 and AP3, thereby is miss targeted during endocytosis and is degraded instead of recycled, accounting for the decrease in Ca2+ channels. This loss of Ca2+ channels in turn decreases [Ca2+]i. Furthermore, disruption of BLOC-1 could disrupt AP3 mediated generation of reserve pool vesicles from which vesicles can later be recruited to the RRP.
It has been proposed that schizophrenia is mainly a synaptic disorder (Frankle et al., 2003; Jentsch and Roth, 1999) and our results show that synaptic mechanisms mediating mobilization and priming of synaptic vesicles are altered in dys −/− mice.
Conclusion
By understanding the neurobiological functions for each of the proteins encoded by the genes involved in schizophrenia risk and the consequence of the functional mutations that associate with schizophrenia phenotypes, it is hoped that a convergent theory of cellular and network dysfunction in schizophrenia can be elucidated. Mechanistically, the data here provide a broader molecular explanation of the mechanisms of action of dysbindin. Decreased expression of dysbindin-1 has been shown to decrease the activity- dependent release of glutamate from pre-synaptic terminals (Chen et al., 2008; Jentsch et al., 2009). Here we show that this phenomenon is potentially of exceptional importance because demonstrate that one of the possible mechanisms of action of dysbindin may be exerted through a set of glutamate-dependent cellular and network processes already heuristically linked to schizophrenia (Harrison and Weinberger 2005).
The precise understanding of the molecular interactions and cellular functions engaged by molecules implicated in schizophrenia, such as dysbindin-1, will shed light into mechanisms that when impaired contribute to disease pathogenesis. However, it is likely that multiple cellular processes in different combinations may be affected in order to trigger schizophrenia. Thus, it is essential to expand our knowledge of multiple molecular candidates of disease susceptibility.
Acknowledgments
Funding source: This research was funded by PHS Grants MH-83269 (TC, JDJ, AL).
Footnotes
Conflict of Interests:
The authors do not have any conflict of interest to disclose
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, Geyer M, Tsuang MT, Salomon D, Schork NJ, Fanous AH, O’Donovan MC, Niculescu AB. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Neyroz P, Bähler M, Masotti L, Greengard P. Time-resolved fluorescence study of the neuron-specific phosphoprotein synapsin I. Evidence for phosphorylation-dependent conformational changes. J Biol Chem. 1990;265(21):12584–12595. [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257(5067):255–9. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS, Ridge RM. Intracellular movements of fluorescently labeled synaptic vesicles in frog motor nerve terminals during nerve stimulation. Neuron. 1992;9(5):805–813. doi: 10.1016/0896-6273(92)90235-6. [DOI] [PubMed] [Google Scholar]
- Castillo MA, Ghose S, Tamminga CA, Ulery-Reynolds PG. Differential phosphorylation of syntaxin and synaptosome-associated protein of 25 kDa (SNAP-25) isoforms. J Neurochem. 1999;72(2):614–24. doi: 10.1046/j.1471-4159.1999.0720614.x. [DOI] [PubMed] [Google Scholar]
- Carlson GC, Talbot K, Halene TB, Gandal MJ, Kazi HA, Schlosser L, Phung QH, Gur RE, Arnold SE, Siegel SJ. Dysbindin-1 mutant mice implicate reduced fast-phasic inhibition as a final common disease mechanism in schizophrenia. Proc Natl Acad Sci U S A. 2011;25;108(43):E962–70. doi: 10.1073/pnas.1109625108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterall WA. Structure and function of neuronal Ca2+ channels and their role in neurotransmitter release. Cell. 1998;24:307–323. doi: 10.1016/s0143-4160(98)90055-0. [DOI] [PubMed] [Google Scholar]
- Catrerall WA. Structure and regulation of voltage-gated Ca2+ channels. Annual Review of Cell and Developmental Biology. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synpasins: key actors of synapse function and plasticity. Progress in Neurobiology. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ Regulates the Interaction between Synaptotagmin and Syntaxin 1. Journal of Biological Chemistry. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- Chapman ER. How Does Synaptotagmin Trigger Neurotransmitter Release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Chen XW, Feng YQ, Hao CJ, Guo XL, He X, Zhou ZY, Guo N, Huang HP, Xiong W, Zheng H, Zuo PL, Zhang CX, Li W, Zhou Z. DTNBP1 a schizophrenia susceptibility gene affects kinetics of transmitter release. J Cell Biol. 2008;181(5):791–801. doi: 10.1083/jcb.200711021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38(1):69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4(12):1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nat Struct Mol Biol. 2008;15(8):827–35. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danglot L, Galli T. What is the function of neuronal AP-3? Biol Cell. 2007;99(7):349–61. doi: 10.1042/BC20070029. [DOI] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Jha A, Ramonet D, Matsui T, Leitges M, Shin HS, Porter JE, Geiger JD, Lei S. Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. J Neurosci. 2010;14;30(15):5136–48. doi: 10.1523/JNEUROSCI.5711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman DK, Davis GW. The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science. 2009;20:326(5956):1127–30. doi: 10.1126/science.1179685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability facilitation and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Frankle WG, Lerma J, Laruelle M. The synaptic hypothesis of schizophrenia. Neuron. 2003;39(2):205–16. doi: 10.1016/s0896-6273(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Feng YQ, Zhou ZY, He X, Wang H, Guo XL, Hao CJ, Guo Y, Zhen XC, Li W. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr Res. 2008;106(2–3):218–28. doi: 10.1016/j.schres.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Duan J. The role of genetics in the etiology of schizophrenia. Psychiatr Clin North Am. 2010;33(1):35–66. doi: 10.1016/j.psc.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiani CA, Starcevic M, Rodriguez-Fernandez IA, Nazarian R, Cheli VT, Chan LN, Malvar JS, de Vellis J, Sabatti C, Dell’Angelica EC. The dysbindin-containing complex (BLOC-1) in brain: developmental regulation, interaction with SNARE proteins and role in neurite outgrowth. Mol Psychiatry. 2010;15(2):115, 204–15. doi: 10.1038/mp.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: Are we measuring the, “right stuff”? Schizophr Bull. 2000;26(1):119–36. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259(5096):780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR. Schizophrenia genes gene expression and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10(1):40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20(3):201–25. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Trantham-Davidson H, Jairl C, Tinsley M, Cannon TD, Lavin A. Dysbindin modulates prefrontal cortical glutamatergic circuits and working memory function in mice. Neuropsychopharmacology. 2009;34(12):2601–8. doi: 10.1038/npp.2009.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Li Q, Schlichter LC, Stanley EF. The transmitter release-site CaV2.2 channel cluster is linked to an endocytosis coat protein complex. Eur J Neurosci. 2007;26(3):560–74. doi: 10.1111/j.1460-9568.2007.05681.x. [DOI] [PubMed] [Google Scholar]
- Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, Lee JD, Talbot K, Pare JF, Smith Y, Faundez V. The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell. 2011;22(24):4854–67. doi: 10.1091/mbc.E11-07-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang Q, Oiso N, Novak EK, Gautam R, O’Brien EP, Tinsley CL, Blake DJ, Spritz RA, Copeland NG, Jenkins NA, Amato D, Roe BA, Starcevic M, Dell’Angelica EC, Elliott RW, Mishra V, Kingsmore SF, Paylor RE, Swank RT. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35(1):84–9. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher BS, Reimers MA, Riley BP, Kendler KS. Allelic heterogeneity in genetic association meta-analysis: an application to DTNBP1 and schizophrenia. Hum Hered. 2010;69(2):71–9. doi: 10.1159/000264445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meffert MK, Premack BA, Schulman H. Nitric oxide stimulates calcium-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- Mullin AP, Gokhale A, Larimore J, Faundez V. Cell biology of the BLOC-1 complex subunit dysbindin, a schizophrenia susceptibility gene. Mol Neurobiol. 2011;44(1):53–64. doi: 10.1007/s12035-011-8183-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Salazar G, Smith Y, Faundez V. Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell. 2009;20(5):1441–53. doi: 10.1091/mbc.E08-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Chintala S, Jenkins S, Pare JF, McGaha L, Smith Y, Faundez V. Hermansky-Pudlak protein complexes, AP-3 and BLOC-1, differentially regulate presynaptic composition in the striatum and hippocampus. J Neurosci. 2010;30(3):820–31. doi: 10.1523/JNEUROSCI.3400-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numakawa T, Yagasaki Y, Ishimoto T, Okada T, Suzuki T, Iwata N, Ozaki N, Taguchi T, Tatsumi M, Kamijima K, Straub RE, Weinberger DR, Kunugi H, Hashimoto R. Evidence of novel neuronal functions of dysbindin a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13:2699–2708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- Pan PY, Tian JH, Sheng ZH. Snapin facilitates the synchronization of synaptic vesicle fusion. Neuron. 2009;61(3):412–24. doi: 10.1016/j.neuron.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risinger C, Bennett MK. Differential phosphorylation of syntaxin and synaptosome- associated protein of 25 kDa (SNAP-25) isoforms. J Neurochem. 1999;72(2):614–24. doi: 10.1046/j.1471-4159.1999.0720614.x. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Rettig J, Takahashi M, Catterall WA. Identification of a syntaxin-binding site on N-type calcium channels. Neuron. 1994;13(6):1303–13. doi: 10.1016/0896-6273(94)90417-0. [DOI] [PubMed] [Google Scholar]
- Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem. 1986;261(18):8383–8390. [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23(2):399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Starcevic M, Dell’Angelica EC. Identification of snapin and three novel proteins (BLOS1 BLOS2 and BLOS3/reduced pigmentation) as subunits of biogenesis of lysosome-related organelles complex-1 (BLOC-1) J Biol Chem. 2004;279:28393–28401. doi: 10.1074/jbc.M402513200. [DOI] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D. A potential vulnerability locus for schizophrenia on chromosome 6p24-22: evidence for genetic heterogeneity. Nat Genet. 1995;11(3):287–93. doi: 10.1038/ng1195-287. [DOI] [PubMed] [Google Scholar]
- Talbot K, Eidem WL, Tinsley CL, Benson MA, Thompson EW, Smith RJ, Hahn CG, Siegel SJ, Trojanowski JQ, Gur RE, Blake DJ, Arnold SE. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest. 2004;113(9):1353–63. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot K, Ong WY, Blake DJ, Tang J, Louvena N, Carlsson GC, Arnold SE. Dysbindin and its protein family. In: Javitt DC, Kantrowitz JT, editors. Handbook of Neurochemistry and Molecular Biology. 3. Springer; 2009. Volume Editors, A Lagtha Editor. [Google Scholar]
- Talbot K, Louneva N, Cohen JW, Kazi H, Blake DJ, Arnold SE. Synaptic dysbindin-1 reductions in schizophrenia occur in an isoform-specific manner indicating their subsynaptic location. PLoS One. 2011;6(3):e16886. doi: 10.1371/journal.pone.0016886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, LeGros RP, Louneva N, Yeh L, Cohen JW, Hahn CG, Blake DJ, Arnold SE, Talbot K. Dysbindin-1 in dorsolateral prefrontal cortex of schizophrenia cases is reduced in an isoform-specific manner unrelated to dysbindin-1 mRNA expression. Hum Mol Genet. 2009;18(20):3851–63. doi: 10.1093/hmg/ddp329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36(6):1127–43.24. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Tian JH, Wu ZX, Unzicker M, Lu L, Cai Q, Li C, Schirra C, Matti U, Stevens D, Deng C, Rettig J, Sheng ZH. The role of Snapin in neurosecretion: snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J Neurosci. 2005;25(45):10546–55. doi: 10.1523/JNEUROSCI.3275-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter MP, Thatcher L, Usen N, Hyde TM, Kleinman JE, Freed WJ. Reduction of synapsin in the hippocampus of patients with bipolar disorder and schizophrenia. Mol Psychiatr. 2002;7:571–578. doi: 10.1038/sj.mp.4001158. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Tatsuno M, Kiuchi Y. Maturational change of KCl-induced Ca2+ increase in the rat brain synaptosomes. Brain Dev. 1998;20(4):234–8. doi: 10.1016/s0387-7604(98)00027-8. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Oho C, Omori A, Kuwahara R, Ito T, Takahashi M. HPC-1 is associated with synaptotagmin and omega-conotoxin receptor. J Biol Chem. 1992;267(35):24925–8. [PubMed] [Google Scholar]