

Introduction
G protein-coupled receptors bind ligands that induce conformational changes in the receptor and facilitate the activation of G protein heterotrimers by exchange of GDP for GTP on the G protein α subunit (Gilman, 1987). Upon activation, the G protein heterotrimer undergoes a conformational change that allows Gα and Gβγ subunits to regulate effector proteins such as adenylyl cyclases (ACs) (Coleman et al., 1994; Lambright et al., 1994; Taussig et al., 1994). The G protein signaling cycle ends by hydrolysis of GTP to GDP on the Gα subunit (Coleman et al., 1994; Mixon et al., 1995). In addition to G protein-coupled receptor modulation, the G protein activation cycle is sensitive to accessory proteins such as regulators of G protein signaling (RGS) or activator of G protein signaling (AGS) proteins (Blumer et al., 2007; Neubig and Siderovski, 2002; Sato et al., 2004). These proteins influence G protein signaling and effector activation by modulating the rate of GTP hydrolysis and the availability and/or stability of Gα or Gβγ subunits (Blumer et al., 2007; Neubig and Siderovski, 2002; Sato et al., 2004)
The activator of G protein signaling 3 (AGS3) is a G protein regulator that has been linked to adaptive behaviors involved with drugs of abuse (Bowers et al., 2008; Bowers et al., 2004; Yao et al., 2005). AGS3 was identified as a receptor-independent G protein activator and is thought to bind Gαi subunits in the GDP-bound state, thereby preventing re-association of Gα and Gβγ subunits (De Vries et al., 2000; Peterson et al., 2000; Takesono et al., 1999). The precise molecular actions of AGS3 have yet to be fully described, however it is hypothesized that AGS3 expression may specifically adjust the landscape of effector activation by modulating Gα and Gβγ subunit signaling. Previous studies examining the actions of AGS3 on G protein-coupled receptor signaling in cell-based assays suggest that the effects of AGS3-like molecules are dependent on the duration of receptor activation. For example, an AGS3 consensus peptide was shown to have no effect on the ability of D2DRs to modulate G protein-regulated inwardly rectifying potassium (GIRK) channels acutely, but promoted functional desensitization of this response following repeated receptor activation (Webb et al., 2005). Furthermore, the actions of AGS3 on α2 adrenergic receptor regulation of cAMP signaling were explored in CHO cells, which predominantly express AC6 (Varga et al., 1998). The acute effects on α2 adrenergic receptor-mediated AC signaling were not altered, but sensitization of AC in response to persistent α2 receptor activation was attenuated by AGS3 expression (Sato et al., 2004). More recent studies in nucleus accumbens/striatal neurons have suggested that opioid receptor-induced expression of AGS3 enhances the activity of AC5 and AC7 in a protein kinase-dependent manner (Fan et al., 2009).
The observations described above suggest a complex mode of AGS3 modulation of receptor-mediated AC signaling. The complexity is further exacerbated by the expression and signaling of multiple AC isoforms that display unique patterns of Gα and Gβγ regulation (Sunahara et al., 1996; Watts and Neve, 2005). The present study was designed to examine the effects of AGS3 on G protein-coupled receptor modulation of two recombinant ACs, AC1 and AC2. AC1 is a member of the Ca2+-stimulated group of ACs and can be activated by the Ca2+ ionophore, A23187, and inhibited by both Gαi and Gβγ subunits (Choi et al., 1992; Cumbay and Watts, 2001; Taussig et al., 1993). AC2 is a member of a group of ACs that are conditionally activated by Gβγ subunits (Federman et al., 1992; Taussig et al., 1993). AC2 is also robustly activated by protein kinase C (PKC) phosphorylation in response to phorbol ester stimulation (e.g. PMA) (Bernard et al., 2001; Shen et al., 2012; Yoshimura and Cooper, 1993). The distinct regulatory properties of AC1 and AC2 provide important tools to selectively study the effects of AGS3 expression on G protein-coupled receptor signaling.
It is widely accepted that alterations in cAMP signaling pathways and enhanced activation of dopamine systems in the brain play central roles in the molecular adaptations associated with drug addiction (Carlezon et al., 2005; McClung and Nestler, 2003; Nestler, 2001). The dopaminergic signaling and cAMP signaling pathways are linked by dopamine receptors that modulate adenylyl cyclases via G protein activation. There are two families of dopamine receptors, D1-like (D1 and D5) that couple to Gαs, and D2-like (D2, D3, and D4) the couple to Gαi/o (Missale et al., 1998). We chose to study D2LDR signaling based on the overlapping tissue distribution of the D2LDR with AC1 and AC2 in the brain, and its well-characterized roles in AC signaling and drug abuse, where AGS3 has also been implicated (Maldonado et al., 1997; Phillips et al., 1998; Ralph et al., 1999; Visel et al., 2006; Weiner et al., 1991). HEK293 cells expressing the D2LDR together with either AC1 or AC2 were used to explore cAMP signaling in the absence or presence of AGS3. Our studies revealed that AGS3 expression had modest, but significant potentiating effects on acute D2LDR modulation of AC1 or AC2 activity. In contrast, AGS3 displayed differential effects on AC regulation following persistent D2LDR activation. These findings, along with those reported in the literature, suggest that AGS3 modulates AC signaling in a manner that is isoform-specific and dependent on the duration of receptor activation.
Materials and methods
Materials
[3H]-cAMP (33 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Spiperone, (±)-quinpirole, A23187, 3-isobutyl-1-methylxanthine (IBMX), G418, Dulbecco's modified Eagle's medium (DMEM), and trichloroacetic acid were purchased from Sigma-Aldrich (St. Louis, MO). Phorbol 12-myristate 13-acetate (PMA) was purchased from Tocris Bioscience (Ellisville, MO). Lipofectamine 2000 and Zeocin were purchased from Invitrogen (Carlsbad, CA). FetalClone I (FCI) serum, bovine calf serum (BCS), and Earle's balanced salt solution (EBSS) were purchased from Hyclone (Logan, UT). Hygromycin B was purchased from Calbiochem (La Jolla, CA).
Cell culture and stable cell line generation
HEK293 cells stably expressing AC1 or AC2 were grown in DMEM supplemented with 5% FCI, 5% BCS, 1 unit/ml penicillin, 1 μg/ml streptomycin, 2.5 ng/ml amphotericin B, and either 100 μg/ml Hygromycin B (HEK-AC1) or 300 μg/ml G418 (HEK-AC2) and were maintained in a humidified incubator at 37°C with 5% CO2. Cell lines were transfected with the dual expression vector pBudCE4 with the gene for the human D2LDR driven by the CMV promoter and either rat AGS3-Venus (long splice variant), AGS3-Q/A-Venus, or no gene behind the EF-1α promoter. Several studies have utilized AGS3 fused with a fluorescent protein at the c-terminus (An et al., 2008; Oner et al., 2010; Oner et al., 2013; Vural et al., 2010) and these studies suggest that the AGS3-fluorescent fusion proteins retain similar cellular localization (An et al., 2008; Blumer et al., 2002; Pizzinat et al., 2001; Vural et al., 2010) and interaction with Gαi subunits (De Vries et al., 2000; Oner et al., 2010; Oner et al., 2013; Peterson et al., 2000) as compared to untagged AGS3. Stable transfections were carried out with Lipofectamine 2000 according to the manufacturer's protocol. Clones were isolated by selection with Zeocin (200 μg/ml) and characterized for specific AC function by cAMP accumulation assays (Online Resource 1), D2LDR expression by radioligand binding (Online Resource 2), and AGS3 expression by fluorescence microscopy (Online Resource 3).
Cyclic AMP accumulation assay
Cells were grown to confluency in 48-well plates and cAMP assays were performed on ice in assay buffer (EBSS containing 15 mM Na+-HEPES, 2% BCS, and 0.02% ascorbic acid). For acute cAMP accumulation experiments, cells were stimulated at 37°C for 15 minutes in the presence of 500 μM IBMX. The stimulation buffer was decanted and cells were lysed with ice-cold 3% trichloroacetic acid. The plate was stored at 4°C for at least 1 h before cAMP quantification. For persistent receptor activation experiments, cells were grown to confluency in 48-well plates and pretreated in assay buffer with either vehicle or 1 μM (±)-quinpirole for 2 h at 37°C and 5% CO2. Cells were washed with assay buffer three times (3 minutes each), and subsequently stimulated as described for acute cAMP accumulation assays. For desensitization assays, pretreatments were carried out as described above, followed by re-activation of the receptor as described for acute cAMP accumulation experiments. For heterologous sensitization experiments, subsequent stimulation was achieved by selective activation of AC isoforms (AC1, 10 μM A23187; AC2, 1 μM PMA) in the presence of 500 μM IBMX and 1 μM spiperone to block residual agonist binding from the pretreatment.
Cyclic AMP quantification
Cyclic AMP was quantified using a competitive binding assay (Przybyla and Watts, 2010). Duplicate samples of lysate from the cAMP accumulation assay were added to reaction tubes, followed sequentially by [3H]-cAMP (~1 nM final concentration), and cAMP-binding protein (~100 μg of crude bovine adrenal extract) in 500 μL of cAMP binding buffer (100 mM Tris-HCl, 100 mM NaCl, 3 mM EDTA, pH 7.4). The assay was carried out at 4°C for 2 hr and harvested by filtration through Millipore FB 96-well filter plates, and radioactivity was quantified on a TopCount NXT scintillation counter (Perkin Elmer). Cyclic AMP concentrations were estimated from a standard curve ranging from 300 pmol to 3 nmol cAMP.
Data analysis
GraphPad Prism software was used to generate all dose-response curves (GraphPad Software, San Diego, CA). All logIC50/logEC50, and maximal activation/inhibition values were calculated in GraphPad Prism using a non-linear regression and sigmoidal dose-response equation and were analyzed by unpaired t-tests.
Results and discussion
AGS3 interacts with the GDP-bound state of Gαi subunits, and it has been suggested that this interaction prevents the re-association of Gαi and Gβγ subunits (De Vries et al., 2000; Peterson et al., 2000). The interaction between AGS3 and Gαi-GDP may cause altered G protein signaling through effectors regulated by Gαi and Gβγ subunits. AC isoforms display differential patterns of regulation by G protein subunits (Sunahara et al., 1996; Watts and Neve, 2005). Given that AGS3 may alter signaling by Gαi and/or Gβγ subunits, and AC isoforms are differentially regulated by G protein subunits, we were interested in studying the effects of AGS3 expression on the regulation of AC isoforms following acute and persistent activation of D2LDRs.
Modulation of acute D2LDR-mediated AC1 and AC2 signaling by AGS3 expression
Our initial studies used HEK293 cells stably expressing AC1 or AC2 and the D2LDR. Recombinant AC1 can be selectively activated in HEK293 cells by Ca2+ using the calcium ionophore, A23187 or by capacitative calcium entry (Choi et al., 1992; Cooper et al., 1994). To observe acute Gαi regulation, AC1 activity was selectively increased using A23187, and inhibition by the D2DR agonist quinpirole was examined. A23187-stimulated AC1 activity was inhibited by 54±2% with a logIC50 of −8.08±0.13 (Fig. 1a). In cells expressing AGS3, modest, but significant increases in maximal inhibition (67±4%) and potency (logIC50 = –8.83±0.19) for D2LDR-mediated inhibition of AC1 activity by quinpirole were observed. AC2 is conditionally stimulated (requiring co-activation of AC2 by Gαs or PKC) by Gβγ subunits following the activation of Gαi/o-coupled receptors (Tsu and Wong, 1996; Yoshimura and Cooper, 1993; Zimmermann and Taussig, 1996). To study acute Gβγ subunit signaling, HEK293 cells stably expressing AC2 and the D2LDR were incubated with PMA to stimulate AC2, and quinpirole to activate the D2LDR (subsequently releasing Gβγ subunits). Maximal activation of the D2LDR by quinpirole resulted in a robust increase in cAMP accumulation through conditional activation of AC2 (240±18% of PMA response) with a logEC50 of −7.05±0.06 (Fig. 1b). AGS3 co-expression did not affect the maximal activation of AC2 (250±20% of PMA response), but a significant shift in the potency (log EC50 = –7.45±0.06) of quinpirole-potentiated cAMP accumulation was observed as compared to cells not expressing AGS3. Though modest, the effects of AGS3 on D2LDR-mediated regulation of AC1 or AC2 are consistent with enhanced Gβγ subunit modulation of each effector. Specifically, Gβγ subunits inhibit signaling mediated by AC1, but increase AC2 activity, and AGS3 expression augments the potency of these effects, presumably by binding Gαi subunits. The effects of AGS3 expression on acute D2LDR-mediated AC1 or AC2 activity are contrary to other cell-based studies examining the effects of AGS3 expression on acute G protein signaling. For example, AGS3 did not alter α2-adrenergic receptor-inhibited AC activity in CHO cells (Sato et al., 2004). Perhaps more relevant to our D2DR-AC2 studies is the observation that AGS3 failed to alter D2SDR-stimulated GIRK channel activity, a Gβγ mediated signaling event (Webb et al., 2005). These findings suggest that AGS3 may display AC-specific or even effector-specific regulation of acute G protein signaling.
Fig. 1.
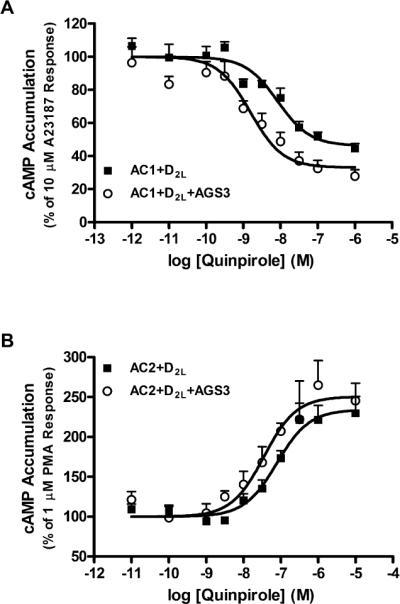
Effect of AGS3 expression on acute regulation of AC isoforms. a. Acute inhibition of A23187 (10 μM)-stimulated cAMP accumulation was measured following incubation with quinpirole as indicated in HEK293 cells stably expressing AC1 and the D2LDR, or AC1 and the D2LDR together with AGS3-Venus. b. Cyclic AMP accumulation was quantified in HEK293 cells stably expressing AC2 and the D2LDR with or without AGS3-Venus following acute treatment with PMA (1 μM) and increasing concentrations of quinpirole as indicated. Data points represent mean ± S.E.M. of at least three independent experiments performed in duplicate
Desensitization of D2LDR-mediated AC1 and AC2 signaling
In addition to studying AC regulation by acute activation of the D2LDR, we were also interested in studying desensitization of the D2LDR. Cells stably expressing AC1 and the D2LDR were treated with quinpirole for 2 h in an effort to promote functional receptor desensitization. Following washing, cells were re-exposed to quinpirole and the subsequent cAMP accumulation was measured. Persistent D2LDR activation by quinpirole failed to induce significant desensitization of D2LDR modulation of AC1 activity. Specifically, quinpirole treatment maximally inhibited A23187-stimulated AC1 activity by 56±9.1% with a log IC50 of −8.0±0.20 following persistent D2LDR activation, as compared to maximum inhibition of 59±1.8% and log IC50 of −8.5±0.20 after vehicle pretreatment (n=2). We then examined the effects of AGS3 expression on D2LDR-modulated AC1 activity following persistent D2LDR stimulation. These experiments revealed that AGS3-expressing cells pretreated with quinpirole displayed a desensitization of D2LDR-modulated AC1 inhibition that was manifested as a significant loss of maximal inhibition (60±6%) compared to vehicle pretreated cells (84±8%), but without a significant change in potency (logIC50 = −8.03±0.39 and −8.64±0.17, respectively) (Fig. 2). The AGS3-promoted desensitization of AC1 inhibition may occur through interactions with Gαi subunits that are involved in the inhibition AC1 activity. However, it has been suggested that Gαi-AGS3 complexes can couple with, and be regulated by Gαi-coupled receptors upon agonist activation, allowing for the possibility of indirect modulation of downstream Gβγ subunit signaling (Oner et al., 2010). Furthermore, an AGS3 consensus peptide disrupted Gβγ subunit activation of GIRK channels following repeated stimulation of D2SDRs (Webb et al., 2005). Given that Gβγ subunits inhibit AC1 activity (Taussig et al., 1993), these data present the possibility that the desensitization of AC1 inhibition is also associated with altered Gβγ subunit signaling.
Fig. 2.
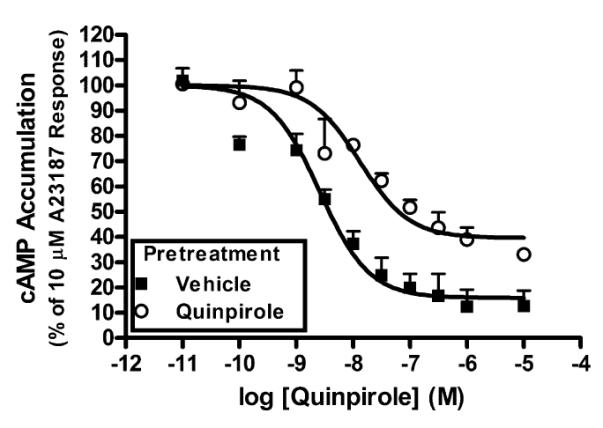
The effect of AGS3 expression on functional desensitization of D2LDR-modulated AC1 activity. HEK293 cells stably expressing AC1, the D2LDR, and AGS3-Venus were treated with quinpirole (1 μM) or vehicle for 2 hrs, followed by selective AC1 stimulation with A23187 (10 μM) and activation of the D2LDR with quinpirole (1 μM). Data represent the mean ± S.E.M. of three independent experiments
In contrast to AC1, a complete desensitization of D2LDR-mediated potentiation of AC2 activation was observed following quinpirole pretreatment in D2LDR-AC2 cells. Specifically, quinpirole pretreatment resulted in 103±11% desensitization of D2LDR-mediated potentiation of AC2 activity as compared to vehicle pretreatment (Fig. 3). These observations suggest potential differences in the general mechanisms for desensitization of D2LDR-modulated AC1 and AC2 activity. Such mechanisms presumably reflect the differential regulatory properties of AC1 and AC2 that involve G protein modulation (Hanoune and Defer, 2001). Subsequent experiments examined the effects of AGS3 on desensitization of D2LDR-potentiated AC2 activity. Surprisingly, expression of AGS3 resulted in significantly less desensitization (46±7%) of D2LDR-mediated potentiation of AC2 activity (Fig. 3). The specificity of this blockade was probed by expressing an AGS3 mutant that does not bind Gαi subunits, AGS3-Q/A (Peterson et al., 2002). AGS3-Q/A failed to significantly alter quinpirole-induced desensitization of D2LDR-AC2 signaling (78±3%), suggesting that AGS3 expression inhibits desensitization of D2LDR signaling through AC2 in a manner that is dependent on the interaction of AGS3 with Gαi.
Fig. 3.
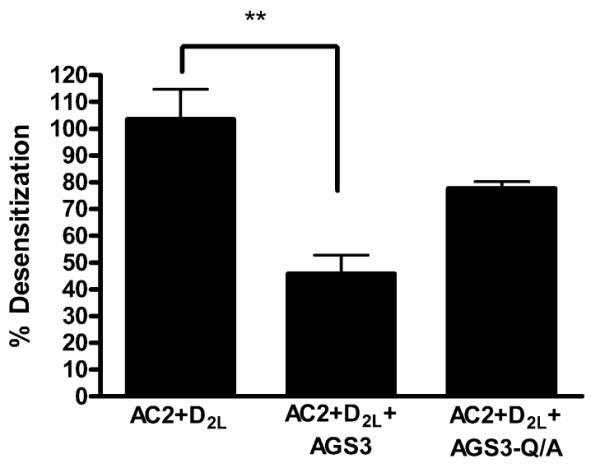
The effect of AGS3 expression on functional desensitization of D2LDR-modulated AC2 activity. HEK293 cells stably expressing AC2 and the D2LDR alone, or together with AGS3-Venus or AGS3-Q/A-Venus were treated with quinpirole (1 μM) or vehicle for 2 hrs, followed by stimulation with PMA (1 μM) and quinpirole (1 μM). Data represent the mean ± S.E.M. of at least three independent experiments. ** p < 0.01, compared with AC2+D2L condition, one way analysis of variance followed by Dunnett's post hoc test
Differential effects of AGS3 expression on adenylyl cyclase signaling
Effects of AGS3 expression on heterologous sensitization of AC1 and AC2
In addition to desensitization, persistent activation of many Gαi/o-coupled receptors (e.g. D2 dopamine and μ-opioid) results in enhanced subsequent AC activation. This heterologous sensitization (a.k.a. superactivation) of cAMP signaling involves both Gαi/o and Gβγ subunits (Watts and Neve, 2005), but the mechanisms of sensitization for AC1 and AC2 appear to differ (Cumbay and Watts, 2001). The functional effects of AGS3 expression on AC sensitization following persistent activation of the D2LDR were examined. For these experiments, cells expressing AC1 and the D2LDR were stimulated with the D2DR agonist quinpirole for 2 h, followed by selective activation of AC1 by A23187. Quinpirole pretreatment resulted in a 235±4% enhancement of A23187-stimulated AC1 activity compared to the vehicle pretreatment (Fig. 4a). Cells co-expressing AGS3 displayed a reduction in subsequent AC1 stimulation (178±12% of vehicle condition) following quinpirole pretreatment, suggesting that AGS3 expression attenuates AC1 sensitization by ~40%. The effects of AGS3 expression on the regulation of AC2 following persistent D2LDR activation were also explored. AC2 displayed an enhanced responsiveness (182±9% of vehicle condition) to stimulation by PMA following quinpirole pretreatment (Fig. 4b). The D2LDR-mediated sensitization of AC2 was nearly eliminated (120±3% of vehicle condition) in AGS3 expressing cells. In contrast, AGS3-Q/A did not alter D2LDR-mediated sensitization of AC2 (202±10% of vehicle response), suggesting that AGS3-Gαi subunit interactions are involved in the inhibition of AC2 sensitization. The ability of AGS3 expression to prevent heterologous sensitization of AC1 and AC2 is similar to that observed by expressing the Gβγ subunit scavenger, βARK-CT ((Koch et al., 1994; Nguyen and Watts, 2005) and data not shown for AC2). These data add support to the hypothesis that AGS3 potentially alters Gβγ subunit signaling following persistent Gi-coupled receptor activation (Oner et al., 2010; Webb et al., 2005)
Fig. 4.
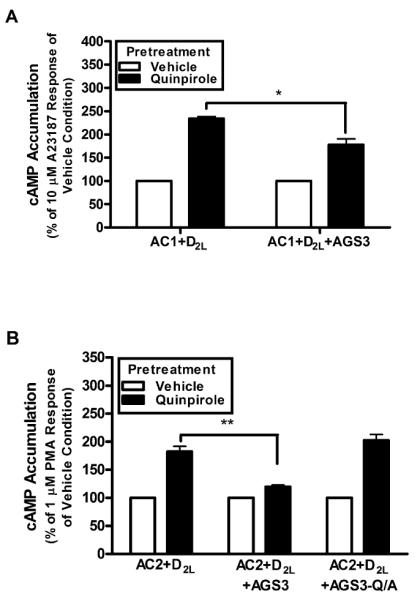
Effect of AGS3 expression on heterologous sensitization of AC isoforms. a. HEK293 cells stably expressing AC1 and D2LDR were stimulated with vehicle or quinpirole (1 μM) for 2 hr, washed, and subsequently stimulated with A23187 (10 μM) as indicated. Data represent mean ± S.E.M. of three independent experiments. * p < 0.05, unpaired t-test. b. HEK293 cells stably expressing AC2 and D2LDR in the absence of, or coexpressed with AGS3-Venus or AGS3-Q/A-Venus as indicated were pretreated with vehicle or quinpirole (1 μM) for 2 hr, washed, and subsequently stimulated with PMA (1 μM). Data represent mean ± S.E.M. of at least three independent experiments. ** p < 0.01, one way analysis of variance followed by Dunnett's post hoc test
The inhibition of D2LDR-mediated sensitization of AC1 and AC2 by AGS3 expression is consistent with a report revealing that AGS3 inhibits α2-adrenergic receptor-mediated sensitization of AC isoforms expressed in CHO cells (predominantly expressing AC6) (Sato et al., 2004; Varga et al., 1998). In contrast, evidence suggests that opioid receptor-induced up-regulation of AGS3 expression mediates sensitization of AC5 and AC7 in nucleus accumbens/striatal neurons (Fan et al., 2009). These differential effects suggest an isoform-dependence for sensitization that may reflect complex and unique isoform-specific mechanisms of AC sensitization involving Gαi, Gβγ, or a combination of the two, and subsequently altered downstream signaling. Alternatively, the isoform-specific effects on AC signaling may reflect differences in receptor types, cell types, or experimental paradigms. The differential effects of AGS3 on AC sensitization, desensitization, and acute D2LDR-mediated signaling are similar to what has been observed for ligands that possess functional selectivity (or promote agonist-directed trafficking) (Kenakin, 1995). Such an observation posits that receptor- and effector-modulating signaling molecules introduce an additional level of complexity for cell signaling studies. For example, AGS3 differentially alters desensitization of D2LDR-mediated modulation of AC1 and AC2, while reducing sensitization of both AC isoforms. These complex mechanisms for regulation by AGS3 in vitro suggest the opportunity for greater signaling diversity in vivo.
Significance of AGS3-regulated AC signaling to drugs of abuse
In vivo studies suggest that AGS3 has a “gatekeeper” role for drug-seeking behavior in response to heroin, cocaine, or ethanol. For example, knocking out AGS3 expression in specific brain regions resulted in a loss of drug-seeking behavior or cocaine-induced locomotor sensitization (Bowers et al., 2008; Bowers et al., 2004; Yao et al., 2005). Interestingly, AGS3 expression is up-regulated in the nucleus accumbens core or prefrontal cortex during withdrawal periods following prolonged ethanol or cocaine self-administration, respectively (Bowers et al., 2008; Bowers et al., 2004). In conjunction with overwhelming evidence that dopamine systems and cAMP signaling pathways are involved in drug addiction (Carlezon et al., 2005; McClung and Nestler, 2003; Nestler, 2001), our data suggest that AGS3 overexpression during periods of withdrawal may change the profile of signaling through specific AC isoforms in response to persistent Gαi/o-coupled receptor activation. In addition, recent studies with AC knockout mice suggest a role for Ca2+-stimulated ACs (AC1 and AC8) in the regulation of cocaine behavioral sensitization (DiRocco et al., 2009). It will be important to study the role of individual AC isoforms in specific brain regions in the context of drug-seeking behavior, where AGS3 may be exerting its effects by fine-tuning D2LDR-mediated signaling through distinct AC isoforms.
Conclusions
Taken as a whole, our data suggest that AGS3 expression alters D2LDR-mediated regulation of effector proteins in a manner that is effector-specific and dependent on the duration of Gαi-coupled receptor activation. The expression of AGS3 has modest, but potency-enhancing effects on effector modulation in response to acute Gαi-coupled receptor stimulation. Expression of AGS3 promoted desensitization of prolonged D2LDR-mediated signaling through AC1, but reduced desensitization of AC2 signaling. In addition, D2LDR-mediated sensitization of AC1 and AC2 signaling was attenuated by AGS3 expression. It is becoming apparent that AGS3 is involved in altering G protein signaling in a complex fashion that is effector-specific.
Supplementary Material
Acknowledgements
We thank Elisabeth Garland and Dr. Karin Ejendal for valuable comments regarding manuscript preparation and Monica Soto Velasquez and Tarsis Brust Fernandes for assistance with confocal microscopy and receptor saturation binding, respectively. We also thank Dr. Stephen Lanier and Dr. Joe Blumer for AGS3 constructs. This work was supported by NIMH grant MH060397 to VJW.
References
- An N, Blumer JB, Bernard ML, Lanier SM. The PDZ and Band 4.1 Containing Protein Frmpd1 Regulates the Subcellular Location of Activator of G-protein Signaling 3 and Its Interaction with G-proteins. Journal of Biological Chemistry. 2008;283:24718–24728. doi: 10.1074/jbc.M803497200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard ML, Peterson YK, Chung P, Jourdan J, Lanier SM. Selective interaction of AGS3 with G-proteins and the influence of AGS3 on the activation state of G-proteins. Journal of Biological Chemistry. 2001;276:1585–1593. doi: 10.1074/jbc.M005291200. [DOI] [PubMed] [Google Scholar]
- Blumer JB, Chandler LJ, Lanier SM. Expression analysis and subcellular distribution of the two G-protein regulators AGS3 and LGN indicate distinct functionality - Localization of LGN to the midbody during cytokinesis. Journal of Biological Chemistry. 2002;277:15897–15903. doi: 10.1074/jbc.M112185200. [DOI] [PubMed] [Google Scholar]
- Blumer JB, Smrcka AV, Lanier SM. Mechanistic pathways and biological roles for receptor-independent activators of G-protein signaling. Pharmacology & Therapeutics. 2007;113:488–506. doi: 10.1016/j.pharmthera.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, Hopf FW, Chou JK, Guillory AM, Chang SJ, Janak PH, Bonci A, Diamond I. Nucleus accumbens AGS3 expression drives ethanol seeking through G beta gamma. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12533–12538. doi: 10.1073/pnas.0706999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, Lanier SM, Kalivas PW. Activator of G protein signaling 3: A gatekeeper of cocaine sensitization and drug seeking. Neuron. 2004;42:269–281. doi: 10.1016/s0896-6273(04)00159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Duman RS, Nestler EJ. The many faces of CREB. Trends in Neurosciences. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Choi EJ, Wong ST, Hinds TR, Storm DR. Calcium and Muscarinic Agonist Stimulation of Type-I Adenylyl Cyclase in Whole Cells. Journal of Biological Chemistry. 1992;267:12440–12442. [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of Active Conformations of G(I-Alpha-1) and the Mechanism of GTP Hydrolysis. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- Cooper DMF, Yoshimura M, Zhang YX, Chiono M, Mahey R. Capacitative Ca2+ Entry Regulates Ca2+-Sensitive Adenylyl Cyclases. Biochemical Journal. 1994;297:437–440. doi: 10.1042/bj2970437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbay MG, Watts VJ. Heterologous sensitization of recombinant adenylate cyclases by activation of D-2 dopamine receptors. Journal of Pharmacology and Experimental Therapeutics. 2001;297:1201–1209. [PubMed] [Google Scholar]
- De Vries L, Fischer T, Tronchere H, Brothers GM, Strockbine B, Siderovski DP, Farquhar MG. Activator of G protein signaling 3 is a guanine dissociation inhibitor for G alpha(i) subunits. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14364–14369. doi: 10.1073/pnas.97.26.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRocco DP, Scheiner ZS, Sindreu CB, Chan GCK, Storm DR. A Role for Calmodulin-Stimulated Adenylyl Cyclases in Cocaine Sensitization. Journal of Neuroscience. 2009;29:2393–2403. doi: 10.1523/JNEUROSCI.4356-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan PD, Jiang Z, Diamond I, Yao LN. Up-Regulation of AGS3 during Morphine Withdrawal Promotes cAMP Superactivation via Adenylyl Cyclase 5 and 7 in Rat Nucleus Accumbens/Striatal Neurons. Molecular Pharmacology. 2009;76:526–533. doi: 10.1124/mol.109.057802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federman AD, Conklin BR, Schrader KA, Reed RR, Bourne HR. Hormonal-Stimulation of Adenylyl Cyclase through Gi-Protein Beta-Gamma-Subunits. Nature. 1992;356:159–161. doi: 10.1038/356159a0. [DOI] [PubMed] [Google Scholar]
- Gilman AG. G-Proteins - Transducers of Receptor-Generated Signals. Annual Review of Biochemistry. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annual Review of Pharmacology and Toxicology. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Agonist-Receptor Efficacy .2. Agonist Trafficking of Receptor Signals. Trends in Pharmacological Sciences. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular Expression of the Carboxyl-Terminus of a G-Protein-Coupled Receptor Kinase Attenuates G(Beta-Gamma)-Mediated Signaling. Journal of Biological Chemistry. 1994;269:6193–6197. [PubMed] [Google Scholar]
- Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural Determinants for Activation of the Alpha-Subunit of a Heterotrimeric G-Protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. 1997;388:586–589. doi: 10.1038/41567. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and Delta FosB. Nature Neuroscience. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: From structure to function. Physiological Reviews. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and Quaternary Structural-Changes in G(I-Alpha-1) Induced by GTP Hydrolysis. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nature Reviews Neuroscience. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Neubig RR, Siderovski DR. Regulators of G-protein signalling as new central nervous system drug targets. Nature Reviews Drug Discovery. 2002;1:187–197. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- Nguyen CH, Watts VJ. Dexras1 blocks receptor-mediated heterologous sensitization of adenylyl cyclase 1. Biochemical and Biophysical Research Communications. 2005;332:913–920. doi: 10.1016/j.bbrc.2005.05.041. [DOI] [PubMed] [Google Scholar]
- Oner SS, An NF, Vural A, Breton B, Bouvier M, Blumer JB, Lanier SM. Regulation of the AGS3-G alpha(i) Signaling Complex by a Seven-transmembrane Span Receptor. Journal of Biological Chemistry. 2010;285:33949–33958. doi: 10.1074/jbc.M110.138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oner SS, Maher EM, Gabay M, Tall GG, Blumer JB, Lanier SM. Regulation of the G-protein Regulatory-G alpha(i) Signaling Complex by Nonreceptor Guanine Nucleotide Exchange Factors. Journal of Biological Chemistry. 2013;288:3003–3015. doi: 10.1074/jbc.M112.418467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson YK, Bernard ML, Ma HZ, Hazard S, Graber SG, Lanier SM. Stabilization of the GDP-bound conformation of Gi alpha by a peptide derived from the G-protein regulatory motif of AGS3. Journal of Biological Chemistry. 2000;275:33193–33196. doi: 10.1074/jbc.C000509200. [DOI] [PubMed] [Google Scholar]
- Peterson YK, Hazard S, Graber SG, Lanier SM. Identification of structural features in the G-protein regulatory motif required for regulation of heterotrimeric G-proteins. Journal of Biological Chemistry. 2002;277:6767–6770. doi: 10.1074/jbc.C100699200. [DOI] [PubMed] [Google Scholar]
- Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M, Grandy DK, Low MJ. Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D-2 receptors. Nature Neuroscience. 1998;1:610–615. doi: 10.1038/2843. [DOI] [PubMed] [Google Scholar]
- Pizzinat N, Takesono A, Lanier SM. Identification of a truncated form of the G-protein regulator AGS3 in heart that lacks the tetratricopeptide repeat domains. Journal of Biological Chemistry. 2001;276:16601–16610. doi: 10.1074/jbc.M007573200. [DOI] [PubMed] [Google Scholar]
- Przybyla JA, Watts VJ. Ligand-Induced Regulation and Localization of Cannabinoid CB1 and Dopamine D-2L Receptor Heterodimers. Journal of Pharmacology and Experimental Therapeutics. 2010;332:710–719. doi: 10.1124/jpet.109.162701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph RJ, Varty GB, Kelly MA, Wang YM, Caron MG, Rubinstein M, Grandy DK, Low MJ, Geyer MA. The dopamine D-2, but not D-3 or D-4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. Journal of Neuroscience. 1999;19:4627–4633. doi: 10.1523/JNEUROSCI.19-11-04627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Gettys TW, Lanier SM. AGS3 and signal integration by G alpha(s)- and G alpha(i)-coupled receptors - AGS3 blocks the sensitization of adenylyl cyclase following prolonged stimulation of a G alpha(i)-coupled receptor by influencing processing of G alpha(i) Journal of Biological Chemistry. 2004;279:13375–13382. doi: 10.1074/jbc.M312660200. [DOI] [PubMed] [Google Scholar]
- Shen J, Wachten S, Halls M, Everett K, Cooper D. Muscarinic receptors stimulate AC2 by novel phosphorylation sites, whereas Gbetagamma subunits exert opposing effects depending on the G-protein source. Biochemical Journal. 2012;447:393–405. doi: 10.1042/BJ20120279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunahara RK, Dessauer CW, Gilman AG. Complexity and diversity of mammalian adenylyl cyclases. Annual Review of Pharmacology and Toxicology. 1996;36:461–480. doi: 10.1146/annurev.pa.36.040196.002333. [DOI] [PubMed] [Google Scholar]
- Takesono A, Cismowski MJ, Ribas C, Bernard M, Chung P, Hazard S, Duzic E, Lanier SM. Receptor-independent activators of heterotrimeric G-protein signaling pathways. Journal of Biological Chemistry. 1999;274:33202–33205. doi: 10.1074/jbc.274.47.33202. [DOI] [PubMed] [Google Scholar]
- Taussig R, Quarmby LM, Gilman AG. Regulation of Purified Type-I and Type-II Adenylyl Cyclases by G-Protein Beta-Gamma-Subunits. Journal of Biological Chemistry. 1993;268:9–12. [PubMed] [Google Scholar]
- Taussig R, Tang WJ, Hepler JR, Gilman AG. Distinct Patterns of Bidirectional Regulation of Mammalian Adenylyl Cyclases. Journal of Biological Chemistry. 1994;269:6093–6100. [PubMed] [Google Scholar]
- Tsu RC, Wong YH. G(i)-mediated stimulation of type II adenylyl cyclase is augmented by G(q)-coupled receptor activation and phorbol ester treatment. Journal of Neuroscience. 1996;16:1317–1323. doi: 10.1523/JNEUROSCI.16-04-01317.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga EV, Stropova D, Rubenzik M, Wang M, Landsman RS, Roeske WR, Yamamura HI. Identification of adenylyl cyclase isoenzymes in CHO and B82 cells. European Journal of Pharmacology. 1998;348:R1–R2. doi: 10.1016/s0014-2999(98)00258-1. [DOI] [PubMed] [Google Scholar]
- Visel A, Alvarez-Bolado G, Thaller C, Eichele G. Comprehensive analysis of the expression patterns of the adenylate cyclase gene family in the developing and adult mouse brain. Journal of Comparative Neurology. 2006;496:684–697. doi: 10.1002/cne.20953. [DOI] [PubMed] [Google Scholar]
- Vural A, Oner S, An N, Simon V, Ma D, Blumer JB, Lanier SM. Distribution of Activator of G-Protein Signaling 3 within the Aggresomal Pathway: Role of Specific Residues in the Tetratricopeptide Repeat Domain and Differential Regulation by the AGS3 Binding Partners Gi alpha and Mammalian Inscuteable. Molecular and Cellular Biology. 2010;30:1528–1540. doi: 10.1128/MCB.01018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. Sensitization of adenylate cyclase by G alpha(i/o)-coupled receptors. Pharmacology & Therapeutics. 2005;106:405–421. doi: 10.1016/j.pharmthera.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Webb CK, McCudden CR, Willard FS, Kimple RJ, Siderovski DP, Oxford GS. D2 dopamine receptor activation of potassium channels is selectively decoupled by G alpha(i)-specific GoLoco motif peptides. Journal of Neurochemistry. 2005;92:1408–1418. doi: 10.1111/j.1471-4159.2004.02997.x. [DOI] [PubMed] [Google Scholar]
- Weiner DM, Levey AI, Sunahara RK, Niznik HB, Odowd BF, Seeman P, Brann MR. D1 and D2 Dopamine Receptor Messenger-Rna in Rat-Brain. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:1859–1863. doi: 10.1073/pnas.88.5.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L, McFarland K, Fan P, Jiang Z, Inoue Y, Diamond I. Activator of G protein signaling 3 regulates opiate activation of protein kinase A signaling and relapse of heroin-seeking behavior. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8746–8751. doi: 10.1073/pnas.0503419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Cooper DMF. Type-Specific Stimulation of Adenylylcyclase by Protein-Kinase-C. Journal of Biological Chemistry. 1993;268:4604–4607. [PubMed] [Google Scholar]
- Zimmermann G, Taussig R. Protein kinase C alters the responsiveness of adenylyl cyclases to G protein alpha and beta gamma subunits. Journal of Biological Chemistry. 1996;271:27161–27166. doi: 10.1074/jbc.271.43.27161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.