

Abstract
Claudins are a family of tight junction (TJ) integral membrane proteins that play a crucial role in maintaining cell polarity, adhesion, and paracellular permeability. Changes in expression levels of claudin proteins have been associated with human lung cancer. Previously, we have reported that claudin‐7 expression is significantly downregulated in human lung carcinomas. To investigate the role of claudin‐7 in lung cancer cells after anti‐cancer drug treatments, we transfected claudin‐7 cDNA into human NCI‐H522 lung cancer cells, which have no detectable expression of claudin‐7 protein. Flow cytometry analysis demonstrated that cells transfected with claudin‐7 had a significantly higher percentage of cell apoptosis when compared to that of vector transfected cell population. The cell viability assayed by MTT and Annexin V was significantly decreased and cell apoptosis was dramatically increased in claudin‐7 transfected cells compared to that of vector transfected cells after cisplatin treatment. Cisplatin is an anti‐cancer drug clinically used to treat tumors in several tissues including lung tumors. Most importantly, after cisplatin treatment, the expression levels of cleaved caspase‐3, ‐8, and poly adenosine 5′‐diphosphate ribose polymerase (PARP) were much higher in claudin‐7 transfected cells than in control cells. Furthermore, using the site‐directed mutagenesis approach, we identified that claudin‐7 was phosphorylated at serine 204 by protein kinase C. Non‐phosphorylated claudin‐7 mutant showed increased cell viability, suggesting that phosphorylation increases chemosensitivity to cisplatin treatment. We concluded that claudin‐7 expression in H522 lung cancer cells increases chemosensitivity to cisplatin through the increased activation of caspase pathway.
Cancer is generally defined as the rapid growth of abnormal cells beyond their usual boundaries, allowing for the spread to other tissues and organs.1 In healthy tissue, epithelial cells are strictly regulated and possess specific cell polarity and organization. Under these conditions, cell growth and motility are regulated by intercellular communication via cell–cell adhesion, cell–matrix adhesion, and gap junction communication.2 Tight junctions (TJs), adheren junctions, and desmosomes form the intercellular junctional complex, which allows the epithelial cell layer to maintain its normal structure.3, 4
The TJ forms a continuous circumferential barrier at the apical end of the lateral membrane in sheets of epithelial cells. Tight junctions create and maintain membrane polarity by restricting the exchange of lipids and proteins in the apical and basolateral membranes, and function as a gatekeeper to the paracellular space by controlling the transfer of water, solutes, and immune cells.5, 6 Claudins are the major structural and functional components of TJs.5 They are a family of tetraspan transmembrane proteins consisting of short amino and carboxyl termini and two extracellular loops. Claudins have a molecular mass of approximately 23 kDa and function in the formation of ion selective pores or barriers and in the adhesion between adjacent cells.7, 8, 9, 10, 11, 12 Phosphorylation of claudins at potential serine and/or threonine phosphorlyation sites in their cytoplasmic carboxyl terminal domain is a known mechanism by which claudins are regulated.4, 13 Recent studies have indicated that WNK4 kinase phosphorylates claudin‐7 in kidney epithelial cells, which increases paracellular Cl− permeability, while protein kinase C (PKC) phosphorylates claudin‐4 to regulate TJ barrier function in ovarian cancer cells.14, 15 In addition to regulating paracellular permeability, claudins are implied to assist in regulating the cell cycle.3, 16, 17 The carboxyl terminus of most claudin proteins ends with tyrosine and valine residues, which bind to the PDZ (PSD95, DLG1, and ZO‐1) domains of zonula occludens (ZO) proteins, ZO‐1, ‐2, and ‐3.18
The expression of claudins in cancerous cells is altered. Claudin‐1 expression is reduced in breast cancer19, 20 and colon cancer.21 Claudin‐7 is downregulated in invasive breast cancer22 as well as head and neck cancers.23 The change in claudin expression supports the idea that tumorigenesis is related to the loss of TJ functions. Loss of TJ functions correlates with the loss of cohesion, invasion, and lack of differentiation observed in cancer cells. Re‐expression of claudins in cancerous cells is hypothesized to reduce cancer development by reducing invasiveness and initiating apoptosis of cancer cells. Claudin‐4 re‐expression has reduced invasiveness in pancreatic cancer cells,24 while claudin‐1 re‐expression in breast cancer cells induced apoptosis.25
Several studies have shown that the reduction of claudin‐7 in breast carcinomas is associated with metastasis.22, 26 Recently, Oshimi et al. have shown that the reduced claudin‐7 gene expression correlates with venous invasion and liver metastasis in colorectal cancer.27 The decrease of claudin‐7 has also been correlated with metastasis in squamous cell carcinomas (SCCs) of the esophagus.28 A recent study by Lioni et al.29 examined the expression of claudin‐7 in the tissue of normal human esophageal cells and esophageal SCCs. Claudin‐7 was expressed in the cell membranes of normal esophageal cells, but in SCCs claudin‐7 was mislocalized to the cytoplasm and its expression was reduced.29 Moldvay et al.30 reported that claudin‐7 expression was upregulated in human lung cancer, though its mRNA expression was downregulated in certain types of lung cancers when compared to normal bronchial epithelial cells. Our recent study showed that claudin‐7 expression was either downregulated or its localization was disrupted in human lung SCCs.31 Clearly, the differential expression of claudin‐7 could be associated with different types or subtypes of cancers as well as different stages of the same type of cancer.
Chemotherapy has been widely used and has played an important role in the treatment of human lung cancer. Cisplatin is a platinum‐based chemotherapeutic drug clinically used to treat various forms of cancers including lung cancer. While downregulation of claudins correlates with increased invasion and metastasis in cancer, it is unclear whether claudins will affect the chemosensitivity to cisplatin treatment. Here, we used NCI‐H522 (H522), a human non‐small cell lung cancer cell line that has no endogenous expression of claudin‐7, to investigate the effect of claudin‐7 on cisplatin treatment by transfection of claudin‐7 into H522 cells. We demonstrate that claudin‐7 increases chemosensitivity to cisplatin in human H522 lung cancer cells through the activation of caspase pathway.
Materials and Methods
Antibodies and reagents
Rabbit anti‐claudin‐7 antibody was obtained from IBL (Immuno‐Biological Laboratories, Fujioka‐Shi, Gunma, Japan). Rabbit anti‐claudin‐1 and ‐3 antibodies were purchased from Invitrogen (Carlsbad, CA, USA). The antibodies against caspase‐3, ‐8, ‐9, and poly adenosine 5′‐diphosphate ribose polymerase (PARP) were from Cell Signaling Technology (Beverly, MA, USA). Horse radish peroxidase‐conjugated anti‐rabbit and anti‐mouse secondary antibodies were purchased from Promega (Madison, WI, USA). Cisplatin, Thiazolyl Blue Tetrazolium Bromide (MTT), and lithium chloride were from Sigma (St. Louis, MO, USA). Annexin V‐FITC and Bisindolylmaleimide I were obtained from Calbiochem (EMD, Darmstadt, Germany). Chelerythrine chloride was from Alomone Labs (Jerusalem, Israel).
Cell culture and transfection
The human lung adenocarcinoma cancer cell line NCI‐H522 was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and grown in RPMI 1640 culture medium supplemented with 10% FBS, 100 units/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified air‐5% CO2 atmosphere.
H522 cells were cultured to 80% confluence and harvested using 0.25% trypsin. The cells were suspended in Nucleofector Solution V (Lonza, Basel, Switzerland) to a final concentration of 1 × 106 cells/100 μL. Claudin‐7 or claudin‐3 cDNA was added to the cell suspension and the transfection was performed using Nucleofector II electroporation device (Lonza). As a control, H522 cells were also transfected with an empty vector. To obtain the stable H522 vector, claudin‐7 and claudin‐3 cell lines, transfected cells were treated with 400 μg/mL geneticin (G418) 48 h after transfection, and passaged using culture medium containing G418. Stable H522 cell lines transfected with claudin‐7 mutants at known phosphorylation sites were generated using the same method.
Cell viability and Annexin V assays
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide cell viability assay was used to analyze the cytotoxic effect of cisplatin on the H522 vector and claudin‐7 transfected cells. Cells were cultured in 24‐well plates and grown to 90% confluence. Various concentrations of cisplatin were prepared from a stock solution of 1.5 mg/mL in 0.9% sodium chloride. Cells were treated with different concentrations of cisplatin for 24 h, and 400 μL of 1 mg/mL MTT solution in serum free RPMI 1640 medium was added to each well and incubated at 37°C for 3 h. After 3 h, the MTT solution was removed and 400 μL of DMSO was added to each well. The plates were placed on a shaker for 3 min and then analyzed with a Synergy HT Microplate Reader and KC4 software (BioTek, Winooski, VT, USA).
Apoptotic and dead cells were analyzed using Annexin V‐FITC kit. H522 vector and claudin‐7 cells were cultured in six‐well plates at 5 × 105 cells per well until cells reached 90% confluence. Cells were treated with cisplatin for 24 h and then resuspended in a binding buffer containing Annexin V‐FITC and propidium iodide (PI). Cells were analyzed by flow cytometry immediately. Fluorescein isothiocyanate was detected at 518 nm and PI was detected at 620 nm. Propidium iodide alone was used as a control to differentiate between viable, early and late apoptotic cells.
Generation of claudin‐7 mutations by site‐directed mutagenesis
Claudin‐7 phosphorylation null mutations were generated by replacing serine with alanine at positions 204 (S204A), 206 (S206A), and 207 (S207A). All mutations were confirmed by DNA sequencing (ECU Genomic Core Facility).15 Mutations were labeled M1 (S207A), M2 (S206A and S207A), and M3 (S204A, S206A, and S207A).
Kinase inhibitor treatment
NetPhos 2.0 was used to examine the possible serine phosphorylation sites, and NetPhosK 1.0 was used to examine the potential kinases for claudin‐7.32 H522 claudin‐7 cells were treated for 12 h with two PKC inhibitors, bisindolylmaleimide I (BIM) and chelerythrine chloride, or with glycogen synthase kinase 3 (GSK‐3) inhibitor, lithium chloride, or with mitogen‐activated protein kinase (MAPK) inhibitor, PD98059, respectively. The treatment concentrations were 5 μM for BIM, 20 μM for chelerythrine chloride, and 10 mM for lithium chloride; all were made in serum free RPMI 1640 media. For the MAP kinase inhibitor treatment, PD98059 was dissolved in DMSO and diluted in serum free RPMI 1640 media to produce a concentration of 50 μM. For the control, an identical volume of DMSO was added to serum free RPMI 1640 media. After the treatment, cells were immediately lysed, and then analyzed by Western blotting.
Statistical analysis
Statistical analysis was performed using Origin 5.0 (OriginLab, MA, USA) software. The differences between two groups were analyzed using the unpaired Student's t‐test. A P‐value of < 0.05 was considered significant.
Results
Claudin‐7‐transfected H522 cells increased chemosensitivity to cisplatin
H522 cells have no detectable expression of claudin‐7. To study the role of claudin‐7 in anti‐cancer drug treatments, H522 cells were stably transfected with claudin‐7 tagged with myc. As indicated in the phase images of Figure 1(a), claudin‐7‐transfected H522 cells (b) showed the nested cell morphology with fewer cellular processes compared to vector‐transfected H522 cells (a). Claudin‐7 was not expressed in vector transfected H522 cells (c) by the immunofluorescent staining method (Data S1), while claudin‐7 transfected cells displayed the claudin‐7 signal that was largely localized at the cell–cell contact area as expected (d, arrows). Western blots confirmed that H522 vector cells did not express claudin‐7, while claudin‐7 cells expressed the claudin‐7 protein as double bands at approximately 22 and 24 kDa (Fig. 1b). The presence of a double band could be due to post‐translational modifications to claudin‐7 such as phosphorylation (see results below). Our previous study reported that serine 206 at the COOH‐terminus of claudin‐7 was phosphorylated by WNK4, a serine/threonine kinase, in kidney epithelial cells.15 We also found that overexpression of claudin‐7 in H522 cells increased the cell–cell adhesion as shown in Figure 1(c). The percentage of cell aggregates present in claudin‐7 cells was significantly higher than that in the vector cells after cell dissociation procedures (Data S1).
Figure 1.
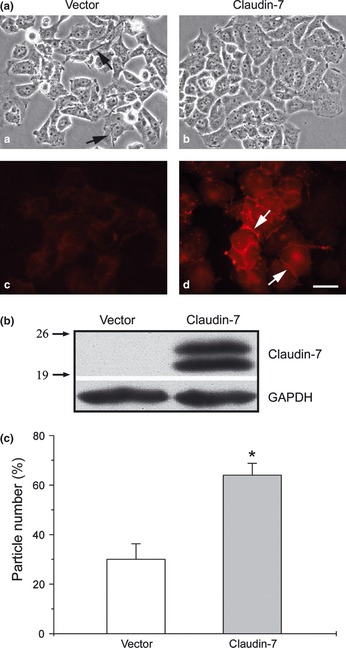
Ectopic expression of Claudin‐7 in human H522 lung cancer cells. (a) H522 parent cells were transfected with vector or myc tagged claudin‐7 cDNA. Stable cell lines were selected by geneticin. Phase images of vector [a] and claudin‐7 [b] cells indicate that vector cells have stretched long cellular processes (arrows). The vector [c] and claudin‐7 cells [d] were immunostained with anti‐myc antibody and claudin‐7 signal was detected at the cell–cell contact area in claudin‐7 cells. Bar: 20 μm. (b) Western blot showed claudin‐7 expression as a doublet at approximately 22 and 24 kDa in claudin‐7 cells. The membrane was blotted against claudin‐7 antibody and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) served as a protein loading control. The numbers on the left indicate the molecular weight in kDa. (c) Cell dissociation assay. H522 vector and claudin‐7 cells were seeded in equal concentration and allowed to grow until confluence. The cells were removed from the culture plates using a cell scraper and dissociated by pipetting. The percentage of particle number was calculated from at least 15 fields from two independent experiments. *P < 0.05, compared to vector cells.
Our results showed that cell death was significantly higher in H522 claudin‐7 cells than in vector cells by flow cytometry analysis (Fig. 2a). H522 vector cells had a 2.08 ± 0.39 mean percentage of cell apoptosis, while H522 claudin‐7 cells had a 6.98 ± 1.22, a 3.3‐fold increase that was statistically significant (Fig. 2a). There was no significant difference in the percentage of cell death between H522 vector and claudin‐3 cells (Fig. 2a, Fig. S1). We did not observe any significant difference in the cell cycle progression, such as G1, S, and G2/M phases between the claudin‐7 and vector cells (data not shown).
Figure 2.
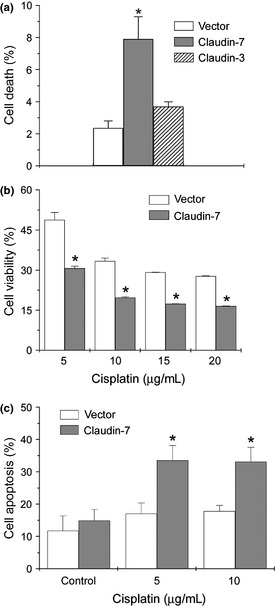
Increased cell apoptosis in H522 cells transfected with claudin‐7. (a) H522 claudin‐7 cells, not H522 claudin‐3 cells, have a significantly higher percentage of cell death than H522 vector cells. The percentage of cell apoptosis was analyzed by flow cytometry analysis. (b) The cell viability measured by MTT assay was decreased in H522 claudin‐7 cells treated with various concentrations of cisplatin. Cisplatin treatment showed a dose‐dependent decrease in cell viability over the 24‐h exposure period. The percentage of cell viability was normalized to vector and claudin‐7 cells without drug treatment. (c) The percentage of cell apoptosis detected by flow cytometry using Annexin V‐fluorescein isothiocyanate (FITC) was significantly higher in claudin‐7 transfected cells after 24 h of cisplatin treatment. *P < 0.05, compared to vector cells.
To investigate whether H522 cells with or without claudin‐7 would respond to anti‐cancer drug treatment differently, claudin‐7 or vector cells were treated with cisplatin, a common chemotherapeutic drug. The MTT assay was used to measure the viability of cells under the treatment with cisplatin. The results demonstrated that H522 claudin‐7 cells had lower cell viability than H522 vector cells did at every concentration of cisplatin treatment (Fig. 2b). To further confirm that H522 claudin‐7 cells increased cell apoptosis after drug treatment, Annexin V apoptosis assay was utilized. Annexin V binds to phosphatidylserine on the surface of the cell membrane when cells undergo apoptosis. We found that there was a significant difference in apoptosis between the H522 vector and claudin‐7 cells (Fig. 2c). H522 claudin‐7 cells had a greater percentage of apoptotic cells at both concentrations of cisplatin treatment compared to H522 vector cells.
Claudin‐7 increased the activation of caspase pathway under cisplatin treatment
To determine whether overexpression of claudin‐7 will affect the expression levels of other claudins, Western blot analysis was performed. Figure 3(a) indicated that claudin‐1 and ‐3 were expressed in vector‐transfected cells (V). However, ectopic expression of claudin‐7 did not affect the protein levels of claudin‐1 and ‐3 in claudin‐7 transfected cells[7]. Claudin‐2 and ‐4 were not expressed in either of the transfected cells (data not shown). Importantly, we found that claudin‐1 (Fig. 3b) and ‐3 (Fig. 3c) were not localized at the cell–cell contact area in vector cells, while upon the transfection of claudin‐7, they both were recruited to the cell membrane and co‐localized with claudin‐7 at the cell–cell contact area in H522 claudin‐7 cells (Fig. 3b and c, arrows). We reported similar results in NCI‐H1299 lung cancer cells in our previous study.31 Co‐immunoprecipitation experiments showed that claudin‐1 interacted with claudin‐7 and formed a protein complex in H522 claudin‐7 cells (Fig. 3d).
Figure 3.
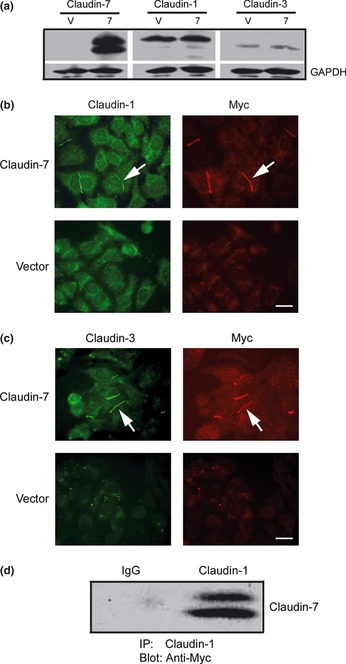
Co‐localization of claudin‐1 and ‐3 with claudin‐7. (a) Claudin‐1 and ‐3 were expressed in H522 vector (V) and claudin‐77 cells. Membranes were probed with anti‐claudin‐1, ‐3, and ‐7 antibodies. (b) Double immunostaining of claudin‐1 with ‐7 (Myc) in claudin‐7 and vector cells. Claudin‐1 was co‐localized with claudin‐7 at the cell‐cell contact area (arrows). (c) Double immunostaining of claudin‐3 with ‐7 (Myc) in claudin‐7 and vector cells. Claudin‐3 was also co‐localized with claudin‐7 at the cell‐cell contact area (arrows). Bar: 20 μm. (d) Co‐immunoprecipitation of claudin‐1 and ‐7. Claudin‐7 transfected cells were lysed in radio immunoprecipitation assay (RIPA) buffer without sodium dodecyl sulfate (SDS) and immunoprecipitated with the anti‐claudin‐1 antibody. The membrane was then blotted with anti‐Myc mouse monoclonal antibody. The IgG served as a negative control.
The results in Figure 2 indicate that overexpression of claudin‐7 increased cell apoptosis in H522 cells after cisplatin treatment. To determine whether caspase pathway has been activated or not after drug treatment, several caspase proteins were examined. Figure 4 illustrates that the signals for cleaved caspase‐3, ‐8, ‐9, and PARP were all stronger in H522 claudin‐7 cells than in the vector cells at both concentrations of cisplatin treatment. The cleavage of caspase‐3, ‐8, and ‐9 represents their activation triggering apoptosis, while the cleavage of PARP represents its inactivation inhibiting the ability of PARP to repair damaged DNA, therefore promoting apoptosis.
Figure 4.
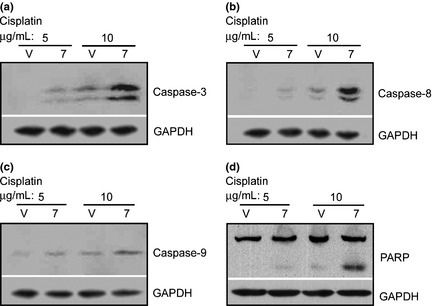
Increased activation of caspase pathway in H522 claudin‐7 cells after cisplatin treatment. Vector (V) and claudin‐7[7] cells were treated with 5 or 10 μg/mL of cisplatin for 24 h at 37°C. Cells were then lysed in radio immunoprecipitation assay (RIPA) buffer, and a total of 20 μg of proteins for each sample was loaded onto the sodium dodecyl sulfate (SDS)‐polyacrylamide gel. The membranes were probed for caspase‐3 (a), caspase‐8 (b), caspase‐9 (c), and PARP (Poly ADP ribose polymerase) (d). Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was used as a loading control.
Phosphorylation of claudin‐7 by protein kinase C at serine 204
To investigate whether the double band of claudin‐7 observed in H522 claudin‐7‐transfected cells (Fig. 1b) was due to protein phosphorylation, we performed experiments using three claudin‐7 phosphorylation mutants as shown in Figure 5(a). The parental H522 cells were transfected with claudin‐7 mutants, in which the serine 204, 206, and 207 were replaced by alanine. The M1 mutant has one point mutation (S207A), M2 contains two point mutations (S206A and S207A), and M3 bears three null‐phosphorylation sites (S204A, S206A, and S207A). The phosphorylation sites were located at the C‐terminus of claudin‐7 on the intracellular side of the plasma membrane (Fig. 5a).
Figure 5.
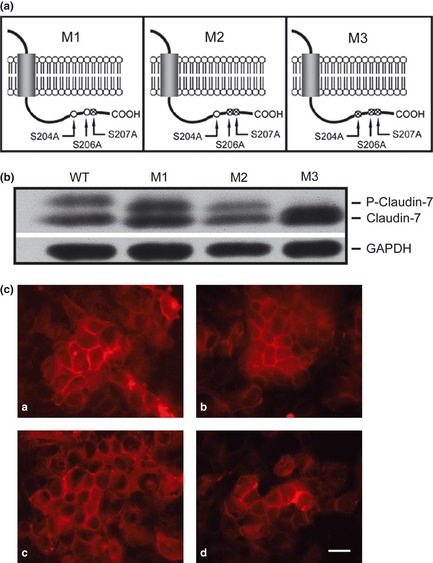
Claudin‐7 was phosphorylated at serine 204 in H522 claudin‐7 cells. (a) Schematic diagram illustrating claudin‐7 constructs with mutations at serine phosphorylation sites located at C‐terminus of claudin‐7. Three claudin‐7 mutants, M1, M2, and M3, were generated to investigate the doublet formation of claudin‐7 and its possible phosphorylation sites. Serine was replaced with alanine to create phosphorylation‐null mutants. M1 has one mutation at serine 207 (S207A); M2 has two mutations at serine 206 and 207 (S206A and S207A); M3 contains three mutations at serine 204, 206, and 207 (S204A, S206A, and S207A). (b) H522 parental cells were stably transfected with claudin‐7 wild type (WT), M1, M2, and M3 constructs. Claudin‐7 doublet was observed in WT, M1, and M2 cells, but not in M3 cells. The top band indicates the phosphorylated claudin‐7. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) served as a loading control. (c) Immunofluorescent staining of claudin‐7 in WT [a], M1 [b], M2 [c], and M3 [d] cells. Cells were grown on glass coverslips and immunostained with anti‐claudin‐7 antibody. Bar: 20 μm.
Western blot analysis showed that the upper band of claudin‐7 was diminished in cells transfected with the M3 mutant while the upper band in the cells transfected with wild‐type (WT), M1, and M2 mutants were present (Fig. 5b). This indicated that claudin‐7 was phosphorylated at serine 204 position. To determine whether phosphorylation‐null mutations affect the localization of claudin‐7, immunofluorecent staining experiments were performed. We found that all three mutants were able to localize to the cell–cell contact area as the WT did (Fig. 5c), suggesting that changing serine 204, 206, and 207 by alanine did not affect the localization of claudin‐7 to the membrane.
To further investigate the phosphorylation of claudin‐7, we used Netphos software to predict the potential protein kinases and possible serine phosphorylation sites.32 The software prediction indicated that PKC and GSK3 were candidates for claudin‐7 phosphorylation at serine 204. Therefore, H522 claudin‐7 cells were treated with PKC and GSK3 inhibitors to investigate their effects on claudin‐7 phosphorylation. Figure 6(a) showed that after the inhibition of PKC by chelerythrine chloride (Chelery‐Cl), the signal intensity for the upper phosphorylated band of claudin‐7 was greatly reduced and the lower dephosphorylated band was greatly enhanced compared to the bisindolylmaleimide I (BIM), lithium chloride (LiCl) and a MAP kinase inhibitor, PD98059 treatments.
Figure 6.
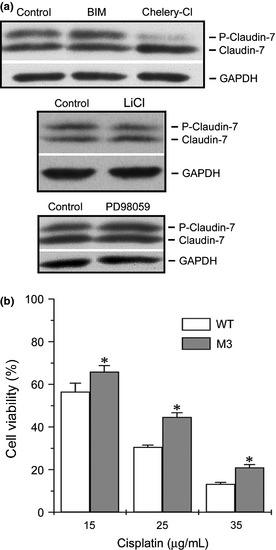
Phosphorylation of claudin‐7 was inhibited by protein kinase C (PKC) inhibitor Chelery‐Cl in H522 claudin‐7 cells. (a) H522 claudin‐7 cells were treated with BIM, Chelery‐Cl, LiCl, and PD98059 for 12 h at 37°C. The membrane was immunoblotted with anti‐claudin‐7 antibody. The top band represented the phosphorylated claudin‐7. After Chelery‐Cl treatment, the phosphorylated form of claudin‐7 was greatly reduced, while the de‐phosphorylated form (lower band) was greatly increased. (b) Increased cell viability in H522 M3 mutant cells after cisplatin treatment. Claudin‐7 WT and M3 cells were treated with 15, 25, and 35 μg/mL of cisplatin for 24 h at 37°C. The MTT assay was used to determine the cell viability. The percentage of cell viability was normalized to claudin‐7 WT and M3 cells without drug treatment. *P < 0.05, compared to WT cells. All experiments were repeated three times with triplicates.
To examine whether claudin‐7 phosphorylation would affect the H522 cell's sensitivity to the drug treatment, cells transfected with M3 mutant were treated with various concentrations of cisplatin. Cisplatin treatments showed a dose‐dependent decrease in cell viability over the 24‐h exposure period for both H522 claudin‐7 WT and M3 mutant cells. However, H522 M3 mutant cells had higher cell viability than claudin‐7 WT cells did (Fig. 6b). It is clear from Figure 6(b) that there is a significant difference between WT and M3 cell viability at every concentration of cisplatin treatment. However, we did not observe a statistically significant difference in the percentage of cell death without cisplatin treatment among H522 claudin‐7 WT and M1‐M3 mutants (Fig. S2). Our data indicates that the phosphorylation of claudin‐7 increased chemosensitivity to cisplatin treatment in the H522 claudin‐7 cells.
Discussion
Claudin proteins play a crucial role in maintaining cell polarity, adhesion, and paracellular permeability, and a loss of these functions is associated with tumorigenesis.33 Previously, we have reported that claudin‐7 inhibited human lung cancer cell migration and invasion.31 In this study, we aimed to study claudin‐7 involvement in lung cancer cell apoptosis after anti‐cancer drug treatment. We found that claudin‐7 increased H522 lung cancer cell apoptosis after cisplatin treatment. The phosphorylation‐null mutant of claudin‐7 reduced this ability compared to the WT claudin‐7. Claudin‐7 induced lung cancer cell apoptosis after drug treatment was through the activation of the caspase pathway.
Our results also showed that overexpression of claudin‐7 did not change the protein expression levels of claudin‐1 and ‐3. However, the immunofluorescent staining illustrated that claudin‐1 and ‐3 were co‐localized at the cell–cell contact area with claudin‐7. It also revealed that claudin‐7 recruited claudin‐1 and claudin‐3 to the cell–cell contact area (Fig. 3b,c). Co‐immunoprecipitation experiments indicated that claudin‐1 and ‐7 interacted with each other and formed a protein complex. The interaction between claudin‐3 and ‐7 was much weaker (data not shown), which was consistent with our previous report on the H1299 lung cancer cell line.31 It has been reported that claudin‐7 can bind to the cell adhesion molecule EpCAM and cell membrane receptors such as CD44.34 It is possible that claudin‐7 forms a protein complex with these membrane‐anchoring proteins and therefore, is stably localized at the cell membrane. Binding of claudin‐7 with claudin‐1 and ‐3 will bring and stabilize claudin‐1 and ‐3 proteins on the cell membrane. The co‐localization and interaction of different claudins could play an important role in exerting their various functions in the cells.
Previously, we reported that claudin‐7 was phosphorylated by WNK4 kinase at serine 206 in kidney epithelial cells.15 In this study, using site‐directed mutagenesis strategy, we showed that phosphorylation of claudin‐7 at serine 204 was inhibited by PKC inhibitor, Chelery‐Cl. It is known that PKC is a family of serine/threonine kinases with at least 10 different isoforms. They are involved in cell signaling, which regulates cell cycle, apoptosis, and differentiation.35, 36, 37 These different PKC subtypes can be inhibited by various inhibitors. This could explain why BIM, another PKC inhibitor, did not prevent the phosphorylation of claudin‐7 since different inhibitors may affect different PKC isoforms. The slight mobility shift of the M1–M3 upper band compared to that of WT in Figure 5(b) could be due to the number of phosphorylation sites since WT, M1, M2, and M3 have 3, 2, 1, and 0 phosphorylation sites, respectively.
It is unclear how the expression of tight junction protein induces cell apoptosis after the drug treatment. Recently, it has been reported that forced expression of tight junction protein occludin in cancer cells exhibits enhanced sensitivity to different apoptotic‐inducing factors via modulation of unique sets of apoptosis‐associated genes.38 Cisplatin‐induced cell apoptosis involves Fas receptor clustering and activation as well as the binding to Fas‐associated death domain protein (FADD).39 It is possible that claudin‐7 could bind to Fas receptor and facilitate its binding to FADD, thereby activating caspase signaling pathway. Phosphorylation of claudin‐7 at serine 204 could potentially make Fas/claudin‐7/FADD interactions more stable. In addition, recruitment of claudin‐1 and ‐3 to the cell membrane by claudin‐7 may also contribute to the increased chemosensitivity in H522 claudin‐7 cells.
Our present study demonstrates that claudin‐7 increases chemosensitivity to cisplatin treatment in H522 lung cancer cells through the activation of the caspase pathway. Claudin‐7 regulation by PKC may transduce downstream signaling pathways in human lung cancer. Analyses of these pathways could provide insight to the mechanisms of lung tumoriogenesis and provide new directions for the detection and therapy of cancer.
Disclosure Statement
The authors have no financial interests in or financial conflict with the subject matter discussed in this manuscript.
Supporting information
Data S1. Supplemental methods.
Fig. S1. Ectopic expression of Claudin‐3 in human H522 lung cancer cells.
Fig. S2. Percentage of cell death in H522 claudin‐7 WT and M1‐M3 mutants.
Acknowledgements
This work was supported by the National Institute of Health grants ES016888 and HL085752. We thank Rodney Tatum, Beverly G. Jeansonne, Dr Ruth Schwalbe, and Kristen Hall for their technical assistance.
(Cancer Sci 2013; 104: 611–618)
References
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59: 225–49. [DOI] [PubMed] [Google Scholar]
- 2. Haass NK, Smalley KS, Herlyn M. The role of altered cell‐cell communication in melanoma progression. J Mol Histol 2004; 35: 309–18. [DOI] [PubMed] [Google Scholar]
- 3. Matter K, Balda MS. Signaling to and from tight junctions. Nat Rev Mol Cell Biol 2003; 4: 225–36. [DOI] [PubMed] [Google Scholar]
- 4. Morin PJ. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res 2005; 65: 9603–6. [DOI] [PubMed] [Google Scholar]
- 5. Heiskala M, Peterson PA, Yang Y. The roles of claudin superfamily proteins in paracellular transport. Traffic 2001; 2: 93–8. [DOI] [PubMed] [Google Scholar]
- 6. Balkovetz DF. Claudins at the gate: determinants of renal epithelial tight junction paracellular permeability. Am J Physiol Renal Physiol 2006; 290: F572–9. [DOI] [PubMed] [Google Scholar]
- 7. Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin‐1 and ‐2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol 1998; 141: 1539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol 2003; 284: C1346–54. [DOI] [PubMed] [Google Scholar]
- 9. Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin‐8 expression in Madin‐Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem 2003; 278: 2350–9. [DOI] [PubMed] [Google Scholar]
- 10. Amasheh S, Meiri N, Gitter AH et al Claudin‐2 expression induces cation‐selective channels in tight junctions of epithelial cells. J Cell Sci 2002; 115: 4969–76. [DOI] [PubMed] [Google Scholar]
- 11. Alexandre MD, Lu Q, Chen YH. Overexpression of claudin‐7 decreases the paracellular Cl‐ conductance and increases the paracellular Na+ conductance in LLC‐PK1 cells. J Cell Sci 2005; 118: 2683–93. [DOI] [PubMed] [Google Scholar]
- 12. Hou J, Renigunta A, Konrad M et al Claudin‐16 and claudin‐19 interact and form a cation‐selective tight junction complex. J Clin Invest 2008; 118: 619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Van Itallie CM, Anderson JM. Claudins and epithelial paracellular transport. Annu Rev Physiol 2006; 68: 403–29. [DOI] [PubMed] [Google Scholar]
- 14. D'Souza T, Indig FE, Morin PJ. Phosphorylation of claudin‐4 by PKCepsilon regulates tight junction barrier function in ovarian cancer cells. Exp Cell Res 2007; 313: 3364–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tatum R, Zhang Y, Lu Q, Kim K, Jeansonne BG, Chen YH. WNK4 phosphorylates ser(206) of claudin‐7 and promotes paracellular Cl(‐) permeability. FEBS Lett 2007; 581: 3887–91. [DOI] [PubMed] [Google Scholar]
- 16. Balda MS, Matter K. Epithelial cell adhesion and the regulation of gene expression. Trends Cell Biol 2003; 13: 310–8. [DOI] [PubMed] [Google Scholar]
- 17. Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 2004; 286: C1213–28. [DOI] [PubMed] [Google Scholar]
- 18. Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four‐transmembrane domain protein components of tight junction strands. Proc Nat Acad Sci USA 1999; 96: 511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kramer F, White K, Kubbies M, Swisshelm K, Weber BH. Genomic organization of claudin‐1 and its assessment in hereditary and sporadic breast cancer. Hum Genet 2000; 107: 249–56. [DOI] [PubMed] [Google Scholar]
- 20. Tokes AM, Kulka J, Paku S et al Claudin‐1, ‐3 and ‐4 proteins and mRNA expression in benign and malignant breast lesions: a research study. Breast Cancer Res 2005; 7: R296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Resnick MB, Konkin T, Routhier J et al Claudin‐1 is a strong prognostic indicator in stage II colonic cancer: a tissue microarray study. Mod Pathol 2005; 18: 511–8. [DOI] [PubMed] [Google Scholar]
- 22. Kominsky SL, Argani P, Korz D et al Loss of the tight junction protein claudin‐7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 2003; 22: 2021–33. [DOI] [PubMed] [Google Scholar]
- 23. Al Moustafa AE, Alaoui‐Jamali MA, Batist G et al Identification of genes associated with head and neck carcinogenesis by cDNA microarray comparison between matched primary normal epithelial and squamous carcinoma cells. Oncogene 2002; 21: 2634–40. [DOI] [PubMed] [Google Scholar]
- 24. Michl P, Barth C, Buchholz M et al Claudin‐4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res 2003; 63: 6265–71. [PubMed] [Google Scholar]
- 25. Hoevel T, Macek R, Swisshelm K, Kubbies M. Reexpression of the TJ protein CLDN1 induces apoptosis in breast tumor spheroids. Int J Cancer 2004; 108: 374–83. [DOI] [PubMed] [Google Scholar]
- 26. Sauer T, Pedersen MK, Ebeltoft K, Naess O. Reduced expression of Claudin‐7 in fine needle aspirates from breast carcinomas correlate with grading and metastatic disease. Cytopathology 2005; 16: 193–8. [DOI] [PubMed] [Google Scholar]
- 27. Oshima T, Kunisaki C, Yoshihara K et al Reduced expression of the claudin‐7 gene correlates with venous invasion and liver metastasis in colorectal cancer. Oncol Rep 2008; 19: 953–9. [PubMed] [Google Scholar]
- 28. Usami Y, Chiba H, Nakayama F et al Reduced expression of claudin‐7 correlates with invasion and metastasis in squamous cell carcinoma of the esophagus. Hum Pathol 2006; 37: 569–77. [DOI] [PubMed] [Google Scholar]
- 29. Lioni M, Brafford P, Andl C et al Dysregulation of claudin‐7 leads to loss of E‐cadherin expression and the increased invasion of esophageal squamous cell carcinoma cells. Am J Pathol 2007; 170: 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moldvay J, Jackel M, Paska C, Soltesz I, Schaff Z, Kiss A. Distinct claudin expression profile in histologic subtypes of lung cancer. Lung Cancer 2007; 57: 159–67. [DOI] [PubMed] [Google Scholar]
- 31. Lu Z, Ding L, Hong H, Hoggard J, Lu Q, Chen YH. Claudin‐7 inhibits human lung cancer cell migration and invasion through ERK/MAPK signaling pathway. Exp Cell Res 2011; 317: 1935–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blom N, Gammeltoft S, Brunak S. Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 1999; 294: 1351–62. [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez‐Mariscal L, Lechuga S, Garay E. Role of tight junctions in cell proliferation and cancer. Prog Histochem Cytochem 2007; 42: 1–57. [DOI] [PubMed] [Google Scholar]
- 34. Kuhn S, Koch M, Nübel T et al A complex of EpCAM, claudin‐7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol Cancer Res 2007; 5: 553–67. [DOI] [PubMed] [Google Scholar]
- 35. Freeley M, Kelleher D, Long A. Regulation of protein kinase C function by phosphorylation on conserved and non‐conserved sites. Cell Signal 2011; 23: 753–62. [DOI] [PubMed] [Google Scholar]
- 36. Clarke H, Marano CW, Peralta Soler A, Mullin JM. Modification of tight junction function by protein kinase C isoforms. Adv Drug Deliv Rev 2000; 41: 283–301. [DOI] [PubMed] [Google Scholar]
- 37. Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab 2010; 298: E395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osanai M, Murata M, Nishikiori N, Chiba H, Kojima T, Sawada N. Epigenetic silencing of occludin promotes tumorigenic and metastatic properties of cancer cells via modulations of unique sets of apoptosis‐associated genes. Cancer Res 2006; 66: 9125–33. [DOI] [PubMed] [Google Scholar]
- 39. Rebillard A, Jouan‐Lanhouet S, Jouan E et al Cisplatin‐induced apoptosis involves a Fas‐ROCK‐ezrin‐dependent actin remodelling in human colon cancer cells. Eur J Cancer 2010; 46: 1445–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Fig. S1. Ectopic expression of Claudin‐3 in human H522 lung cancer cells.
Fig. S2. Percentage of cell death in H522 claudin‐7 WT and M1‐M3 mutants.