

Abstract
The current study is to determine the regulatory role of VEGF-A in cardiac angiogenesis following myocardial infarction (MI). Cardiac angiogenic response and temporal/spatial expression of VEGF-A/VEGF receptors (VEGFR) were examined at 1, 2, 6, 12 hours and 1, 2, 3, 4, 7, 14, and 28 days postMI. We found that following MI, newly formed vessels first appeared at the border zone between noninfarcted and infarcted myocardium as early as day 3 and subsequently in the infarcted myocardium. Vascular density in the infarcted myocardium peaked at day 7 and then gradually declined. VEGF-A mRNA started to increase at the border zone at 2 hours postMI, reached peak at 12 hours, declined at day 1, and returned to normal levels at day 2 and thereafter. VEGF-A protein levels at the border zone were only increased during day 1 postMI. VEGF-A within the infarcted myocardium levels, however, were persistently suppressed postMI. VEGFR expression was significantly increased only at the border zone at day 1, but not in the later stages. The expression of VEGF-A/VEGFR remained unchanged in the noninfarcted myocardium. Thus, the early rise of VEGF-A/VEGFR at the border zone suggests that VEGF-A initiates the cardiac angiogenic response postMI, but short-lived VEGF-A/VEGFR activation at the border zone and consistently suppressed VEGF-A within the infarcted myocardium suggests that VEGF-A may not be crucial to the later stages of angiogenesis.
Keywords: Myocardial infarction, Angiogenesis, VEGF-A, VEGFR
INTRODUCTION
Myocardial infarction (MI) has emerged as a major health problem during the past two decades. Following MI, cardiac repair occurring at the site of myocyte loss preserves the structural integrity and is essential to the heart’s recovery. Angiogenesis is central to cardiac repair and impaired angiogenesis may delay cardiac repair and cause cardiac rupture or immature scar tissue formation (Barandon et al., 2004). Numerous studies have demonstrated that stimulation of angiogenesis is beneficial to ischemic and infarcted heart (Ahn et al., 2008; Boodhwani et al., 2006; Nelson et al., 2000; Pandya et al., 2006).
Angiogenesis is the formation of new capillary blood vessels from existent microvessels. It plays a critical role in various biological processes such as wound healing, embryological development, the menstrual cycle, and inflammation and the pathogenesis of various diseases such as cancer, diabetic retinopathy, and rheumatoid arthritis. Promoting angiogenesis can aid in accelerating various physiological processes requiring increased vascularization such as the healing of wounds, fractures, and burns, and the treatment of inflammatory diseases, ischemia, peripheral vascular disease, and MI (Kang et al., 2002; Ren et al., 2003; Timar et al., 2001). Conversely, inhibition of angiogenesis can aid in the treatment of diseases such as cancer, diabetic retinopathy, and rheumatoid arthritis, where increased vascularization contributes to their progression (Roccaro et al., 2005; Wheatley-Price and Shepherd, 2008).
The sequence of events that lead to the sprouting of new vessels is fairly well documented. Initially, a degradation of basement membrane associated with an increase in vessel permeability and proteolysis of the surrounding extracellular matrix leads, upon action of angiogenic factors, to initiate endothelial cell migration and proliferation. As endothelial cells proliferate and assemble, they receive instructions to mature the newly formed sprouts and form a lumen. Angiogenesis is a tightly regulated process requiring the homeostatic balance of inducers and inhibitors. Vascular endothelial growth factors (VEGFs), a family of growth factors, are important signaling proteins involved in angiogenesis containing at least 7 members with a homodimeric structure. VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor are encoded in the mammalian genome (Ferrara and Gerber, 2001). VEGF-A, usually referred to as VEGF, acts as a key player in vasculogenesis and angiogenesis. There are three major isoforms of VEGF-A in the rat (VEGF120, VEGF164, and VEGF188) (McColm et al., 2004), arising from alternative splicing of mRNA, and the relative abundance of each isoform varies depending on the organ. VEGF-A exerts its biologic effect through interaction with cell surface receptors, VEGFR-1 and VEGFR-2 selectively expressed on vascular endothelial cells (Ria et al., 2003). VEGFR-2 appears to mediate almost all of the known cellular responses to VEGF-A. The function of VEGFR-1 is not well-defined, although it is thought to modulate VEGFR-2 signaling. It has been shown that blockage of VEGF impairs angiogenesis in various models (Neufeld et al., 1999).
The importance of VEGF-A in angiogenesis in the infarcted heart is not well understood. The expression of VEGF in the infarcted heart has been addressed in a few studies, but the findings are contradictory. Heba et al. reported that VEGF mRNA was not detectable in the normal rat heart, but was highly expressed in the infarcted myocardium, lasting over the course of 6 weeks (Heba et al., 2001). However, Li et al reported that VEGF was expressed in the normal heart (Li et al., 1996), and its expression was significantly increased in the infarcted myocardium only in the early stage of MI. To better understand the role of VEGF-A in cardiac angiogenesis following MI, in the current study we used the molecular and cellular imaging for the measurement of temporal and spatial responses of cardiac VEGF-A and VEGFR expression in rats following experimental MI.
METHODS
Animal Model
The study follows the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health and was approved by University of Tennessee Health Science Center Animal Care and Use Committee. Left ventricular anterior transmural MI was created in 8-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN) via permanent ligation of left coronary artery. Animals were anesthetized with 1.5% isoflurane, intubated and ventilated with a rodent respirator. The heart was exposed via a left thoractomy and the left anterior descending artery was ligated with a 6-0 silk suture. The chest was then closed and lungs reinflated using positive end-expiratory pressure (Sun and Weber, 1994). Only hearts with large free wall infarction (40–45% of left ventricle) were used in the study. To estimate infarct area at hour 2, 6 and 12 postMI, hearts were collected and flushed with cold saline through aorta. The color of noninfarcted myocardium became pale, while the color of infarcted myocardium remained unchanged. In addition, infarcted myocardium became thinner than normal left ventricle at day 1 and beyond. Age and sex matched rats without undergoing surgery served as controls (n = 8). Sham-operated rats were not used as controls, since sham- operation causes pericardial injury/angiogenesis, thus affects data interpretation. Rats with MI were sacrificed at postoperative times 1, 2, 6, and 12 hours, 1, 2, 3, 4, 7, 14, and 28 days (n = 8 for each time point). Hearts were removed and kept frozen at −80 °C until use.
Immunohistochemistry
Immunohistochemical labeling of CD31, specifically expressed by endothelial cells, was used as a marker of angiogenesis. Cryostat coronal sections (6 μm) were air-dried, fixed in 10% buffered formalin for 10 min, and washed in phosphate-buffered saline (PBS). Endogenous peroxidase activity was blocked by immersion of the sections in 0.3% H2O2 for 10 minutes at room temperature. Sections were blocked with 5% goat serum in PBS. Tissues were incubated with primary antibodies to CD31 (BD Biosciences, San Jose, CA) for 1 hour at room temperature. Sections were then incubated with peroxidase conjugated secondary antibody (Sigma, St. Louis, MO) for 1 hour at room temperature, washed in PBS for 10 minutes, and incubated with 0.5 mg/ml diaminobenzidine tetrahydrochloride 2-hydrate + H2O2 for 1 minutes. Negative control sections were incubated with secondary antibody alone. Sections were then dehydrated, mounted, and viewed by light microscopy (Zhao et al., 2009).
Quantification of Microvascular Density
Microvascular density was quantitated in CD31 stained sections with a computerized image analyzing system. The methods generally comprise creating a digital image of a coronal section of the heart, determining the surface area of microvessels in the infarcted myocardium, determining the total area of the infarcted myocardium, and calculating the ratio of the microvascular area to the total infarcted area. The digital image of the heart was created using image processing software (NIH image 1.60). CD31 staining in large vessels was disregarded and microvascular density is expressed as microvascular volume fraction (Samoszuk et al., 2002).
Quantitative In Situ Hybridization
The localization and density of cardiac VEGF-A mRNA was analyzed by quantitative in situ hybridization. Briefly, coronal cryostat sections (16 μm) were fixed in 4% formaldehyde for 10 minutes and incubated in 0.25% acetic anhydride in 0.1M TE-HCL for 10 min. Sections were then hybridized (overnight at 56°C) with a random primed 35S-UTP-labeled VEGF-A RNA probe. The antisense probe recognizes all spliced forms of VEGF-A. Sections were washed, dried, and subsequently exposed to Kodak Biomax X-ray film. Exposure time was based on signal intensity. Quantification of mRNA optical density was performed using a computer image analyzing system (NIH Image, 1.60) (Sun et al., 1998).
Western Blotting
VEGF-A protein levels were measured by western blot. Briefly, the normal, noninfarcted left myocardium (septum), border zone and infarcted myocardium were dissected and homogenized. The supernatant was collected and separated by 10% SDS-PAGE. After electrophoresis, samples were transferred to PVDF membranes and incubated with antibody against VEGF-A (sc-152) (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were subsequently incubated with peroxidase-conjugated secondary antibody. After washing, the blots were developed with the enhanced chemiluminescence method. The amount of protein detected was assessed by means of quantitative densitometry analysis with a computer image analyzing system (Kamalov et al., 2005).
Quantitative In Vitro Autoradiography
The localization and density of VEGFR were detected by quantitative in vitro autoradiography. Cryostat 16 μm coronal sections of heart were preincubated for 15 minutes in PBS buffer containing 0.2% bovine serum albumin, then incubated for 1 hour in a fresh volume of the same buffer containing 0.2 μCi/mL 125I-VEGF (PerkinElmer, Waltham, MA). Nonspecific binding was measured in the presence of 1 μM unlabeled VEGF-A. After incubation, sections were washed, dried and exposed to Kodak NMB-6 film. Quantification of VEGFR, includingVEGFR-1 and VEGFR-2 binding density was performed using a computer image analysis system. Specific VEGFR binding was obtained by subtraction of nonspecific binding from total binding and is expressed as optical density (Sun and Weber, 1994).
Statistical Analysis
Statistical analysis of data obtained from in situ hybridization, microvascular density, western blot and in vitro autoradiography was performed using analysis of variance. Values are expressed as mean ± SEM with P<0.05 considered significant. Multiple group comparisons among controls and each group were made by Scheffe’s F-test.
RESULTS
Angiogenesis in the Infarcted Heart
As detected by immunohistochemical CD31 staining (Figure 1), we observed abundant microvessels in the normal myocardium that include arterioles, capillaries and venules. At day 1 following MI, microvessels underwent necrosis in the infarcted myocardium. Newly formed microvessels appeared at the border zone between infarcted and noninfarcted myocardium as early as day 3, and in the infarcted myocardium by day 4. Microvascular density reached a peak in the infarcted myocardium at day 7, but was decreased at days 14 and 28 (not shown). Abundant large vessels are also formed in the infarcted myocardium. However, only microvessels are considered angiogenesis. Quantitative microvascular volume in the infarcted myocardium is shown in figure 2.
Figure 1.
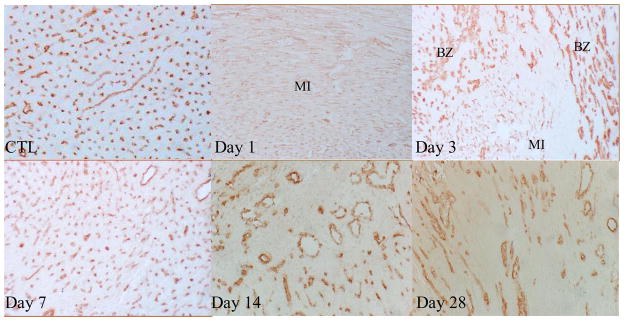
Angiogenesis in the infarcted heart: As detected by immunohistochemical CD31 labeling, normal myocardium (CTL) was rich in capillaries (brown). Following MI, pre-existing vessels were totally lost in the infarcted myocardium at day 1, newly formed microvessels appeared at the border zone at day 3. Abundant new microvessels were seen in the infarcted myocardium at day 7. Vascular density was then declined at day 14 and 28. x200
Figure 2.
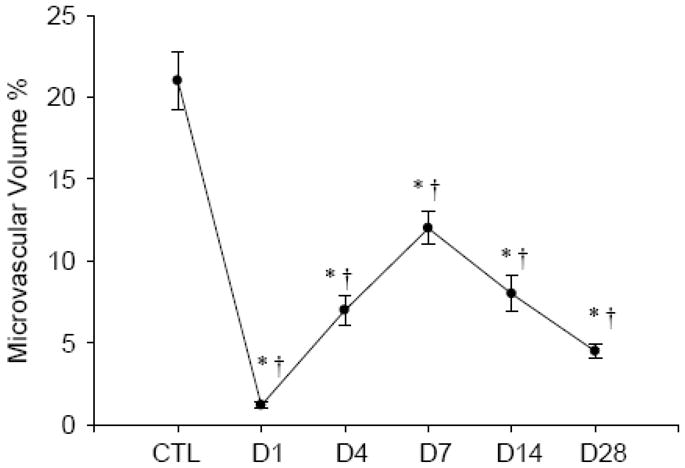
Temporal changes of microvascular density in the infarcted myocardium. *p<0.05 vs CTL; †p<0.05 vs previous time point.
Localization and Density of Cardiac VEGF-A Gene Expression
As detected by quantitative in situ hybridization (Figures 3 and 4), VEGF-A mRNA was found evenly distributed in the normal heart with a relatively low density. An increase in VEGF-A mRNA was detectable at the border zone and endocardium at 2 hours postMI, became more evident at 6 hours, peaked at 12 hours, declined at 24 hours, but still remained significantly elevated compared with controls, and then returned to controls levels at day 2 and beyond. But VEGF-A mRNA levels in the infarcted myocardium started to decline at 2 hours postMI and remained decreased for over the course of 4 weeks. VEGF-A mRNA in the noninfarcted myocardium remained unchanged.
Figure 3.
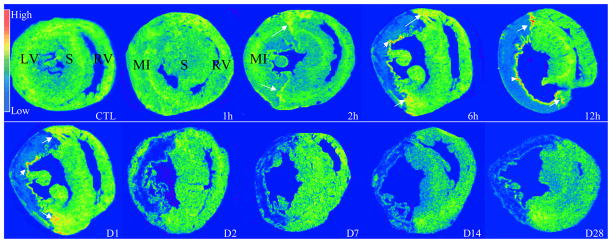
Temporal and spatial gene expression of VEGF-A in the infarcted heart: As detected by in situ hybridization, VEGF-A mRNA was normally present in both left and right ventricles (RV, LV). Following MI, VEGF-A mRNA levels remained unchanged in the infarcted myocardium at 1 hour. VEGF-A mRNA levels were increased at the border zone (arrows) and endocardium (arrowheads) at 2 hours, became more evident at 6 hours, reached peak at 12 hours, declined, but was still higher than controls at day 1 and returned to normal level at day 2. VEGF-A mRNA was increased constantly reduced in the infarcted myocardium for over the course of 4 weeks. VEGF-A mRNA levels remained unchanged in the noninfarcted septum (S).
Figure 4.
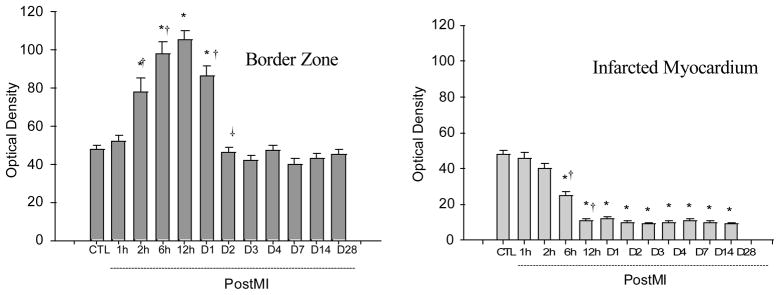
Quantitative VEGF-A mRNA levels in the border zone and infarcted myocardium at different time points postMI.
Cardiac VEGF-A Protein Levels
Western blot analysis was performed to detect the VEGF-A protein levels in the normal myocardium and various regions of the infarcted heart including border zone, infarcted myocardium and noninfarcted myocardium at different time points postMI. Both VEGF188 and VEGF164 were expressed in the normal rat heart with VEGF164 levels being significantly higher than VEGF188 (Figure 5). VEGF120 expression was not evident in the normal myocardium. Compared to controls, both VEGF188 and VEGF164 protein levels at the border zone was significantly increased at 2 hours postMI compared to controls, however, the increase in VEGF-A only lasted for one day. VEGF188 then returned to control levels at day 2 and thereafter, while VEGF164 levels were significantly lower than controls at day 2 and afterward (Figure 5). In the infarcted myocardium, VEGF-A protein levels gradually declined in the first day postMI and remained reduced over the course of 4 weeks (Figure 6). In noninfarcted myocardium (septum), VEGF-A protein levels remained unchanged in both early and late stages of MI (not shown).
Figure 5.

Temporal changes of VEGF-A protein levels in the border zone postMI: As detected by western blot, VEGF188 and VEGF164 were normally expressed in the rat heart. Following MI, both VEGF188 and VEGF164 were significantly increased at 2, 6, 12 and 24 hours. VEGF188 declined to the normal level at day 2 and thereafter, while VEGF164 levels were significantly lower than controls at day 2 and beyond.
Figure 6.
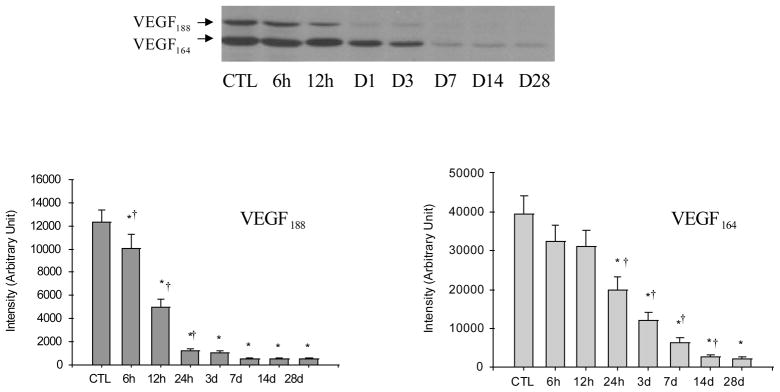
Temporal changes of VEGF-A protein levels in the infarcted myocardium: Compared to controls, VEGF188 and VEGF166 gradually declined in the infarcted myocardium for over the course of 4 weeks postMI.
The Localization and Density of Cardiac VEGFR
The temporal and spatial response of VEGFR in the infarcted heart was detected by quantitative in vitro autoradiography. In the normal heart, we found that VEGFR were uniformly distributed in the left and right ventricles (Figures 7 and 8). Following MI, VEGFR binding density was significantly increased at the border zone at day 1 postMI, returned to the normal level at day 3 and for the following weeks. However, VEGFR binding density was significantly reduced in the infarcted myocardium at day 1 and gradually returned to the normal level thereafter.
Figure 7.

Cardiac VEGFR expression: As detected by in vitro autoradiography, VEGFR binding was normally expressed in both LV and RV. Following MI, VEGFR binding was greatly increased at border zone (arrows) at day 1, but not in the later stages. VEGFR was largely decreased in the infarcted myocardium at day 1 postMI and returned to the control level at day 7. VEGFR binding density remained unchanged in the noninfarcted septum and right ventricle compared to controls.
Figure 8.
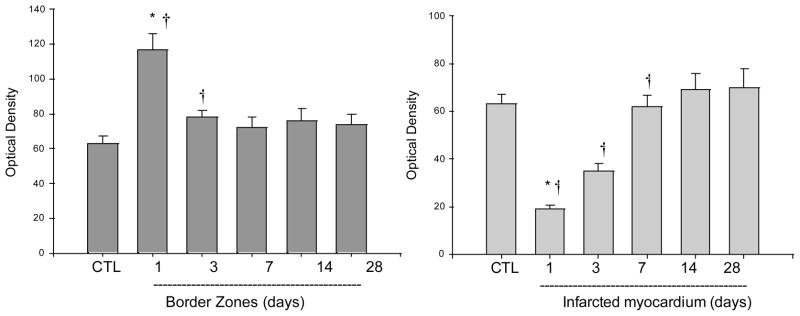
Quantitative specific VEGFR binding density in the border zone and infarcted myocardium.
DISSCUSSION
To more completely determine the role of VEGF-A in the cardiac angiogenesis after MI, we explored the spatial and temporal expression of VEGF-A and VEGFR in the infarcted rat heart.
Firstly, we examined the angiogenic response in the infarcted heart. Following acute MI, myocytes/interstitial cells and existing blood vessels in the infarcted myocardium undergo necrosis. Since the myocardium can barely regenerate after it is injured, the lost myocardium is replaced by scar tissue to preserve the structural integrity of the heart. Angiogenesis and inflammatory/fibrogenic responses are central to cardiac repair and scar tissue formation. Our study has shown that in rat experimental MI, newly formed vessels were seen first in the border zones and then extended into the infarcted myocardium. Microvascular density was most evident in the infarcted myocardium at day 7 postMI, subsequently declined at day 14 and beyond. These findings suggest that angiogenesis is most active in the early stage of cardiac repair following MI.
Secondly, we studied the involvement of VEGF-A in cardiac angiogenesis following MI. The proangiogenic role of VEGF-A has been mostly investigated in tumors (Gerstner et al., 2009; Pourgholami and Morris, 2008). Across most, if not all, cancers, sustained angiogenesis is considered to be one of central hallmarks of cancer. VEGF-A is highly expressed in cancers and serves as the predominant regulator of this pathological angiogenic process, in which VEGF-A has been identified to stimulate vascular endothelial cell growth, survival, and proliferation. It has been also shown to facilitate survival of existing vessels (Machein and Plate, 2004). However, the expression and the role of VEGF-A in the infarcted heart is not well understood. In the current study, we observed rapidly increased VEGF-A mRNA at the border zone, starting as early as 2 hours postMI. However, the elevation of VEGF-A mRNA in the same region was short-lived and lasted for only one day. The change in VEGF-A protein levels at the border zone showed a similar pattern. VEGF-A mRNA was also enhanced in the endocardium of infarcted myocardium. This is because certain amount of myocytes in endocardium are survived due to oxygen transport from left ventricle cavity. Factors inducing the activation of VEGF-A production have been well documented. Hypoxia is a major stimulator of VEGF expression (Forsythe et al., 1996). Hypoxia-induced transcription of VEGF mRNA is mediated by hypoxia-inducible factor 1 (HIF-1), a transcription factor that responds to changing intracellular oxygen concentration. During hypoxia, HIF rapidly accumulates and is transported to the nucleus where it induces expression of a wide variety of target gene products, including VEGF. VEGF production can be also stimulated by other factors that include other growth factors, cytokines and reactive oxygen species (Dong et al., 2004; Joo et al., 2003; Kosmidou et al., 2001). In contrast to the previous report (Heba et al., 2001; Li et al., 1996), our study has shown consistently reduced VEGF-A mRNA in the infarcted myocardium in both early and late stages of cardiac repair. The finding is further confirmed with a persistent reduction in VEGF-A protein levels in the infarcted myocardium as detected by western blot. The potential reasons for the contradictory findings between studies in our and previous reports may be largely due to the different techniques used. Quantitative in situ hybridization and in vitro autoradiography with computer image analyzing system used in the current study are more specific and sensitive techniques in detecting the changes in targeted mRNA receptors at different locations. Our findings suggest that the early rise of VEGF-A expression at the border zone may trigger the cardiac angiogenic response following MI. However, the constant attenuation of VEGF-A expression in the infarcted myocardium suggests that VEGF-A may not be critical to the later events of the angiogenesis cascade. The withdrawl of VEGF-A may be also related to the later vascular stabilization in the infarcted myocardium.
Thirdly, we mapped the localization and density of VEGFR in the normal heart and the temporal changes of cardiac VEGFR following MI. VEGF-A stimulates cellular responses by binding to VEGFR on the cell surface, causing them to become activated through transphosphorylation. VEGF-A binds to VEGFR-1 and VEGFR-2, which are expressed almost exclusively in endothelial cells (Rahimi, 2006). A third receptor has been reported (VEGFR-3), however, VEGF-A is not a ligand for this receptor (Valtola et al., 1999). Using 125I-VEGF-A as a ligand (Cooper et al., 1999), we observed a low level of VEGFR binding in the normal heart, which was uniformly distributed in both left and right ventricles. VEGFR in the normal heart should mediate the role of VEGF in the survival of existing vessels. Following MI, VEGFR binding density was greatly increased at the border zone at day 1 postMI and then returns to the normal level. The pattern of VEGFR expression at the border zone is coincident with enhanced VEGF-A production, which further supports the role of VEGF-A in triggering cardiac angiogenesis following MI. VEGFR binding, however, was largely reduced in the infarcted myocardium at day 1 postMI, related to the necrosis of existing vessels following MI. However, unlike VEGF-A, which was consistently reduced in the infarcted myocardium, VEGFR binding density gradually increased to the normal level in the infarcted myocardium in the following weeks. The data suggest that VEGFR is constitutively expressed in the newly formed vessels in the infarcted myocardium irrespective of local VEGF-A levels.
In summary, the current study has explored the spatial and temporal expression of VEGF-A (mRNA and protein) and VEGFR in the infarcted rat heart. VEGF-A and VEGF receptors were found significantly increased at the border zone only in the first day postMI, but not in the later stages. However, VEGF-A expression was consistently suppressed in the infarcted myocardium, even during the first week when angiogenesis is most active. These data suggest that VEGF-A plays a prominent role in triggering the cardiac angiogenic response following acute MI, which is initiated at the border zone, but is not integral for the later events of the angiogenesis cascade in the infarcted myocardium. Further study is required to determine the regulatory role of VEGF-A in cardiac angiogenesis postMI.
Acknowledgments
This work was supported by NIH Heart, Blood, and Lung Institute (RO1-HL77668, Yao Sun)
Footnotes
Authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn A, et al. Therapeutic angiogenesis: a new treatment approach for ischemic heart disease--Part II. Cardiol Rev. 2008;16:219–29. doi: 10.1097/CRD.0b013e3181620e50. [DOI] [PubMed] [Google Scholar]
- Barandon L, et al. Frizzled A, a novel angiogenic factor: promises for cardiac repair. Eur J Cardiothorac Surg. 2004;25:76–83. doi: 10.1016/s1010-7940(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Boodhwani M, et al. The future of therapeutic myocardial angiogenesis. Shock. 2006;26:332–41. doi: 10.1097/01.shk.0000225318.08681.a7. [DOI] [PubMed] [Google Scholar]
- Cooper ME, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–39. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- Dong J, et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. Embo J. 2004;23:2800–10. doi: 10.1038/sj.emboj.7600289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP. The role of vascular endothelial growth factor in angiogenesis. Acta Haematol. 2001;106:148–56. doi: 10.1159/000046610. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–13. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner ER, et al. Anti-vascular endothelial growth factor therapy for malignant glioma. Curr Neurol Neurosci Rep. 2009;9:254–62. doi: 10.1007/s11910-009-0037-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heba G, et al. Relation between expression of TNF alpha, iNOS, VEGF mRNA and development of heart failure after experimental myocardial infarction in rats. J Physiol Pharmacol. 2001;52:39–52. [PubMed] [Google Scholar]
- Joo YE, et al. Cyclooxygenase-2 overexpression correlates with vascular endothelial growth factor expression and tumor angiogenesis in gastric cancer. J Clin Gastroenterol. 2003;37:28–33. doi: 10.1097/00004836-200307000-00009. [DOI] [PubMed] [Google Scholar]
- Kamalov G, et al. Expression of the multifunctional Y-box protein, YB-1, in myofibroblasts of the infarcted rat heart. Biochem Biophys Res Commun. 2005;334:239–44. doi: 10.1016/j.bbrc.2005.06.082. [DOI] [PubMed] [Google Scholar]
- Kang DH, et al. Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 2002;13:806–16. doi: 10.1681/ASN.V133806. [DOI] [PubMed] [Google Scholar]
- Kosmidou I, et al. Reactive oxygen species stimulate VEGF production from C(2)C(12) skeletal myotubes through a PI3K/Akt pathway. Am J Physiol Lung Cell Mol Physiol. 2001;280:L585–92. doi: 10.1152/ajplung.2001.280.4.L585. [DOI] [PubMed] [Google Scholar]
- Li J, et al. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- Machein MR, Plate KH. Role of VEGF in developmental angiogenesis and in tumor angiogenesis in the brain. Cancer Treat Res. 2004;117:191–218. doi: 10.1007/978-1-4419-8871-3_13. [DOI] [PubMed] [Google Scholar]
- McColm JR, et al. VEGF isoforms and their expression after a single episode of hypoxia or repeated fluctuations between hyperoxia and hypoxia: relevance to clinical ROP. Mol Vis. 2004;10:512–20. [PMC free article] [PubMed] [Google Scholar]
- Nelson MA, et al. Therapeutic angiogenesis: a new treatment modality for ischemic heart disease. Heart Dis. 2000;2:314–25. [PubMed] [Google Scholar]
- Neufeld G, et al. Vascular endothelial growth factor (VEGF) and its receptors. Faseb J. 1999;13:9–22. [PubMed] [Google Scholar]
- Pandya NM, et al. Angiogenesis--a new target for future therapy. Vascul Pharmacol. 2006;44:265–74. doi: 10.1016/j.vph.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Pourgholami MH, Morris DL. Inhibitors of vascular endothelial growth factor in cancer. Cardiovasc Hematol Agents Med Chem. 2008;6:343–7. doi: 10.2174/187152508785909528. [DOI] [PubMed] [Google Scholar]
- Rahimi N. VEGFR-1 and VEGFR-2: two non-identical twins with a unique physiognomy. Front Biosci. 2006;11:818–29. doi: 10.2741/1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, et al. Inflammatory mechanisms in myocardial infarction. Curr Drug Targets Inflamm Allergy. 2003;2:242–56. doi: 10.2174/1568010033484098. [DOI] [PubMed] [Google Scholar]
- Ria R, et al. Vascular endothelial growth factor and its receptors in multiple myeloma. Leukemia. 2003;17:1961–6. doi: 10.1038/sj.leu.2403076. [DOI] [PubMed] [Google Scholar]
- Roccaro AM, et al. Antiangiogenesis for rheumatoid arthritis. Curr Drug Targets Inflamm Allergy. 2005;4:27–30. doi: 10.2174/1568010053622911. [DOI] [PubMed] [Google Scholar]
- Samoszuk M, et al. Measuring microvascular density in tumors by digital dissection. Anal Quant Cytol Histol. 2002;24:15–22. [PubMed] [Google Scholar]
- Sun Y, Weber KT. Angiotensin II receptor binding following myocardial infarction in the rat. Cardiovasc Res. 1994;28:1623–8. doi: 10.1093/cvr/28.11.1623. [DOI] [PubMed] [Google Scholar]
- Sun Y, et al. Angiotensin II, transforming growth factor-beta1 and repair in the infarcted heart. J Mol Cell Cardiol. 1998;30:1559–69. doi: 10.1006/jmcc.1998.0721. [DOI] [PubMed] [Google Scholar]
- Timar J, et al. Angiogenesis-dependent diseases and angiogenesis therapy. Pathol Oncol Res. 2001;7:85–94. doi: 10.1007/BF03032573. [DOI] [PubMed] [Google Scholar]
- Valtola R, et al. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol. 1999;154:1381–90. doi: 10.1016/S0002-9440(10)65392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley-Price P, Shepherd FA. Targeting angiogenesis in the treatment of lung cancer. J Thorac Oncol. 2008;3:1173–84. doi: 10.1097/JTO.0b013e318187220f. [DOI] [PubMed] [Google Scholar]
- Zhao W, et al. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol. 2009;90:621–629. doi: 10.1111/j.1365-2613.2009.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]