

Abstract
Clavulanic acid is a psychoactive compound that has been shown to modulate central nervous system activity. Importantly, in neurotoxin-induced animal models, clavulanic acid has been shown to improve motor function (Huh et al., 2010) suggesting that it can be neuroprotective; however, the mechanism as how clavulanic acid can induce neuroprotection is not known. We demonstrate here that clavulanic acid abrogates the effects of the neurotoxin 1-methyl-4-phenylpyridinium (MPP+) which mimics Parkinson’s disease (PD) by inducing neurodegeneration. To further establish the mechanism we identified that clavulanic acid inhibits neurotoxin-induced loss of mitochondrial membrane potential and ROS production. Consistent with these results, neurotoxin-induced increase in Bax levels was also decreased in clavulanic acid treated cells. Importantly, neurotoxin-induced release of cytochrome c levels as well as caspase activation was also inhibited in clavulanic acid treated cells. In addition, Bcl-xl levels were also restored and the Bcl-xl/Bax ratio that is critical for inducing apoptosis was increased in clavulanic acid treated cells. Overall, these results suggest that clavulanic acid is intimately involved in inhibiting neurotoxin-induced loss of mitochondrial function and induction of apoptosis that contributes towards neuronal survival.
Keywords: Clavulanic acid, MPTP, Neuroprotection, Parkinson’s disease (PD), Mitochondria, Apoptosis
1. Introduction
Neurodegenerative disorders, such as Parkinson’s disease (PD), affect over 6 million patients in the US and 44 million worldwide (Dorsey et al., 2007). These diseases are characterized by neurodegeneration and eventual neuronal cell death. Currently, approved drugs to treat these diseases provide symptomatic relief only, rather than slowing neuronal cell death. Therefore, discovering and developing drugs that inhibit neuronal death is critical (Lohle and Reichmann, 2010). However, despite numerous approaches in drug development, there are no available neuroprotective agents to stop neurodegeneration (Voss and Ravina, 2008).
Clavulanic acid is previously known as a non-competitive inhibitor of lactamase and augments other β-lactam family antibiotics. The compound itself has negligible intrinsic antibacterial activity (Payne et al., 1994). Recent studies demonstrate that clavulanic acid possesses strong CNS modulating effects. According to Kim et al. (2009), clavulanic acid reduces anxiety in rodent and primate models (Kim et al., 2009). Clavulanic acid readily crosses the blood-brain barrier, rendering viable CNS drug properties. Accordingly, the distribution ratio of clavulanic acid between human cerebrospinal fluid and plasma is 0.25, suggesting considerably higher level of brain penetration than most other small molecules (Nakagawa et al., 1994).
Our recently published studies indicate that clavulanic acid protects hippocampal and dopaminergic neurons from toxin-induced acute death in vivo. In these studies, clavulanic acid protected neurons in rodents exposed to kainic acid or MPTP. Furthermore, we observed that clavulanic acid improved motor function in MPTP-treated mice in a behavioral test (Huh et al., 2010). It is believed that PD is caused by the progressive degeneration and eventual death of dopaminergic neurons of the substantianigra (SN) (Samii et al., 2004). In addition, numerous studies have suggested a role for apoptosis in the pathology of PD. At the cellular level, it is believed that mitochondrial dysfunction leads to apoptotic cell death in dopaminergic neurons of the SN in PD (Burke, 1998). Our previous studies indicate that clavulanic acid may have neuroprotective effects and warrants further investigation of its therapeutic use in CNS disorders, such as PD.
Therefore, in this report, we investigated the neuroprotective effects of clavulanic acid in neuronal cell culture after MPP+ induced toxicity. We further characterized the anti-apoptotic effects of clavulanic acid after MPP+ exposure. Our data indicate that clavulanic acid protects neuronal cells from MPP+- induced cell death and enhances its potential as a neuroprotective drug.
2. Results
2.1. Clavulanic acid protects neuronal cells from MPP+ induced cell death
MPTP is a known mitochondrial toxin that induces apoptosis and has been used to model dopaminergic neuronal death in PD (Burns et al., 1983; Langston et al., 1986; Smeyne and Jackson-Lewis, 2005). To examine whether clavulanic acid protects against MPP+ (the active metabolite of MPTP)-induced cell death, neuronally differentiated SH-SY5Y cells were treated with clavulanic acid and exposed to MPP+. The addition of retinoic acid inhibited cell division and lead to the differentiation of dopaminergic cells. Intracellular dopamine levels were measured (data not shown) which showed that differentiated cells had higher intracellular dopamine levels than undifferentiated cells. Cell viability was measured using the MTT assay. Exposure to 250 μM MPP+ for 24 h significantly reduced cell survival (Fig. 1). Importantly, pretreatment of cells with increasing concentrations of clavulanic acid (1, 10, 100, and 500 μM) displayed a trend of increasing cell survival in the presence of MPP+ with 100 and 500 μM clavulanic acid significantly increasing cell survival in the presence of MPP+ (Fig. 1). Similar results were seen with 500 μM MPP+ exposure, where again a significant increase in cell survival was observed in cells that were pretreated with 100 and 500 μM clavulanic acid. Overall, these results suggest that clavulanic acid significantly protected SH-SY5Y cells from MPP+-induced neuron cell death.
Fig. 1.

The protective effect of clavulanic acid against MPP+ induced toxicity in SH-SY5Y cells: SH-SY5Y cells were treated with 250 μM MPP+ or 500 μM MPP+ in the presence or absence of clavulanic acid for 24 h. Cell viability was assessed by using the MTT assay. The viability of untreated controls was defined as 100%. Data are expressed as means ±SD (n=6). *p<0.05 compared with respective controls.
2.2. Clavulanic acid protects against MPP+-induced decrease in mitochondrial membrane potential and ROS generation
The effect of clavulanic acid on MPP+-induced ROS was investigated. Intracellular ROS generation of cells was examined using 2′,7′-dichlorfluorescein-diacetate (DCFH-DA), which is a well-established compound to detect and quantify ROS produced intracellularly (LeBel et al., 1992). MPP+ alone induced a significant increase in intracellular ROS in differentiated SH-SY5Y cells. In contrast, cells pretreated with 500 μM of clavulanic acid significantly decreased ROS generation that was induced by MPP+ (Fig. 2A).
Fig. 2.

Clavulanic acid protects against MPP+ induced ROS generation and loss in mitochondrial membrane potential: (A) SH-SY5Y cells were treated with 500 μM MPP+ in the presence or absence of clavulanic acid for 24 h and then loaded with DCFH-DA to measure ROS generation. (B) Mitochondrial membrane potential was measured by using Rhodamine 123. Cells were loaded with Rhodamine 123 after the respective treatment to measure the mitochondrial membrane potential. Data are expressed as means ±SD (n=6). *p<0.05 compared with respective controls.
Due to the increase in ROS produced by MPP+ treatment, we investigated mitochondrial permeability transition (MPT) and mitochondrial dysfunction which is an early event induced by MPP+. It is known that disruption of mitochondrial membrane permeability can lead to the generation of ROS as a result of opening the MPT pore (Susin et al., 1998; Tan et al., 1998). We investigated MPP+ mediated loss of mitochondrial membrane potential by measuring the fluorescent cationic dye rhodamine 123. The uptake of rhodamine 123 into the mitochondria is a function of mitochondrial transmembrane potential and therefore depolarization of the mitochondrial membrane potential results in the loss of rhodamine 123 from the mitochondria and a decrease in intracellular fluorescence (Ferreiro et al., 2008). As expected, MPP+ treatment resulted in the reduction of rhodamine 123 fluorescence as compared with control untreated cells (Fig. 2B). Interestingly, cells treated with clavulanic acid and exposed to MPP+ displayed a significant increase in mitochondrial membrane potential when compared to MPP+ treated cells alone (Fig. 2B).
2.3. Clavulanic acid protects SH-SY5Y cells from MPP+-induced apoptosis
MPP+ disrupts mitochondrial membrane potential which alters the expression/localization of pro- and anti-apoptotic proteins resulting in apoptosis (Selvaraj et al., 2009). Therefore, we sought to confirm the anti-apoptotic effects of clavulanic acid by studying the proteins involved in the mitochondrial-mediated apoptotic process. The apoptotic pathway is largely mediated through Bcl2 family proteins, which include both pro-apoptotic members such as Bax and Bak that promote mitochondrial permeability and anti-apoptotic members such as Bcl2 and Bcl-xl that inhibit their effects (Antonsson et al., 1997; Desagherand Martinou, 2000). Treatment with MPP+ induced an increase in pro-apoptotic Bax protein in the mitochondrial fraction. Consistent with this, a decrease in anti-apoptotic Bcl-xl protein was also observed in the mitochondrial fraction. The disruption of mitochondrial transmembrane potential and the subsequent cytochrome c release have been considered as an early event in the apoptotic process. As expected a decrease in the amount of cytochrome c levels was observed in the mitochondria of MPP+ treated cells (Fig. 3A). Interestingly, clavulanic acid treatment in the presence of MPP+ resulted in a significant decrease in Bax protein to the mitochondria (Fig. 3B). Moreover, clavulanic acid significantly increased the amount of cytochrome c present in the mitochondria after MPP+ treatment (Fig. 3C) as well as significantly increased the amount of anti-apoptotic Bcl-xl protein associated with the mitochondria (Fig. 3D). Quantitative analysis of the Bcl-xl to Bax protein ratio associated with the mitochondria shows that treatment with MPP+ significantly decreased the Bcl-xl:Bax ratio which was significantly attenuated by clavulanic acid treatment (Fig. 3D).
Fig. 3.

Clavulanic acid inhibits the MPP+ induced mitochondrial mediated apoptotic pathway: SH-SY5Y cells were treated with 500 μM MPP+ in the presence or absence of clavulanic acid for 24 h. Cells were harvested; mitochondrial fractions were prepared by using mitochondrial isolation kit (Sigma-Aldrich) and subjected to western blotting using indicated antibodies (Fig. 3A). Quantification of relative expression of Bax (B), cytochrome c (C), and Bcl-xl (D) is provided in the bar graph. E indicates the ratio of Bcl-xl to Bax in control, 500 μM MPP+ in the presence or absence of clavulanic acid. *p<0.05 compared with respective controls.
In the cytoplasm, disruption of the mitochondrial membrane potential resulted in MPP+-induced release of cytochrome c into the cytoplasm (Fig. 4B) from the mitochondria (Fig. 3C) whereas treatment with clavulanic acid attenuated the MPP+-induced release of cytochrome c from the mitochondria to the cytoplasm (Fig. 4B). Upon release of cytochrome c from the mitochondria, initiator caspase 9 is cleaved and activated and in turn cleaves procaspase 3 (Budihardjo et al., 1999). Therefore, we investigated the expression of cleaved caspase 9 and 3 after MPP+ and clavulanic acid treatment. As expected, MPP+ treatment resulted in cleaved caspase 9 and 3 expression in the cytoplasm (Fig. 4C and D). In contrast, treatment with clavulanic acid decreased the amount of cleaved caspase 9 and 3 produced by MPP+ treatment (Fig. 4C and D). Taken together, these data indicate that clavulanic acid protects against MPP+-induced mitochondrial mediated apoptosis.
Fig. 4.

Clavulanic acid inhibits MPP+-mediated apoptotic pathway by caspase activation: SH-SY5Y cells were treated with 500 μM MPP+ in the presence or absence of clavulanic acid for 24 h. For whole cell lysates, cells were harvested, lysed in RIPA buffer (Millipore) and subjected to western blotting using individual antibodies as described in the figure (A). Quantification of relative expression of cytochrome c in the cytosol (B), caspase 3 (C), and caspase 9 (D) is provided as bar graph. *p<0.05 compared with respective controls.
3. Discussion
The intrinsic apoptotic pathway is characterized by disruption of the mitochondrial inner transmembrane potential, an increase in the mitochondrial membrane permeability, release of cytochrome c into the cytoplasm, and activation of the caspase cascade. In addition to the release of mitochondrial mediated apoptotic factors, reactive oxygen species (ROS) are generated (Zamzami et al., 1995), leading to oxidation of lipids, proteins, and nucleic acids, and further increasing the changes in the mitochondrial inner membrane potential (Marchetti et al., 1997; Kroemer and Reed, 2000). The neurodegenerative consequences of PD are produced by the selective loss of dopaminergic neurons in the substantia nigra and it is believed that progressive mitochondrial changes in dopaminergic cells, leading to apoptosis may underlie the root cause of PD. Although the exact mechanism of PD induced neuronal death is still vague, it is known that the loss of dopaminergic neurons involves multiple cell death pathways and therefore therapies that inhibit apoptosis may be of enormous potential in treating PD.
The MPP+ model has been used as a paradigm for PD since it has been shown to mimic dopaminergic cell death as seen in PD (Sonsalla and Heikkila, 1986;Schober, 2004). Our studies indicate that clavulanic acid protects against cell death caused by MPP+ by attenuating apoptosis. Apoptosis is associated with MPTP-induced neurotoxicity (Javitch et al., 1985; Smeyne and Jackson-Lewis, 2005) and it has been shown that apoptosis may be involved in the etiology of PD. MPP+ toxicity has been shown to activate caspase 9 and 3 in many cell types and is also associated with apoptosis and mitochondrial mediated cell death pathway.
Since our previous in vivo studies indicated that clavulanic acid protected hippocampal and dopaminergic neurons in rodents exposed to kainic acid or MPTP and improved motor function in MPTP-treated mice in a behavioral test (Huh et al., 2010), we wanted to investigate the effects of clavulanic acid on apoptosis at the cellular level. Our studies verify that MPP+ toxicity in SH-SY5Y cells induces apoptosis, which is characterized by a decrease in mitochondrial membrane potential, generation of ROS, release of cytochrome c into the cytoplasm, translocation of Bax and Bcl-xl, and activation of caspase 9 and 3. As depicted in Fig. 5, clavulanic acid attenuates MPP+-induced apoptosis. Treatment with clavulanic acid increases cell survival after MPP+ treatment. Moreover, clavulanic acid maintains the mitochondrial membrane potential and significantly decreases the production of ROS by MPP+. The ability of clavulanic acid to enhance cell survival is further characterized by a decrease in cytochrome c release into the cytoplasm, maintenance of Bcl-xl localization to the mitochondria, and a decrease in cleaved caspase 9 and 3 after MPP+ treatment. Taken together, these data indicate that clavulanic acid protects against MPP+-induced apoptosis in SH-SY5Y cells.
Fig. 5.
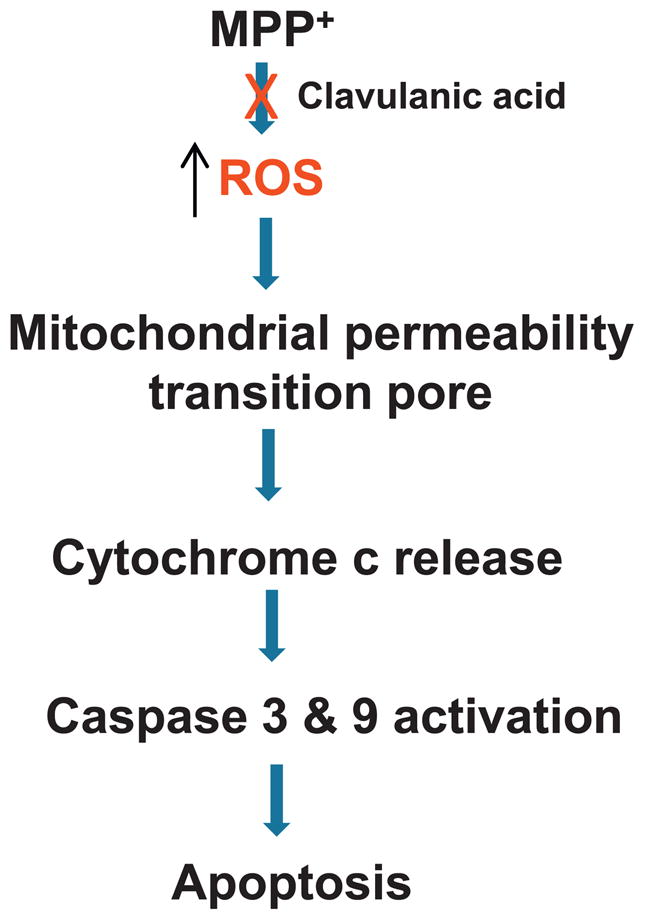
Schematic of neuroprotection by clavulanic acid: Model for MPP+ induced dopaminergic loss and clavulanic acid mediated neuroprotection. MPP+ increases ROS production and induces mitochondrial dysfunction. This leads to the opening of the mitochondrial transition pore, release of cytochrome c and caspase activation. In contrast, pretreatment with clavulanic acid inhibits ROS production, restores mitochondrial function and inhibits apoptosis in dopaminergic cells that lead to increased neuronal survival.
PD and other neurodegenerative diseases such as AD are characterized by neurodegeneration. It has been hypothesized that PD is specifically characterized by the accumulation of mitochondrial damage and dysregulation, leading to apoptosis (Burke, 1998;Samii et al., 2004). The MPTP model has been used to mimic neuronal apoptosis in order to investigate PD. We propose that clavulanic acid inhibits the ability of MPP+ to generate an increase in the release of ROS from the mitochondrial permeability transition pore, leading to a decrease in mitochondrial membrane potential, release of cytochrome c from the mitochondria to the cytoplasm, activation of caspase 9 and 3 through alterations in Bax and Bcl-xl leading to apoptosis as depicted in Fig. 5. We find that clavulanic acid protects against apoptosis caused by MPP+ in neuronal cells as well as in vivo animal models, indicating that clavulanic acid’s potential as a therapy against neurodegenerative diseases such as PD.
4. Experimental procedure
4.1. SH-SY5Y culture and MTT assays
SH-SY5Y cells were cultured in a medium containing DMEM, Hanks’ balanced salt solution (HBSS), F-12 medium (2:1:1) with 10% heat inactivated fetal bovine serum (Invitrogen, CA) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin in a water-saturated atmosphere of 5% CO2 at 37 °C. Cells were differentiated into neuronal cells by the addition of retinoic acid (as described in Ammer and Schulz, 1994). Differentiated cells were treated with 250 μM MPP+ or 500 μM MPP+ in the presence or absence of different concentrations of clavulanic acid (1 μM or 10 μM or 100 μM or 500 μM) for 24 h. Cell viability was measured by using the Vybrant MTT cell proliferation assay kit (Molecular Probes, OR). Absorbance was read at 570 nm (630 nm as a reference) on a microplate reader (Molecular Device, CA). Cell viability was expressed as a percentage where control mock treated cells were considered as 100%.
4.2. Measurement of mitochondrial membrane potential
Mitochondrial transmembrane potential was measured using fluorescent cationic dye rhodamine 123. It has been shown that the uptake of rhodamine 123 into mitochondria is a function of mitochondrial transmembrane potential (Ferreiro et al., 2008). Depolarization of mitochondrial membrane potential results in the loss of Rh123 from the mitochondria and a decrease in intracellular fluorescence. SH-SY5Y cells were incubated with MPP+ (24 h) in the presence or absence of 500 μM clavulanic acid followed by 20 min incubation with 10 μM of rhodamine 123 at 37 °C in DMEM media. The fluorescence signals of the dye were measured immediately at an excitation wavelength of 488 nm and an emission wavelength of 510 nm using a fluorescence microplate reader. The results were expressed as percentage of increase or decrease above control fluorescence.
4.3. Measurement of intracellular reactive oxygen species (ROS)
Cells were treated with 500 μM MPP+ in the presence or absence of 500 μM clavulanic acid for 24 h and then incubated with 10 μM 2′,7′-dichlorodihydrofluorescein diacetate(DCFH-DA) (Molecular Probes Inc., OR) for 30 min at 37 °C. Cells were washed twice with PBS and the fluorescence intensity was captured using a microplate reader with an excitation wavelength of 485 nm and emission wavelength of 520 nm.
4.4. Western blot analyses and assessment of apoptosis
To perform western blot analyses, cytosolic and mitochondrial fractions were prepared by using mitochondrial isolation kits (Sigma-Aldrich, MO) in control mock treated, MPP+ treated and clavulanic acid pretreated along with MPP+ treatment. Protein concentrations were determined using the DC protein assay kit (Bio-Rad, CA). Samples were resolved on NuPAGE 4–12% Bis-Tris gel (Invitrogen, CA), transferred to polyvinylidenedifluoride (PVDF) membranes and probed with respective antibodies as labeled in the figure. Peroxidase-conjugated respective secondary antibodies were used to label the proteins and detected using ECL reagent and Bio-Rad Quantity One (CA)system.
Acknowledgments
We specially thank UND for their help and duly acknowledge grant support from the NIH (DE017102, 5P20RR017699) awarded to BBS.
Footnotes
This study was supported by Rexahn Pharmaceuticals, Inc.
References
- Ammer H, Schulz R. Retinoic acid-induced differentiation of human neuroblastoma SH-SY5Y cells is associated with changes in the abundance of G proteins. J Neurochem. 1994 Apr;62 (4):1310–1318. doi: 10.1046/j.1471-4159.1994.62041310.x. [DOI] [PubMed] [Google Scholar]
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. doi: 10.1126/science.277.5324.370. [DOI] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Burke RE. Programmed cell death and Parkinson’s disease. Mov Disord. 1998;13 (Suppl 1):17–23. [PubMed] [Google Scholar]
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA. 1983;80:4546–4550. doi: 10.1073/pnas.80.14.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- Ferreiro E, Oliveira CR, Pereira CMF. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol Dis. 2008;30:331–342. doi: 10.1016/j.nbd.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Huh Y, Ju MS, Park H, Han S, Bang YM, Ferris CF, Koppell GA, King JA, Kim ML, Kim DJ, Ahn CH, Oh MS. Clavulanic acid protects neurons in pharmacological models of neurodegenerative diseases. Drug Develop Res. 2010;71:351–357. [Google Scholar]
- Javitch JA, D’Amato RJ, Strittmatter SM, Snyder SH. Parkinsonism-inducing neurotoxin, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: uptake of the metabolite N-methyl-4-phenylpyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA. 1985;82:2173–2177. doi: 10.1073/pnas.82.7.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DJ, King JA, Zuccarelli L, Ferris CF, Koppel GA, Snowdon CT, Ahn CH. Clavulanic acid: a competitive inhibitor of beta-lactamases with novel anxiolytic-like activity and minimal side effects. Pharmacol Biochem Behav. 2009;93 (2):112–120. doi: 10.1016/j.pbb.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6 (5):513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Langston JW, Irwin I. MPTP: current concepts and controversies. Clin Neuropharmacol. 1986;9:485–507. [PubMed] [Google Scholar]
- LeBel CP, Ischiropoulos H, Bondy SC. Evaluation of the probe 2′,7′-dichlorofluorecein as a indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol. 1992;5:227–231. doi: 10.1021/tx00026a012. [DOI] [PubMed] [Google Scholar]
- Lohle M, Reichmann H. Clinical neuroprotection in Parkinson’s disease—still waiting for the breakthrough. J Neurol Sci. 2010;289:104–114. doi: 10.1016/j.jns.2009.08.025. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA, Kroemer G. Redox regulation of apoptosis: impact of thiol oxidation status on mitochondrial function. Eur J Immunol. 1997;27 (1):289–296. doi: 10.1002/eji.1830270142. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Yamada M, Tokiyoshi K, Miyawaki Y, Kanayama T. Penetration of potassium clavulanate/ticarcillin sodium into cerebrospinalfluid in neurosurgical patients. Jpn J Antibiot. 1994;47:93–101. [PubMed] [Google Scholar]
- Payne DJ, Cramp R, Winstanley DJ, Knowles DJ. Comparative activities of clavulanic acid, sulbactam, and tazobactam against clinically important beta-lactamases. Anti-microb Agents Chemother. 1994;38:767–772. doi: 10.1128/aac.38.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- Schober A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004;318:215–224. doi: 10.1007/s00441-004-0938-y. [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Watt JA, Singh BB. TRPC1 inhibits apoptotic cell degeneration induced by dopaminergic neurotoxin MPTP/MPP(+) Cell Calcium. 2009 Sep;46 (3):209–218. doi: 10.1016/j.ceca.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res. 2005;134:57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Heikkila RE. The influence of dose and dosing interval on MPTP-induced dopaminergic neurotoxicity in mice. Eur J Pharmacol. 1986;129:339–345. doi: 10.1016/0014-2999(86)90444-9. [DOI] [PubMed] [Google Scholar]
- Susin SA, Zamzami N, Kroemer G. Biochim. Biophys Acta. 1998;1366:151–165. doi: 10.1016/s0005-2728(98)00110-8. [DOI] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Mather P, Schubert D. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss T, Ravina B. Neuroprotection in Parkinson’s disease:myth or reality? Curr Neurol Neuro Sci Rep. 2008;8:304–309. doi: 10.1007/s11910-008-0047-5. [DOI] [PubMed] [Google Scholar]