

Summary
Newer therapies are needed for the treatment of status epilepticus (SE) refractory to benzodiazepines. Enhanced glutamatergic neurotransmission leads to SE, and AMPA receptors are modified during SE. Reducing glutamate release during SE is a potential approach to terminate SE. The neuropeptide somatostatin (SST) is proposed to diminish presynaptic glutamate release by activating SST type-2 receptors (SST2R). SST exerts an anticonvulsant action in some experimental models of seizures. Here, we investigated the mechanism of action of SST on excitatory synaptic transmission at the Schaffer collateral-CA1 synapses and the ability of SST to treat SE in rats using patch-clamp electrophysiology and video-EEG monitoring of seizures. SST reduced action potential-dependent EPSCs (sEPSCs) at Schaffer collateral-CA1 synapses at concentrations up to 1 μM; higher concentrations had no effect or increased the sEPSC frequency. SST also prevented paired-pulse facilitation of evoked EPSCs and did not alter action-potential-independent miniature EPSCs (mEPSCs). The effect of SST on EPSCs was inhibited by the SST2R antagonist cyanamid-154806 and was mimicked by the SST2R agonists, octreotide and lanreotide. Both SST and octreotide reduced the firing rate of CA1 pyramidal neurons. Intraventricular administration of SST, within a range of doses, either prevented or attenuated pilocarpine-induced SE or delayed the median time to the first grade 5 seizure by 11 min. Similarly, octreotide or lanreotide prevented or attenuated SE in more than 65% of animals. Compared to the pilocarpine model, octreotide was highly potent in preventing or attenuating continuous hippocampal stimulation-induced SE in all animals within 60 min of SE onset. Our results demonstrate that SST, through the activation of SST2Rs, diminishes presynaptic glutamate release and attenuates SE.
Keywords: Somatostatin, somatostatin type 2 receptors, glutamate, synaptic transmission, status epilepticus
Introduction
Novel therapies are needed for the treatment of status epilepticus (SE), which is currently treated with benzodiazepines, barbiturates, and anesthetics such as propofol and midazolam, all of which enhance GABAergic inhibition (Meierkord et al., 2010). However, GABAergic inhibition is compromised during SE (Kapur and Macdonald, 1997; Goodkin et al., 2005; Naylor et al., 2005; Goodkin et al., 2008), and pharmacoresistance to benzodiazepines develops (Treiman et al., 1998; Meierkord et al., 2010; Neligan and Shorvon, 2010). An alternate approach to treat SE is to reduce excitatory neurotransmission. The activation of muscarinic receptors increases glutamate release from presynaptic terminals of hippocampal principal neurons (Olivos and Artalejo, 2008; Kozhemyakin et al., 2010; Sun and Kapur, 2012) and can cause SE in humans and experimental animals (McDonough, Jr. and Shih, 1997; Okumura et al., 1996; Turski et al., 1989). Furthermore, AMPA receptors (AMPARs) expressed on CA1 pyramidal neurons of the hippocampus are modified during SE (Rajasekaran et al., 2012). The blockade of AMPARs can terminate benzodiazepine-refractory SE (Fritsch et al., 2010; Rajasekaran et al., 2012), suggesting that inhibiting postsynaptic excitatory receptors can terminate SE. A similar approach to reduce the strength of glutamatergic transmission during SE is to reduce the release of neurotransmitter.
Somatostatin (SST) is proposed to reduce glutamatergic neurotransmission (Boehm and Betz, 1997). However, studies on SST and glutamatergic neurotransmission were performed on cultured hippocampal neurons at autaptic synapses (Boehm and Betz, 1997), which do not occur in intact hippocampal preparations. Furthermore, it is difficult to clearly differentiate between pre- and post-synaptic effects in autaptic preparations. The actions of SST are mediated by SST receptor (SSTR) subtypes 1-5, which are widely distributed throughout the brain (Hoyer et al., 1995; Schulz et al., 2000). Immunocytochemical studies demonstrate that SST2R is located on the soma, dendrites, and axons of hippocampal neurons, suggesting that SST2R may mediate both the presynaptic and postsynaptic effects of SST (Dournaud et al., 1996). Indeed, SST2R agonists mimic and antagonists block the inhibitory effect of SST on glutamate release from cortical synaptosomes (Grilli et al., 2004). However, whether SST2R mediates the effects of SST at hippocampal synapses remains unexplored.
SST suppresses kindled seizures (Monno et al., 1993; Mazarati and Telegdy, 1992; Mazarati and Wasterlain, 2002; Zafar et al., 2012) and brief epileptiform discharges in hippocampal slices (Tallent and Siggins, 1999). These anticonvulsant actions are proposed to be mediated by SST2R and SST4R in a species-specific manner (Vezzani et al., 1991; Moneta et al., 2002; Cammalleri et al., 2004; Stragier et al., 2006; Qiu et al., 2008; Aourz et al., 2011). However, SE differs from isolated seizures because the prolonged seizures of SE cause rapid plasticity of neurotransmission. There is some evidence that SST could transiently terminate electrically-induced SE (eg. Mazarati and Wasterlain, 2002); however, the efficacy of SST and its analogs in terminating SE is not well characterized.
We tested whether SST suppresses glutamate release by activating SST2Rs. Further, we tested the ability of SST and SST2R agonists to treat SE.
Methods
All experimental procedures were performed according to a protocol approved by the University of Virginia Animal Care and Use Committee. Adult male Sprague-Dawley rats (220-250 g, 60 – 90 days) were used for the experiments.
Electrophysiology
The animals were anesthetized with isoflurane prior to decapitation. Brains were removed and immersed in cold (2-4°C) slicing ACSF composed of (in mM) 5.5 NaCl, 2 KCl, 5 MgSO4, 1.1 KH2PO4, 1 CaCl2, 10 dextrose, 25 NaHCO3 and 113 sucrose (osmolarity 300 mOsm) saturated with 95%O2-5%CO2. 300-μM-thick horizontal dorsal hippocampal slices were cut and maintained in continuously oxygenated ACSF containing (in mM) 127 NaCl, 2 KCl, 1.5 MgSO4, 25.7 NaHCO3, 10 dextrose, and 1.5 CaCl2 (pH 7.4; 300 mOsm). Whole-cell patch-clamp recordings were obtained from visually identified CA1 pyramidal neurons (CA1 neurons) at room temperature.
To record excitatory postsynaptic currents (EPSCs), patch electrodes (final resistance 3 - 6 MΩ) were filled with a filtered internal recording solution consisting of (in mM): 117.5 CsMeSO4, 10 2-Hydroxyethyl] piperazine-N - [2-ethansulfonic acid] (HEPES), 0.3 N-[and glycol-bis (a-aminoethyl ether) N,N,N,N -tetraacetic acid (EGTA), 15.5 CsCl, and 1.0 MgCl2, pH 7.3 (with CsOH); the osmolarity was 290 - 300 mOsm. Current-clamp recordings were performed using a recording solution containing (in mM) 100 potassium gluconate, 5 KCl, 10 HEPES, 1 EGTA, 2 MgCl2, and 0.1 CaCl2, pH 7.2 (with KOH). The electrode shank contained (in mM): 4 ATP Mg2+ salt, 0.3 GTP Na+ salt and, for voltage clamp studies, 5 QX-314. Neurons were voltage clamped at -65 mV for the duration of the EPSC recordings. Whole-cell capacitance and series resistance (baseline 10-20 MΩ) were compensated by 80% at a 10 ms lag. Recordings were performed when the series resistance after compensation was less than 20 MΩ. The access resistance was monitored with a 10 ms, -5 mV test pulse and the recording was terminated if the series resistance increased by 25% any time during the experiment. Currents were filtered at 5 kHz, digitized using a Digidata 1322 digitizer (Molecular Devices, Sunnyvale, CA) and acquired using Clampex 8.2 software (Molecular Devices, Sunnyvale, CA).
Spontaneous EPSCs (sEPSCs) were recorded from CA1 neurons after blocking GABAA receptors with the antagonist picrotoxin (50 μM). Action-potential independent EPSCs (mEPSCs) were recorded by blocking action potentials with 1 μM tetrodotoxin (TTX, Alomone labs, Jerusalem, Israel). Evoked EPSCs (eEPSCs) were obtained by stimulating (2-8 V, 10 μs duration, and 0.07Hz) the Schaffer collateral pathway using a glass electrode filled with ACSF to evoke visually identifiable EPSCs. eEPSCs were analyzed only if they were not contaminated by spontaneous events. 30-50 eEPSCs were recorded at baseline and following drug application. They are individually analyzed using Clampfit 8.2 software (Molecular Devices, Sunnyvale, CA).
Current-clamp recordings were performed using the I = 0 (bridge mode) setting on the Axopatch 200B amplifier. The recordings were obtained only from cells whose baseline resting membrane potential (RMP) was -65 mV or lower. The firing rate of the neurons was studied using loose-patch recordings under conditions of elevated potassium (3 mM). The recording solution in these experiments was the same as that of the external perfusion solution.
The digitized current traces were analyzed with MiniAnalysis (Synaptosoft, Decatur, GA) as previously described (Kozhemyakin et al., 2009, Rajasekaran et al., 2007, 2009). The parameters used to detect EPSCs in the study were: Threshold = RMS*3, period to search for local maximum 10000 μs, time before peak for a baseline 80000 μs, period to search for a decay 250000 μs, fraction of peak to find decay time 0.001, period to average a baseline 5000 μs. In sEPSC studies, a minimum of 150 events were detected at baseline and following drug application. For current-clamp recordings, the changes in RMP were determined using Clampfit 8.2 software (Molecular Devices, Sunnyvale, CA). For in vitro experiments, slices were obtained from at least 3-5 rats unless otherwise stated. The Kolmogorov-Smirnov (K-S) test was used to compare cumulative distributions of frequency and amplitude of continuously recorded EPSCs. For the population, drug application data were analyzed using paired t-tests as these values represented repeated measures on the same neurons. Where more than 2 drugs were tested, a One Way ANOVA with Tukey’s post-hoc test was performed. Statistical analysis was performed using GraphPad Prism 5 software (GraphPad, Mountain View, CA). Data values are expressed as the mean ± SEM unless noted otherwise, and p < 0.05 was considered significant.
In vivo studies
Bipolar metal electrodes and an intraventricular cannula were implanted in the hippocampus and cortex as previously described (Martin and Kapur, 2008; Todorovic et al., 2012). Electrode headsets were connected to a Grass 7D amplifier and then to a Stellate digital system for electroencephalographic (EEG) recording.
SST, octreotide and lanreotide (Sigma, St. Louis, MO) were suspended in sterile saline and infused into the ventral hippocampus via a 28-gauge injection needle extending 1 mm past the cannula guide. The needle was connected to a 1.0 ml syringe, which was driven by an infusion pump (KD Scientific, Portland, OR) at a rate of 2.0 μl/min. Saline, SST, octreotide, or lanreotide were administered intracerebroventricularly (i.c.v.) at a rate of 120 μL/hr for 3 hours. The infusions were initiated 90 minutes before pilocarpine injection (50 mg/kg) or the beginning of hippocampal stimulation. EEG and video monitoring began 10 min prior to drug infusion and continued for up to 6 h after infusion was completed. Brief electrographic discharges (20-120 s duration) occurred during the initial infusion of saline or drugs and stopped prior to pilocarpine administration.
SE was induced by a combination of lithium-pilocarpine as previously described (Martin and Kapur, 2008). Animals were initially protected from the peripheral effects of cholinergic stimulation by the administration of scopolamine. However, in an initial study i.c.v. infusion of saline (70 μl) prevented SE in 57% (12/21) of the animals suggesting that the presence of the i.c.v. cannula sufficiently breached the blood brain barrier (BBB) to allow entry of scopolamine into the brain, preventing cholinergic-stimulation-induced SE. Scopolamine dose was subsequently eliminated, and all animals developed seizures and SE. In some animals, SE was induced by continuous hippocampal stimulation by stimulating the left ventral hippocampus with 10s trains of 50 Hz, 1 ms, 400 mA biphasic square wave current pulses delivered every 13 s for 90 min (Lothman et al., 1989; Borris et al., 2000).
EEG recordings were reviewed for the presence of seizures, which were defined as rhythmic spike and wave discharges with a frequency of at least 2 Hz and an amplitude at least 3 times that of the baseline EEG. The EEG criterion for SE was the occurrence of continuous epileptiform activity for 30 minutes during which spike frequencies did not drop below 1 Hz or intermittent seizures lasting 30 minutes with brief periods of spiking lasting less than 5 minutes. The termination of these events was recorded when the spike frequency dropped below 1 Hz for more than 30 minutes. Seizures were scored behaviorally from video recordings as per Racine criteria (Racine, 1972).
Results
SST diminished action potential-dependent glutamate release at Schaffer collateral-CA1 pyramidal neuron synapses
SST is proposed to inhibit glutamate release from presynaptic terminals. We studied the effect of SST on the modulation of synaptic transmission at Schaffer collateral-CA1 pyramidal neuron glutamatergic synapses by recording spontaneous EPSCs from CA1 pyramidal neurons in transverse hippocampal slices. After recording sEPSCs for 10 minutes, 1μM SST was bath-applied to a slice. SST decreased the mean frequency of sEPSC in the slice from 1.06 ± 0.14Hz to 0.62 ± 0.11Hz (p < 0.05; n=16; paired t-test, Fig 1A). The inter-event intervals during the baseline recording were compared to those after SST application using a cumulative frequency plot. In each of the recordings, SST caused a significant right shift in the cumulative frequency plot (p < 0.05, KS test, Fig 1B). The mean amplitude (17.11 ± 1.06 vs. 17.13 ± 0.96 pA), rise time (6.27± 0.33 ms vs. 6.81 ± 0.38 ms) and decay time constant (23.54 ± 3.84 ms vs. 23.29 ± 3.22 ms) of sEPSCs were not altered by SST (p > 0.05, paired t-test).
Fig 1.
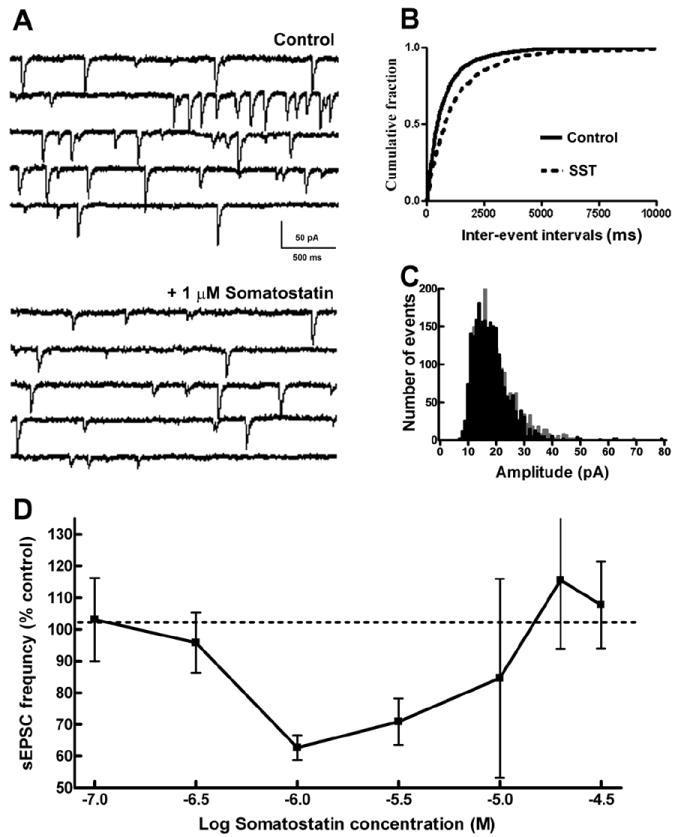
Somatostatin (SST) reduced the frequency of sEPSCs at Schaffer collateral-CA1 synapses. (A), Representative sEPSC recordings obtained from CA1 pyramidal neurons (CA1 neurons) before (Control) and after the application of 1 μM SST. (B), Cumulative probability plots of sEPSC frequency obtained by pooling data from 16 neurons before (—) and after (- - -) application of SST. (C), Distribution of sEPSC amplitudes binned by 1pA before (grey) and after (black) application of SST. Note that SST reduced the number of large amplitude sEPSCs. (D), Relationship of SST concentration to sEPSC frequency. Note that SST diminished the frequency of sEPSCs at lower concentrations but enhanced them at higher concentrations. The arithmetic equivalent of log dose used and their respective sample size are: 100 nM (n=3), 316 nM (n=3), 1μM (n=4), 3.16μM (n=4), 10μM (n=4), 19.9μM (n=3) and 31.6μM (n=3).
Diminished sEPSC frequency with unaltered sEPSC amplitude suggested a presynaptic action of SST. The amplitude distribution of sEPSCs at baseline and 10 min following 1 μM SST application were compared by pooling 2305 consecutive events from all neurons (events per cell ranging from 123 to 368). As demonstrated in Fig 1C, SST application reduced the number of larger-amplitude sEPSCs (p < 0.05, KS test), suggesting that SST inhibited multiquantal release.
To characterize the concentration dependence of the effects of SST on sEPSC frequency, we tested 7 different concentrations of SST (n= 24) ranging from 100 nM to 30 μM. A concentration-dependent reduction of sEPSC frequency was observed when SST was applied in concentrations ranging from 100 nM to 1 μM with an IC50 of 0.8 μM. However, no further reduction in SST frequency occurred when higher concentrations of SST were applied. In fact, SST enhanced sEPSCs when applied at concentration of 20 or 30 μM (Fig 1D). This range of SST concentrations (100 nM to 30 μM) did not significantly change the amplitude of sEPSCs (range 92-114% of control). Thus, SST had a U-shaped concentration-response relationship in suppressing glutamatergic transmission.
Inhibition of multiquantal release suggested that SST affected presynaptic mechanisms. We therefore studied the effect of SST on paired-pulse responses to electrical stimulation of Schaffer collateral afferents (eEPSCs). The stimulation intensity was set to consistently evoked EPSCs. SST decreased the mean eEPSC amplitude from 167.9 ± 21.12 pA to 146.2 ± 13.51pA (n=6; p < 0.05, paired t-test, Fig 2A, B). The rise time and decay time of eEPSCs were unaltered. In paired-pulse studies, under baseline conditions, the response to the second stimulus was larger than the response to the first (paired-pulse facilitation, PPF, Fig 2C). At baseline, the mean peak eEPSC amplitude in response to the first stimulus was 159 ± 43.3 pA and it was 210.2 ± 52.2 pA in response to the second stimulus (n = 5, p < 0.05, paired t-test). In the presence of SST (1μM), the peak amplitude in response to the first stimulus was less than at baseline (baseline vs. SST, 159 ± 43.3 vs. 130.9 ± 41.6 pA, p < 0.05, paired t-test), and the response to the second stimulus similar to that following the first stimulus (114.7 ± 31.8 vs. 130.9 ± 41.6 pA, p > 0.05, paired t-test, Fig 2D). Thus, SST (1 μM) abolished PPF of eEPSCs and paired pulse response was diminished from 136.5± 4.7% to 91.2 ± 3.2%.
Fig 2.
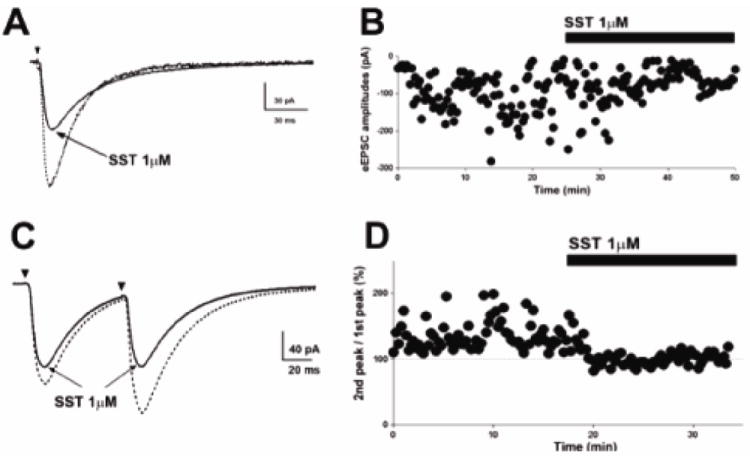
SST inhibits presynaptic glutamate release. (A), Representative averaged peak eEPSC before (dotted trace) and after (solid trace) SST application reveals diminished synaptic potency of eEPSCs following SST application. (B), Time course change in eEPSC amplitude following SST application in cell shown in (A). (C), Representative averaged eEPSCs obtained by paired stimuli before and after application of SST show that SST application abolished paired-pulse facilitation. (D), Time course change in the paired-pulse response following SST application. Dotted lines at 100% indicate the normalized amplitude of eEPSCs evoked by the 1st stimulus.
SST did not alter the action potential-independent (miniature) glutamate release. mEPSCs were recorded in the presence of 1 μM tetrodotoxin. In 11 cells, SST (1 μM) did not change the frequency (0.32 ± 0.07 Hz vs. 0.38 ± 0.1 Hz) or amplitude (16.22 ± 0.86 pA vs. 16.87 ± 0.94 pA) of mEPSCs (p > 0.05, paired t-test), suggesting that SST does not alter AMPARs.
SST inhibition of glutamate release at Schaffer collateral-CA1 pyramidal neuron synapses is mediated by SST2R
SST modulation of presynaptic glutamate release is thought to be mediated by SST2R (Dournaud et al., 1996; Boehm and Betz, 1997). We therefore tested the effect of SST2R modulation on glutamatergic synaptic transmission by selectively blocking SST2R. The SST2R antagonist cyanamid-154806 (1 μM, Hicks et al., 1998) occluded the effect of SST on sEPSCs (Fig 3). The application of cyanamid-154806 did not alter baseline sEPSC frequency (0.65 ± 0.16 Hz vs. 0.56 ± 0.1 Hz, n = 7, p > 0.05, ANOVA, Fig 3A, B); however, in the presence of cyanamid-154806, SST (1 μM) did not depress sEPSC frequency (0.53 ± 0.11 Hz, p > 0.05, ANOVA; n=7, Fig 3C, D). Further, SST did not change the amplitude of eEPSCs in the presence of cyanamid-154806 (p > 0.05; ANOVA; n=4, data not shown). These observations suggest that SST2R mimics the action of SST on glutamatergic transmission.
Fig 3.
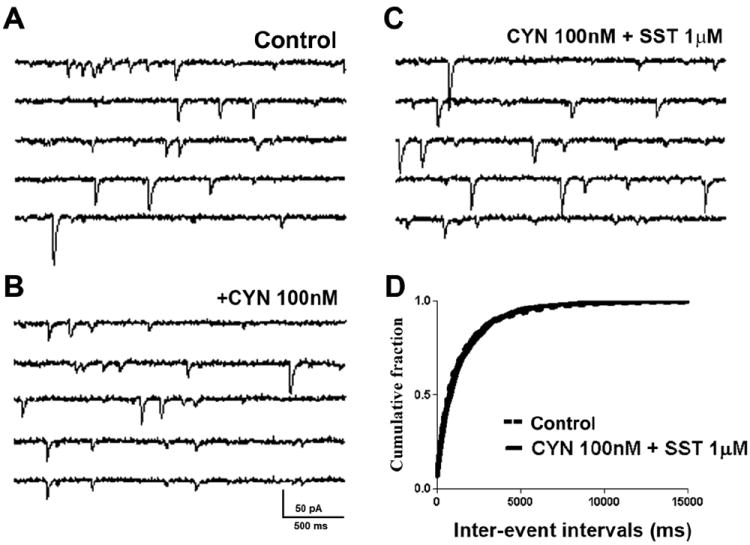
The SST2R antagonist cyanamid-154806 (CYN) blocks SST actions on sEPSCs. (A-C), Representative sEPSC recordings obtained from a CA1 neuron before (A) and after (B) the application of CYN (100 nM) and (C) SST (1 μM) in CYN-pretreated slices. (D), Cumulative probability histogram of sEPSC frequency at baseline (control, dotted line) and after application of SST in CYN pretreated slices (solid line) obtained by pooling data from 7 cells. CYN alone did not alter eEPSC frequency (cumulative probability not shown).
Next, the effect of the clinically available SST2R agonist octreotide on EPSCs was investigated. Octreotide (1 μM) decreased sEPSC frequency from 0.71 ± 0.11 Hz to 0.41 ± 0.06 Hz (p < 0.05, paired t-test, n = 10, Fig 4A) and increased the inter-event intervals, as demonstrated by a representative cumulative probability plot (p < 0.05, KS test, Fig 4B). Interestingly, octreotide also diminished the amplitude of sEPSCs from 25.17 ± 2.6 pA to 20.46 ± 1.27 pA (p < 0.05, paired t-test, n = 10, Fig 4C). Octreotide did not change the rise and decay time constants of sEPSCs. The sEPSC amplitude distribution histogram obtained by pooling 787 consecutive events from all neurons (events per cell ranging from 100 to 190) revealed a reduction in proportion of large-amplitude events 10 min after octreotide application (p < 0.05, KS test, Fig 4D, E). These findings suggested that octreotide, similar to SST, diminished action-potential dependent release of glutamate
Fig 4.
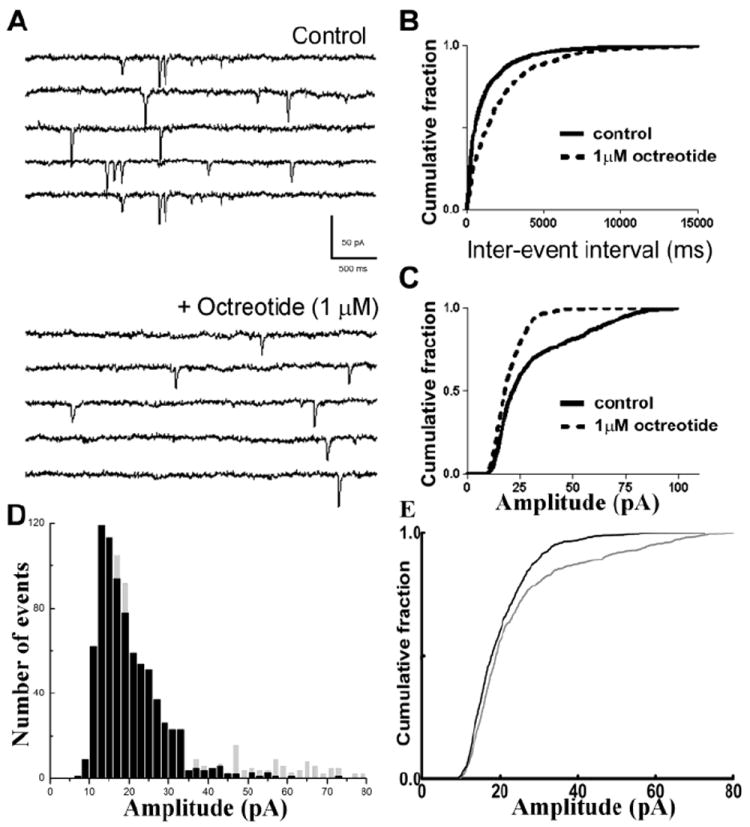
The SST2R antagonist octreotide reduced the frequency and amplitude of sEPSCs at CA1-schaffer collateral synapses. (A), Representative sEPSC recordings obtained from CA1 neurons before (Control) and after application of 1 μM octreotide. (B, C), Cumulative probability plots of sEPSC frequency (B) and amplitude (C) before (—) and after (- - -) octreotide application. (D), Amplitude distribution histograms obtained from sEPSCs recorded at baseline (grey) and after the application of octreotide (black) revealed reduced number of large amplitude events after octreotide application. (E), Cumulative amplitude distribution of sEPSCs in (D) reveals a left shift in sEPSC amplitude following octreotide application (p < 0.05, KS test).
Similar to SST, the application of octreotide also diminished paired-pulse responses. At baseline, the peak amplitude to first stimulus was -142.7 ± 23.5 pA; this was enhanced to -181.8 ± 17 pA in response to the second stimulus (n = 5, p < 0.05, paired t-test). Octreotide abolished the PPF observed at baseline conditions (Fig 5A, B). The peak amplitude of first stimulus (-136.2 ± 18.4 pA) and that of the second stimulus (-130.2 ± 18.3 pA) were similar (n = 5, p > 0.05, paired t-test). Octreotide also decreased the mean amplitude of the eEPSCs from 175.1 ± 11.7 pA to 162.0 ± 9.51 pA (n = 6, p <0.05; paired t-test). These observations indicate that SST2R activation inhibited presynaptic glutamate release. At higher concentrations, octreotide, similar to SST, increased sEPSC frequency. Perfusion with 10 μM octreotide increased sEPSC frequency from 1.08 ± 0.2 to 1.42 ± 0.4 Hz (p < 0.05, paired t-test, n = 6). However, the mean amplitude of the sEPSCs was unchanged (18.2 ± 2.1 pA vs. 17.3 ± 2 pA).
Fig 5.
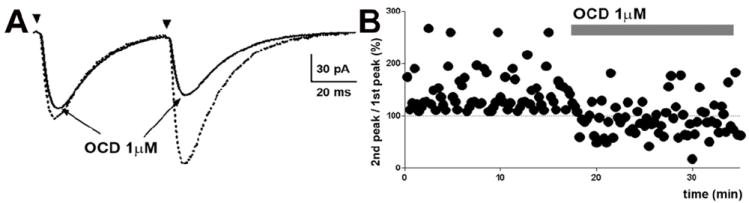
Octreotide diminished the amplitude of electrically evoked EPSCs (eEPSCs) and paired-pulse facilitation (PPF). (A), Representative averaged eEPSCs obtained by paired stimuli before and after the application of octreotide. (B), Time course change in paired-pulse responses following the application of octreotide. The dotted lines at 100% indicate the normalized amplitude of eEPSCs evoked by the 1st stimulus.
SST and SST2R agonists dampen the in vitro firing activity of CA1 pyramidal neurons
The effect of octreotide on the firing rates of CA1 neurons was determined (Fig 6). The loose-patch cell-attached configuration was used to record action potentials without disturbing the intracellular contents. After recording a stable baseline under conditions of elevated potassium, the application of octreotide (1 μM) significantly reduced the frequency of spontaneous spiking activity from 3.26 ± 0.39 Hz to 1.06 ± 0.44 Hz (n = 5, p < 0.05, paired t-test) (Fig 6A, C, E). The decrease in firing rate could be due membrane hyperpolarization. However, because the intracellular potential is uncontrolled in this configuration, the measures of membrane potential are not reliable. We therefore performed whole-cell current-clamp recordings on CA1 neurons to test whether octreotide altered the membrane potential and input resistance. The application of 1 μM octreotide did not significantly alter the resting membrane potential (-60.2 ± 1 mV vs. -60.3 ± 0.9 mV; p = 0.8, paired-t test, n = 5) or input resistance (60.24 ± 1.32 MΩ vs. 60.83 ± 1.27 MΩ; p = 0.40, paired-t test) of CA1 neurons. In contrast, the application of 10 μM octreotide depolarized CA1 neurons from -60.5 ± 1.6 mV to -56 ± 2 mV (p < 0.05, paired-t test, n = 7) without altering input resistance (59.34 ± 0.86 vs. 59.05 ± 0.88 MΩ; p > 0.05, paired-t test). Similar to octreotide, SST (1 μM) also depressed the firing rate of CA1 neurons (1.28 ± 0.60 Hz to 0.32 ± 0.16 Hz, n = 5, p < 0.05, paired t-test) (Fig 6B, D, E).
Fig 6.
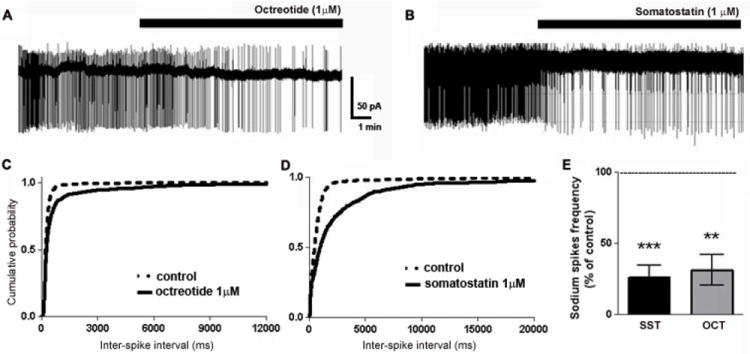
Octreotide and SST reduced the firing rate of CA1 pyramidal neurons. (A, B), Representative trace of a loose-patch recording from CA1 neurons in ACSF containing elevated potassium before and after the application of octreotide (A) or SST (B). (C, D), Cumulative probability plots of neuronal firing before (- - -) or after (—) octreotide (C) or SST (D). (E), Bar graph demonstrating the significant decrease in neuronal firing rate following the application of octreotide or SST.
SST and SST2R agonists prevented or attenuated status epilepticus
We tested whether SST and SST2R agonists can treat pilocarpine-induced SE (Treiman et al., 1990; Wang et al., 2009). The animals in the control group (n = 9) treated with saline administered i.c.v. exhibited recurrent or continuous seizure activity for more than 5 hrs after pilocarpine treatment (Fig 7A). These animals also exhibited behavioral seizures, including head bobbing, forelimb clonus, rearing, and rearing and falling seizures. The median time interval from pilocarpine injection to the first grade 5 (Racine scale) seizure was 63 minutes.
Fig 7.
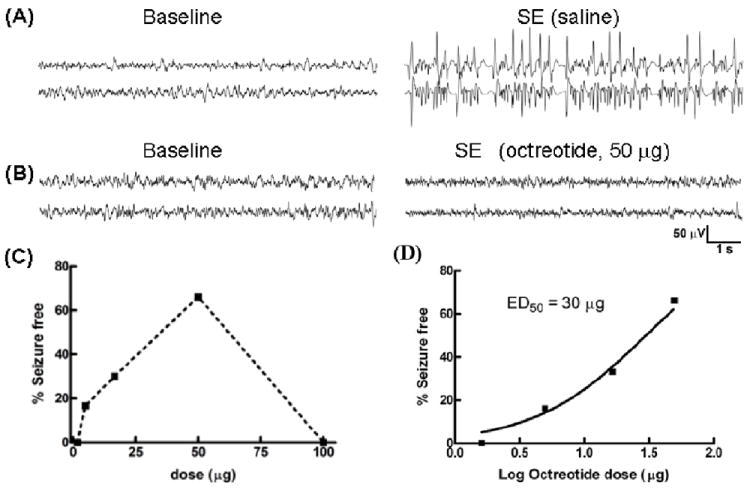
The SST2R agonist octreotide prevented pilocarpine-induced status epilepticus (SE). (A, B), Representative EEG traces from the hippocampus of animals pretreated with i.c.v. saline (A) or octreotide (50 μg) (B) during pilocarpine-induced SE. (C), The relationship between various doses of octreotide and the percentage of seizure-free animals at 1 h is displayed. (D), The relationship in (C) was best approximated by a sigmoidal function (with minimum fixed to 0 and maximum set at 100%); the ED50 value of octreotide to prevent SE was 30 μg. The arithmetic doses of the log doses plotted in (D) are: 1.6μg (0.204), 5.0μg (0.699), 16.6μg (1.22), 50μg (1.699).
The animals that underwent SST infusion exhibited an all-or-none behavior: SST treatment either prevented SE or had no effect at all. SST (72 μg) was infused i.c.v. over 3 hours into 11 animals. Five of the 11 animals did not develop SE, whereas all saline-treated animals developed SE (p < 0.05, two sided Chi-squared test). These 5 animals displayed normal baseline EEG activity for 5 hours of recording. The remaining 6 animals displayed continuous seizure activity within minutes of pilocarpine administration that lasted for 3- 5 hours. The median time interval to the first grade 5 seizure in these animals was 74 minutes. To test whether higher doses of SST would be more effective, 144 μg of the drug was infused in the ventricles, and SE was induced. In all 5 animals tested, EEG and behavioral seizures continued unabated, and the median latency to the first grade 5 seizure was 27 minutes.
We then tested the efficacy of SST2R agonists to treat SE. Octreotide doses from 1.6 to 100 μg (n= 25 total) were tested to define the most effective dose. At a low dose of 1.6 μg, electrographic seizures were unaffected in all of the animals tested (n=3), but behavioral seizures were suppressed. Two of the 3 animals did not exhibit any grade 5 seizures, and the remaining animal took 111 minutes for the first grade 5 seizure to occur. SE in this animal lasted for more than 5 hours. A higher dose 16.6 μg prevented electrographic SE in 4 of 10 animals tested. In the remaining animals, electrographic seizures continued for more than 4 hours; however, behavioral seizures appeared to progress slowly, and the median latency to the first grade 5 seizure was 100 minutes. Octreotide was most efficacious at a dose of 50 μg; it prevented 66.6% of animals (4/6) from going into SE (Fig 7B). In the remaining 2 animals, seizures lasted for 5 hours, and the latency to the first grade 5 seizure was 42 minutes. A higher dose (100 μg) of octreotide failed to prevent SE in any of the 3 animals tested. When a dose-response relationship was calculated for the termination of EEG seizures, a clear dose-response relationship emerged in the dose range from 1.6 μg to 50 μg. The data could be fit to an equation for a sigmoidal dose response curve with an ED50 of 30 μg (Fig 7C).
Lanreotide, another SST2 receptor agonist, was compared to octreotide in its ability to treat cholinergic SE. Several doses of lanreotide were tested. Low doses of 5 μg (n=6) and 7.8 μg (n=6) were minimally effective. 5 μg prevented SE in 1/6 (17%), of animals tested and increased the latency to the first grade 5 seizure to 85 minutes. Three of 6 animals treated with 7.8 μg did not have any grade 5 seizures and the median time to first grade 5 seizure was 83 minutes. A dose of 10 μg was most effective, as it prevented SE in 5/7 animals (71%), and 6/7 animals in this group did not show behavioral seizures. At a dose of 16.6 μg, lanreotide was ineffective in preventing or delaying SE (n = 6). However, this dose abolished behavioral seizures in 4/6 animals. At 50 μg dose, lanreotide was ineffective in preventing or delaying both behavioral and electrographic SE in any of the 6 animals tested. Thus, within a range of concentration, both octreotide and lanreotide were equally efficacious in preventing or attenuating SE; the duration of SE was however unaltered.
Because the effects of drugs on experimental SE can be model dependent, it is important to confirm the effectiveness of these drugs on SE in multiple models. We therefore induced SE by continuous hippocampal stimulation (CHS) (Lothman et al., 1989) and monitored seizures by video and EEG observations every 15 minutes for the first hour and then every hour for five hours post-treatment (Fig 8A). After electrical stimulation, seizures consisting of rhythmic spiking at a frequency of 1.5 - 3 Hz continued for 3-12 hours. Behaviorally, the animals exhibited predominantly grade 2 seizures, with very few grade 3-5 seizures. Nevertheless, in the control group, 60% of the animals continued to have seizure activity for longer than 5 hours, and 80% of the animals had continuous seizure activity that lasted for longer than 3 hours. The animals treated with octreotide 1.5 hrs prior to CHS (50 μg) prevented CHS-SE within 30 minutes in 3/5 animals and within 60 minutes in all animals (p < 0.05, Chi-square test, Fig 8B, D). The animals treated with octreotide were visibly sedated during and after treatment, and those that showed extended electrographic seizure activity lacked any of the grade 2 behavioral seizures that the animals in the control group displayed.
Fig 8.
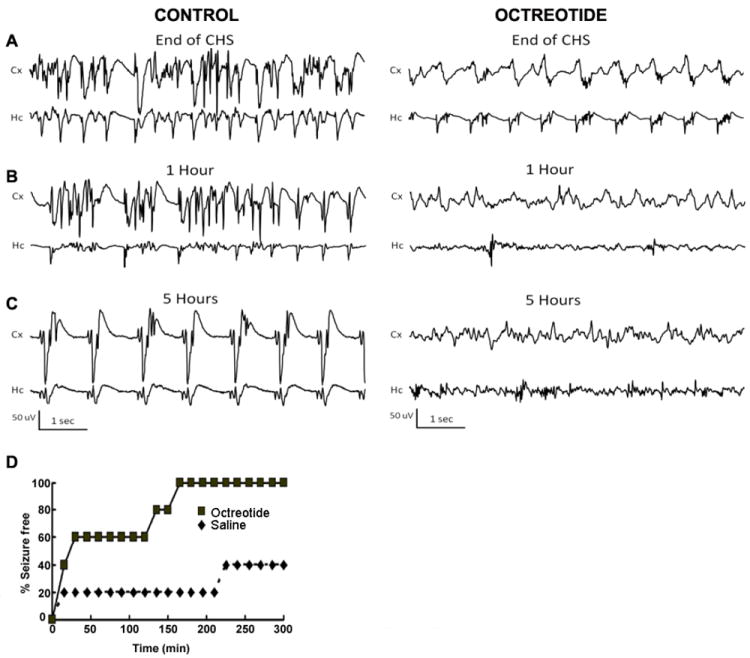
The SST2R agonist octreotide prevented SE induced by continuous hippocampal stimulation (CHS). (A, B, C), Representative EEG traces from the cortex (Cx) and hippocampus (Hc) at the end of CHS (A) and 1 hr (B) and 5 hr (C) after the end of CHS in animals treated with i.c.v.-administered saline or octreotide (50 μg) 1.5 hr before electrical stimulation. (D), The time course of octreotide control of electrographic seizure is displayed. The onset and persistence of seizure freedom in octreotide-pretreated animals was studied by analyzing seizure freedom at various time points up to 5 hr following SE.
Discussion
This study demonstrates that (1) SST diminished presynaptic glutamate release at CA1-Schaffer collateral synapses, (2) SST actions are mediated by SST2R, and (3) SST and its analogs octreotide and lanreotide attenuate SE within a range of doses. This study provides the first direct evidence that the actions of SST in the rat CA1-Schaffer collateral synapses are mediated by SST2R and that SST inhibition of glutamate release can be a novel therapeutic strategy to treat SE.
The loss of large amplitude sEPSCs indicates that SST and SST2R agonists diminished presynaptic glutamate release. The mechanism by which SST and SST2R reduce presynaptic glutamate release remains to be investigated. Based on our finding that SST and octreotide application abolished PPF and diminished the amplitude of eEPSCs, it can be speculated that SST/SST2R agonists likely affected calcium dynamics and presynaptic release mechanisms. Indeed, SST and SST2R activation target presynaptic VGCCs by activating pertussis toxin-sensitive G-proteins on presynaptic terminals (Tallent and Siggins, 1997) and depress vesicular exocytosis. The pertussis toxin-sensitive Gi/Go family of G-proteins causes a positive shift in the voltage dependent activation of calcium currents (Bean, 1989; Ikeda and Dunlap, 1999; Mirotznik et al., 2000; Dolphin, 2003), resulting in the inhibition of calcium currents from VGCCs. This inhibition of calcium currents reduces presynaptic release. An inhibitory action on store calcium release and calcium-induced calcium release (CICR) may also account for SST/SST2R actions. Blockade of CICR by ryanodine or thapsiagargin decreased release probability, synaptic potency (i.e. the average peak EPSC amplitude of successfully evoked events, Stevens and Wang, 1994) and PPF (50-75 ms ISI) at Schaffer collateral-CA1 synapses (Emptage et al., 2001; Cabezas and Buno, 2006). A detailed characterization of the action of SST and SST2Rs on calcium dynamics during short-term plasticity is necessary to conclusively determine their effect on release probability.
Prior studies have proposed that SST inhibits presynaptic glutamate release (Boehm and Betz, 1997; Tallent and Siggins, 1997; Grilli et al., 2004). Boehm and Betz first reported that SST reduced excitatory autaptic currents and mEPSCs in cultured hippocampal neurons and suggested that this reduction was likely mediated by presynaptically located SSTRs. However, cultured neurons do not retain the anatomical connectivity observed in intact hippocampal preparations. For instance, cultured neurons lack the laminar organization of afferent connectivity and the dendritic arbor alignment found in organotypic hippocampal slices (Kaech et al., 2012). Likewise, in autaptic cultures, all synapses arise from the same type of neuron; this does not mimic the heterogeneity of synapses observed in hippocampal slices. mEPSCs and sEPSCs may arise from different subsets of synapses in hippocampal slices; however, in an autaptic cell, they arise from the same population of synapses. These differences can potentially confound extrapolation of the results from cultured neurons to a more intact preparation, such as hippocampal slices. Using intracellular sharp electrode recordings, another study demonstrated that SST depressed the amplitude of eEPSCs mediated by AMPA/KA or NMDA receptors at CA1 pyramidal neurons in acute hippocampal slices (Tallent and Siggins, 1997); however, this study did not examine whether the spontaneous release of glutamate was altered. Furthermore, because the amplitude of eEPSCs can be modified by both pre- and post-synaptic factors, the locus of action of SST remained unclear.
We conclude that the effects of SST are mediated by SST2R because blockade of SST2R with cyanamid-154806 prevented the effects of SST on EPSCs recorded from CA1 neurons and both octreotide and lanreotide mimicked the actions of SST in inhibiting glutamate release. Our studies provide direct evidence in support of earlier findings that the SST-induced reduction of potassium-evoked glutamate release in mouse synaptosomes was mimicked by octreotide and prevented by cyanamid 154806 (Grilli et al., 2004).
Activation of SST2R by SST decreased the firing rate of CA1 neurons, suggesting that diminished presynaptic glutamate release likely reduced CA1 pyramidal neuron excitability. Thus, SST could potentially dampen epileptiform activity in the hippocampus by suppressing glutamate release. Indeed, SST blocked in vitro epileptiform activity in CA3 and CA1 pyramidal neurons (Tallent and Siggins, 1999).
Using both EEG and video to analyze seizures, the current study demonstrated that SST and an SST2R agonist could attenuate SE. Thus, blocking excitatory neurotransmission by reducing glutamate release could be a novel therapeutic option to treat SE. Prior studies on the anti-seizure effects of SST largely relied on behavioral analysis of seizures or did not study a wide range of doses (Mazarati and Telegdy, 1992; Monno et al., 1993; Mazarati and Wasterlain, 2002; Stragier et al., 2006; Aourz et al., 2011; Zafar et al., 2012). As demonstrated in this study, electrographic SE can continue in many animals where behavioral seizures have been attenuated.
SST can augment muscarinic M-currents in hippocampal pyramidal neurons through the activation of SST4Rs to produce anticonvulsant effects (Moore et al., 1998, Qiu et al., 2008). However, activation of M-currents is unlikely to mediate the anticonvulsive action of SST in the present study because SST2R agonists, octreotide and lanreotide prevented pilocarpine-induced SE; SST2Rs do not couple to M-channels (Qiu et al., 2008). Also, SST2Rs could prevent electrically-induced SE. The expression of SST2R is greater in the hippocampus of rats whereas SST4R expression is predominant in the mouse hippocampus (Perez et al., 1994; Piwko et al., 1997; Hannon et al., 2002). These differences suggest that the anticonvulsant action of SST might be mediated by SSTRs in a species-specific manner. Consequently, the depression of excitatory neurotransmission and the anticonvulsant action of SST is largely mediated by SST2R in rats and SST4R in mice (Boehm and Betz, 1997; Moneta et al., 2002; Qiu et al., 2008).
Despite the ability of SST2R agonists to prevent SE, there are multiple limitations that currently limit the use of these drugs. Both SST and octreotide had limited efficacy, peaking at 60-70% at the most effective dose. These observations are consistent with our in vitro functional studies where the drugs failed to suppress presynaptic glutamate release at higher concentrations. One factor that could contribute to this partial efficacy of SST and octreotide may be the rapid desensitization of the SST2R (Dournaud et al., 1998). Ligand-induced SST2R internalization is implicated in SST2R desensitization via reduction in the fraction of surface-expressed receptors (Hipkin et al., 1997; Beaumont et al., 1998). Interestingly, agonist-induced internalization of SST2R in rat brain slices appears to be rapid (<40 min post-exposure) and more pronounced at higher rather than lower concentrations (Boudin et al., 2000). Thus, a reduction in the amount of SST2R at the cell surface likely limits the anticonvulsant efficacy of SST or its analogs at higher concentrations.
Currently available SST analogs do not permeate the BBB or if they do, at rates equivalent to the absorption of albumin (Banks et al., 1990) but their accumulation is prevented by the ABC transporters in the BBB (Fricker et al., 2002). However, it has been suggested that the BBB breaks down during cholinergic SE (Uva et al., 2008; Marchi et al., 2009; Marchi et al., 2007). Octreotide is actively transported across the BBB (Fricker et al., 2002), but it is unclear whether the barrier is sufficiently breached to allow the peripheral administration of octreotide. In preliminary experiments with 5 animals in cholinergic SE, peripheral administration of octreotide was not effective in attenuating SE. Thus, the development of agonists capable of penetrating the BBB and activating SST2Rs is necessary. In addition, it is necessary to design agonists that have a minimal impact on the cell surface availability of the receptors (Koenig et al., 1997;Liu et al., 2005) and improved bioavailability. Thus production of enzymatically resistant analogs with long circulation times can result in significant CNS accumulation even when BBB permeability is low (Banks, 2006). Indeed, the recently developed highly selective SST2R agonists such as L-054,522 and its further optimized molecule, compound 1, have been found to have effective oral bioavailability (Robertson et al., 2010). In addition, alternate drug delivery strategies such as intra-nasal delivery, convection-enhanced diffusion, use of nanospheres that are employed for delivery of other peptide drugs can be employed to deliver SST analogs across the BBB.
In summary, this demonstrates that SST and SST2R agonists prevent SE and that SST2Rs are novel targets to treat SE.
Highlights.
Glutamatergic neurotransmission is enhanced during status epilepticus (SE).
Somatostatin (SST) diminished glutamate release onto CA1 pyramidal neurons.
SST type 2 receptors (SST2R) mediated SST diminution of glutamate release.
SST and SST2R agonists prevented pilocarpine- or electrically-induced SE.
SST inhibition of glutamate release can be a novel therapeutic strategy to treat SE.
Acknowledgments
This study was supported by NIH National Institute of Neurological Disorders and Stroke grants RO1 NS 040337, RO1 NS 044370, and the CounterACT Program, NIH Office of the Director (NIH OD) and NINDS, UO1 NS 58204.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aourz N, De Bundel D, Stragier B, Clinckers R, Portelli J, Michotte Y, Smolders I. Rat hippocampal somatostatin SST3 and SST4 receptors mediate anticonvulsive effects in vivo: Indications of functional interactions with SST2 receptors. Neuropharmacology. 2011;61:1327–1333. doi: 10.1016/j.neuropharm.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Banks WA. The CNS as a target for peptide and peptide-based drugs. Expert Opin Drug Deliv. 2006;3:707–712. doi: 10.1517/17425247.3.6.707. [DOI] [PubMed] [Google Scholar]
- Banks WA, Schally AV, Barrera CM, Fasold MB, Durham DA, Csernus VJ, Groot K, Kastin AJ. Permeability of the murine blood-brain barrier to some octapeptide analogs of somatostatin. Proc Natl Acad Sci USA. 1990;87:6762–6766. doi: 10.1073/pnas.87.17.6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Hepworth MB, Luty JS, Kelly E, Henderson G. Somatostatin receptor desensitization in NG108-15 cells. A consequence of receptor sequestration. J Biol Chem. 1998;273:33174–33183. doi: 10.1074/jbc.273.50.33174. [DOI] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. J Neurosci. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borris DJ, Bertram EH, Kapur J. Ketamine controls prolonged status epilepticus. Epilepsy Res. 2000;42:117–122. doi: 10.1016/s0920-1211(00)00175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudin H, Sarret P, Mazella J, Schonbrunn A, Beaudet A. Somatostatin-induced regulation of SST2A receptor expression and cell surface availability in central neurons: role of receptor internalization. J Neurosci. 2000;20:5932–5939. doi: 10.1523/JNEUROSCI.20-16-05932.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammalleri M, Cervia D, Langenegger D, Liu Y, Dal Monte M, Hoyer D, Bagnoli P. Somatostatin receptors differentially affect spontaneous epileptiform activity in mouse hippocampal slices. Eur J Neurosci. 2004;20:2711–2721. doi: 10.1111/j.1460-9568.2004.03741.x. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Dournaud P, Boudin H, Schonbrunn A, Tannenbaum GS, Beaudet A. Interrelationships between somatostatin SST2A receptors and somatostatin-containing axons in rat brain: evidence for regulation of cell surface receptors by endogenous somatostatin. J Neurosci. 1998;18:1056–1071. doi: 10.1523/JNEUROSCI.18-03-01056.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dournaud P, Gu YZ, Schonbrunn A, Mazella J, Tannenbaum GS, Beaudet A. Localization of the somatostatin receptor SST2A in rat brain using a specific anti-peptide antibody. J Neurosci. 1996;16:4468–4478. doi: 10.1523/JNEUROSCI.16-14-04468.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker G, Nobmann S, Miller DS. Permeability of porcine blood brain barrier to somatostatin analogues. Br J Pharmacol. 2002;135:1308–1314. doi: 10.1038/sj.bjp.0704557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch B, Stott JJ, Joelle DJ, Rogawski MA. Treatment of early and late kainic acid-induced status epilepticus with the noncompetitive AMPA receptor antagonist GYKI 52466. Epilepsia. 2010;51:108–117. doi: 10.1111/j.1528-1167.2009.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Yeh JL, Kapur J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25:5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-Specific Trafficking of GABAA receptors during status epilepticus. J Neurosci. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli M, Raiteri L, Pittaluga A. Somatostatin inhibits glutamate release from mouse cerebrocortical nerve endings through presynaptic SST2 receptors linked to the adenylyl cyclase-protein kinase A pathway. Neuropharmacology. 2004;46:388–396. doi: 10.1016/j.neuropharm.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Hannon JP, Petrucci C, Fehlmann D, Viollet C, Epelbaum J, Hoyer D. Somatostatin SST2 receptor knock-out mice: localisation of SST1-5 receptor mRNA and binding in mouse brain by semi-quantitative RT-PCR, in situ hybridisation histochemistry and receptor autoradiography. Neuropharmacology. 2002;42:396–413. doi: 10.1016/s0028-3908(01)00186-1. [DOI] [PubMed] [Google Scholar]
- Hicks GA, Feniuk W, Humphrey PPA. Outward current produced by somatostatin (SIRF) in rat anterior cingulated pyramidal cells in vitro. Br J Pharmacol. 1998;124:252–258. doi: 10.1038/sj.bjp.0701824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipkin RW, Friedman J, Clark RB, Eppler CM, Schonbrunn A. Agonist-induced desensitization, internalization, and phosphorylation of the SST2A somatostatin receptor. J Biol Chem. 1997;272:13869–13876. doi: 10.1074/jbc.272.21.13869. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Bell GI, Berelowitz M, Epelbaum J, Feniuk W, Humphrey PP, O’Carroll AM, Patel YC, Schonbrunn A, Taylor JE. Classification and nomenclature of somatostatin receptors. Trends Pharmacol Sci. 1995;16:86–88. doi: 10.1016/s0165-6147(00)88988-9. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci. 1997;17:7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JA, Edwardson JM, Humphrey PP. Somatostatin receptors in Neuro2A neuroblastoma cells: ligand internalization. Br J Pharmacol. 1997;120:52–59. doi: 10.1038/sj.bjp.0700859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozhemyakin M, Rajasekaran K, Kapur J. Central cholinesterase inhibition enhances glutamatergic synaptic transmission. J Neurophysiol. 2010;103:1748–1757. doi: 10.1152/jn.00949.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Cescato R, Dewi DA, Rivier J, Reubi JC, Schonbrunn A. Receptor signaling and endocytosis are differentially regulated by somatostatin analogs. Mol Pharmacol. 2005;68:90–101. doi: 10.1124/mol.105.011767. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, Bekenstein JW, Perlin JB. Self-sustaining limbic status epilepticus induced by ‘continuous’ hippocampal stimulation: electrographic and behavioral characteristics. Epilepsy Res. 1989;3:107–119. doi: 10.1016/0920-1211(89)90038-7. [DOI] [PubMed] [Google Scholar]
- Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G, Batra A, Carlton E, Najm I, Granata T, Janigro D. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. 2009;33:171–181. doi: 10.1016/j.nbd.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi N, Oby E, Batra A, Uva L, De Curtis M, Hernandez N, Boxel-Dezaire A, Najm I, Janigro D. In vivo and in vitro effects of pilocarpine: relevance to ictogenesis. Epilepsia. 2007;48:1934–1946. doi: 10.1111/j.1528-1167.2007.01185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BS, Kapur J. A combination of ketamine and diazepam synergistically controls refractory status epilepticus induced by cholinergic stimulation. Epilepsia. 2008;49:248–255. doi: 10.1111/j.1528-1167.2007.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazarati A, Wasterlain CG. Anticonvulsant effects of four neuropeptides in the rat hippocampus during self-sustaining status epilepticus. Neurosci Lett. 2002;331:123–127. doi: 10.1016/s0304-3940(02)00847-9. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Telegdy G. Effects of somatostatin and anti-somatostatin serum on picrotoxin-kindled seizures. Neuropharmacology. 1992;31:793–797. doi: 10.1016/0028-3908(92)90043-o. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev. 1997;21:559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- Meierkord H, Boon P, Engelsen B, Gocke K, Shorvon S, Tinuper P, Holtkamp M. EFNS guideline on the management of status epilepticus in adults. Eur J Neurol. 2010;17:348–355. doi: 10.1111/j.1468-1331.2009.02917.x. [DOI] [PubMed] [Google Scholar]
- Mirotznik RR, Zheng X, Stanley EF. G-Protein types involved in calcium channel inhibition at a presynaptic nerve terminal. J Neurosci. 2000;20:7614–7621. doi: 10.1523/JNEUROSCI.20-20-07614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moneta D, Richichi C, Aliprandi M, Dournaud P, Dutar P, Billard JM, Carlo AS, Viollet C, Hannon JP, Fehlmann D, Nunn C, Hoyer D, Epelbaum J, Vezzani A. Somatostatin receptor subtypes 2 and 4 affect seizure susceptibility and hippocampal excitatory neurotransmission in mice. Eur J Neurosci. 2002;16:843–849. doi: 10.1046/j.1460-9568.2002.02146.x. [DOI] [PubMed] [Google Scholar]
- Monno A, Rizzi M, Samanin R, Vezzani A. Anti-somatostatin antibody enhances the rate of hippocampal kindling in rats. Brain Res. 1993;602:148–152. doi: 10.1016/0006-8993(93)90255-l. [DOI] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neligan A, Shorvon SD. Frequency and prognosis of convulsive status epilepticus of different causes: a systematic review. Arch Neurol. 2010;67:931–940. doi: 10.1001/archneurol.2010.169. [DOI] [PubMed] [Google Scholar]
- Okumura T, Takasu N, Ishimatsu S, Miyanoki S, Mitsuhashi A, Kumada K, Tanaka K, Hinohara S. Report on 640 victims of the Tokyo subway sarin attack. Ann Emerg Med. 1996;28:129–135. doi: 10.1016/s0196-0644(96)70052-5. [DOI] [PubMed] [Google Scholar]
- Olivos L, Artalejo AR. Muscarinic excitation-secretion coupling in chromaffin cells. Acta Physiol (Oxf) 2008;192:213–220. doi: 10.1111/j.1748-1716.2007.01816.x. [DOI] [PubMed] [Google Scholar]
- Peineau S, Potier B, Petit F, Dournaud P, Epelbaum J, Gardette R. AMPA-SST2 somatostatin receptor interaction in rat hypothalamus requires activation of NMDA and/or metabotropic glutamate receptors and depends on intracellular calcium. J Physiol. 2003;546:101–117. doi: 10.1113/jphysiol.2002.025890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J, Rigo M, Kaupmann K, Bruns C, Yasuda K, Bell GI, Lubbert H, Hoyer D. Localization of somatostatin (SRIF) SSTR-1, SSTR-2 and SSTR-3 receptor mRNA in rat brain by in situ hybridization. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:145–160. doi: 10.1007/BF00169831. [DOI] [PubMed] [Google Scholar]
- Piwko C, Thoss VS, Probst A, Hoyer D. The elusive nature of cerebellar somatostatin receptors: studies in rat, monkey and human cerebellum. J Recept Signal Transduct Res. 1997;17:385–405. doi: 10.3109/10799899709036616. [DOI] [PubMed] [Google Scholar]
- Qiu C, Zeyda T, Johnson B, Hochgeschwender U, de Lecea L, Tallent MK. Somatostatin receptor subtype 4 couples to the M-current to regulate seizures. J Neurosci. 2008;28:3567–3576. doi: 10.1523/JNEUROSCI.4679-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rajasekaran K, Todorovic M, Kapur J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann Neurol. 2012;72:91–102. doi: 10.1002/ana.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CR, Flynn SP, White HS, Bulaj G. Anticonvulsant neuropeptides as drug leads for neurological diseases. Nat Prod Rep. 2011;28:741–762. doi: 10.1039/c0np00048e. [DOI] [PubMed] [Google Scholar]
- Schulz S, Handel M, Schreff M, Schmidt H, Hollt V. Localization of five somatostatin receptors in the rat central nervous system using subtype-specific antibodies. J Physiol Paris. 2000;94:259–264. doi: 10.1016/s0928-4257(00)00212-6. [DOI] [PubMed] [Google Scholar]
- Stragier B, Clinckers R, Meurs A, De Bundel D, Sarre S, Ebinger G, Michotte Y, Smolders I. Involvement of the somatostatin-2 receptor in the anti-convulsant effect of angiotensin IV against pilocarpine-induced limbic seizures in rats. J Neurochem. 2006;98:1100–1113. doi: 10.1111/j.1471-4159.2006.03942.x. [DOI] [PubMed] [Google Scholar]
- Sun J, Kapur J. M-type potassium channels modulate Schaffer-collateral CA1 glutamatergic synaptic transmission. J Physiol. 2012 doi: 10.1113/jphysiol.2012.235820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin depresses excitatory but not inhibitory neurotransmission in rat CA1 hippocampus. J Neurophysiol. 1997;78:3008–3018. doi: 10.1152/jn.1997.78.6.3008. [DOI] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol. 1999;81:1626–1635. doi: 10.1152/jn.1999.81.4.1626. [DOI] [PubMed] [Google Scholar]
- Todorovic MS, Cowan ML, Balint CA, Sun C, Kapur J. Characterization of status epilepticus induced by two organophosphates in rats. Epilepsy Res. 2012;101:268–276. doi: 10.1016/j.eplepsyres.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med. 1998;339:792–798. doi: 10.1056/NEJM199809173391202. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Walton NY, Kendrick C. A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res. 1990;5:49–60. doi: 10.1016/0920-1211(90)90065-4. [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- Uva L, Librizzi L, Marchi N, Noe F, Bongiovanni R, Vezzani A, Janigro D, De Curtis M. Acute induction of epileptiform discharges by pilocarpine in the in vitro isolated guinea-pig brain requires enhancement of blood-brain barrier permeability. Neuroscience. 2008;151:303–312. doi: 10.1016/j.neuroscience.2007.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Serafini R, Stasi MA, Vigano G, Rizzi M, Samanin R. A peptidase-resistant cyclic octapeptide analogue of somatostatin (SMS 201-995) modulates seizures induced by quinolinic and kainic acids differently in the rat hippocampus. Neuropharmacology. 1991;30:345–352. doi: 10.1016/0028-3908(91)90059-k. [DOI] [PubMed] [Google Scholar]
- Wang NC, Good LB, Marsh ST, Treiman DM. EEG stages predict treatment response in experimental status epilepticus. Epilepsia. 2009;50:949–952. doi: 10.1111/j.1528-1167.2008.01911.x. [DOI] [PubMed] [Google Scholar]
- Zafar R, King MA, Carney PR. Adeno-associated viral vector-mediated expression of somatostatin in rat hippocampus suppresses seizure development. Neurosci Lett. 2012;509:87–91. doi: 10.1016/j.neulet.2011.12.035. [DOI] [PubMed] [Google Scholar]