

Abstract
Human DNA repair glycosylases must encounter and inspect each DNA base in the genome in order to discover damaged bases that may be present at a density of less than one in ten million normal base pairs. This remarkable example of specific molecular recognition requires a reduced dimensionality search process (facilitated diffusion) that involves both hopping and sliding along the DNA chain. Despite the widely accepted importance of facilitated diffusion in protein-DNA interactions, the molecular features of DNA that influence hopping and sliding are poorly understood. Here we explore the role of the charged DNA phosphate backbone in sliding and hopping by human uracil DNA glycosylase (hUNG), which is an exemplar that efficiently locates rare uracil bases in both dsDNA and ssDNA. Substitution of neutral methylphosphonate groups for anionic DNA phosphate groups weakened nonspecific DNA binding affinity by 0.4–0.5 kcal/mole per substitution. In contrast, sliding of hUNG between uracil sites embedded in duplex and single stranded DNA substrates persisted unabated when multiple methylphosphonate linkages were inserted between the sites. Thus a continuous phosphodiester backbone negative charge is not essential for sliding over nonspecific DNA binding sites. We consider several alternative mechanisms for these results. A model consistent with previous structural and NMR dynamic results invokes the presence of open and closed conformational states of hUNG. The open state is short-lived and has weak or nonexistent interactions with the DNA backbone that are conducive for sliding, and the populated closed state has stronger interactions with the phosphate backbone. These data suggest that the fleeting sliding form of hUNG is a distinct weakly interacting state that facilitates rapid movement along the DNA chain and resembles the transition state for DNA dissociation.
The integrity of the information content of genomic DNA depends on efficient and accurate repair of damaged DNA bases. In many cases, this task is initiated by base excision repair DNA glycosylases, which locate and cleave the glycosidic bond of rare mutagenic bases in DNA (1, 2). Unlike transcription factors or other DNA binding proteins, these unique repair glycosylases must rapidly encounter and inspect each base in the genome in the process of efficiently locating their damage targets. This unique search requirement, which is driven by the evolutionary necessity to patrol the genome, places stringent restraints on the thermodynamic and kinetic aspects of the enzyme-nucleic acid interaction that almost certainly differ from typical DNA binding proteins. If the glycosylase interacts too strongly with nonspecific DNA, then it spends too much time at non-target sites, if it interacts too weakly or moves too fast, then its residence time is not long enough to allow detection of DNA damage when it is encountered. These properties of an efficient damage search are one example of what has been called the “search-speed/stability” paradox (3, 4).
To resolve the paradox, DNA glycosylases have harnessed the most favorable mechanistic features of two distinct modes of facilitated diffusion: DNA hopping and sliding (2, 3, 5, 6). Frequent dissociation from the DNA chain most often results in reassociation at a nearby DNA segment (hopping), keeping the enzyme from wasting time unproductively searching regions where there is no DNA and allowing it to bypass bound proteins (7, 8). Once the enzyme has encountered a new DNA segment, it then has an opportunity to remain in contact with the chain and move along it in a one-dimensional sliding mode (3, 5, 6). An upper limit on the length of DNA over which sliding can occur is determined by the residence time of the enzyme on nonspecific DNA and the 1D diffusion constant (6). The importance of sliding, even over short segments of the DNA chain, is that the enzyme remains in contact with its substrate, thereby expanding the number of bases that can be inspected during each binding event. These two general modes of the search have been observed (or inferred) for many DNA glycosylases and other site-specific DNA binding proteins (7–20).
Although the fundamental importance of hopping and sliding in the damage search is well appreciated, a quantitative mechanistic understanding of the molecular features of the DNA chain that influence an enzyme's ability to hop and slide are poorly understood. In this regard, it is widely believed that the polyanion character of the DNA phosphate backbone provides an important nonspecific electrostatic handle allowing engagement of positively charged side chains on the enzyme. Such interactions may play a role in both hopping and sliding along nonspecific DNA, but also in other steps of the reaction such as specific recognition, making it challenging to sort out these individual effects (21–23). Specifically, electrostatic tracking along the phosphate backbone is often invoked as the primary translocation mode for DNA sliding, but a direct test of this mechanism has been absent. Here we investigate the role of charged DNA phosphate groups in the ability of human uracil DNA glycosylase (hUNG) to hop and slide along DNA during its search for uracil bases. The results show that a continuous backbone charge is not required for hUNG to track efficiently along a DNA strand, and that the transient sliding state has features that resemble the transition state for DNA dissociation.
Materials and Methods
Protein and Oligonucleotide Reagents
hUNG was purified as previously described (9). Protein concentrations were determined by absorbance measurements at 280 nm using an extinction coefficient of 33.68 mM−1 cm−1. Oligonucleotides except for those containing methylphosphonate linkages were ordered from Integrated DNA Technologies (www.IDTDNA.com) in the crude desalted form and purified by denaturing PAGE. All oligonucleotide sequences are reported in the Supplemental Methods and concentrations were determined by UV absorption at 260 nm using extinction coefficients calculated from nearest neighbor parameters.
Experimental conditions
All measurements in this paper and the accompanying paper (insert reference upon publication) were made at 37 °C in a standard reaction buffer consisting of 20 mM HEPES pH 7.5, 0.002% Brij 35 detergeant (Sigma Aldrich), 3 mM EDTA (added from a 0.5 M pH 8.0 stock), and 1 mM DTT unless otherwise noted.
Synthesis of oligonucleotides containing methylphosphonate linkages
Oligonucleotides containing methylphosphonate linkages were synthesized using standard phosphoramidite synthesis procedures on an Applied Biosystems 390 DNA/RNA synthesizer. Nucleoside phosphoramidites and methylphosphonamidites were purchased from Glenn Research (Sterling, VA). After synthesis, the DNA was deprotected and cleaved from the silica support by the addition of 0.5 mL 45:45:10 acetonitrile/ethanol/ammonium hydroxide and allowed to incubate at room temperature for 30 minutes. 0.5 mL of ethylenediamine was then added and the DNA containing solution was allowed to sit overnight at room temperature. The DNA containing solution was separated from the silica support and dried under vacuum. After resuspension in 25 mM Tris-HCl pH 7.5 (Buffer A) the DNA was then purified from the failure products by HPLC by injection onto a Dionex™ DNA Pac anion exchange column and eluted with a linear gradient from 10% Buffer A to 90% Buffer B (25 mM Tris-HCl pH 7.5, 1M NaCl).
90mer oligonucleotide substrates used in the site transfer assays containing methylphosphonate linkages were first synthesized as smaller precursors and then a 3-piece ligation was performed to create the final product (Supplemental Fig. S1). Sequences of the precursor oligonucleotides are listed in the Supplemental Methods. For ligation, piece 1 (1.5 nanomoles) and piece 2 containing methylphosphonate linkages (2 nanomoles) were first phosphorylated at the 5′ end by incubation with T4 PNK (New England Biolabs) at 37 °C in a single reaction mixture (~200 μL volume in 1× DNA ligase buffer, New England Biolabs™). After inactivation of T4 PNK, Piece 3 (2 nmoles) and the Splint (2 nanomoles) were then added to the reaction mix and hybridized by heating to 95 °C for 5 minutes and allowed to cool slowly to room temperature by placing the heat block on the bench top. Fresh ATP was then added to 1 mM final concentration along with T4 DNA ligase (New England Biolabs) and the reaction mixture was incubated at 37 °C overnight. The reaction was then mixed with 50% formamide (final concentration) and the ligated product was purified by denaturing PAGE (Supplemental Fig. S1).
Determination of DNA Dissociation Constants by Fluorescence Anisotropy
Binding of hUNG to non-specific DNA was determined using fluorescence anisotropy in a Spex Fluormax 3 fluorimeter at 37 °C. Concentrated hUNG in the standard reaction buffer containing 50 nM labeled DNA was titrated into a cuvette containing 50 nM labeled DNA in reaction buffer in order to avoid dilution of the DNA during the titration. After each addition the cuvette was placed in the fluorimeter and allowed to equilibrate for 2 minutes as the reading was found to stabilize after 60–90 seconds. For dissociation constants greater than 5 μM, KD values were determined by diluting a concentrated solution of hUNG in reaction buffer and 50 nM labeled DNA with a solution of labeled DNA only. The forward titration was found to overlay titrations performed by dilution indicating that anisotropy values were determined at equilibrium. Data were then fitted to a single site binding isotherm (anisotropy = Bmax × [hUNG]free/(KD + [hUNG]free) + Bmin), where Bmax and Bmin are the maximal and minimum anisotropies, and it was assumed that the free DNA concentration equals the total (which is a valid assumption given that the KD >> [DNA]total).
Intramolecular Site Transfer Assay
Site transfer measurements were performed identically as before (9, 11) with some modifications in the steps after reaction quenching to account for substrate and buffer differences. The DNA concentration in all site transfer measurements was 40 nM and the hUNG concentration ranged from 10 – 20 pM under the standard reaction conditions, and for the data presented in Fig. 6 from 300 pM to 1.5 nM.
Figure 6.

Site transfer measurements of hUNG under approximated physiological ionic strength conditions (140 mM potassium glutamate, 10 mM Na-HEPES pH 7.5, 200 μM MgSO4). (a) Gel images of the raw site transfer measurements in the presence and absence of uracil. (b) Determination of Ptrans by linear extrapolation. The values are the same in the presence and absence of uracil indicating that site transfer occurs entirely by sliding under these high salt conditions.
For methylphosphonate containing duplexes (S5M and S6M), 30 picomoles of the top and bottom DNA strands were 5′-end labeled with 33P by incubation with T4 polynucleotide kinase and [γ33P] ATP in separate reactions. The reactions were then mixed and the strands hybridized by heating to 95 °C for 10 minutes in a dry heat block followed by slow cooling to room temperature by placing the heat block on the bench top. The hybridized DNA was then separated from the unicorporated [γ33P] ATP by gel filtration using P30 resin (BioRad™) and then desalted using P6 resin (BioRad™). Samples obtained before and after gel filtration were analyzed by native gel electrophoresis, where percent recovery was calculated from imaging of the band densities, and completeness of hybridization was confirmed. In general, the percent recovery was at least 80%. After reaction with hUNG and quenching by uracil DNA glycosylase inhibitor protein (UGI, New England Biolabs™) each individual reaction was treated with the nicking enzyme Nt.BbvCI and APE1 endonuclease as previously described (9, 11) resulting in discrete double-stranded fragments corresponding to the hUNG reaction products. Each sample was then separated by electrophoresis on a 0.5 millimeter thick 10% native gel (19:1 bis:acrylamide) run in 1× TBE buffer at 20 Watts in a model S2 sequencing gel for 1 hour and 40 minutes without pre-running the gel.
For S6Mss the 5′ and 3′ ends were labeled by incubation with [γ32P] ATP and 3′-deoxyadenosine 5′-triphosphate (cordycepin 5'-triphosphate)-[α-32P] using polynucleotide kinase and terminal transferase (New England Biolabs), respectively. Similarly as above for the duplex substrates, after radiolabeling the unincorporated nucleotides and excess salts were removed by gel filtration using P30 and P6 resins (Biorad™). After reaction with hUNG and quenching, the resulting abasic sites were cleaved by the addition of 0.25 M ethylenediamine pH 8.0 (final concentration) followed by immediate heating to 95 °C for 5 minutes. Formamide containing both xylene cyanol and bromphenol blue was then added to 65% final concentration and the samples were loaded onto a 10% denaturing gel (19:1 bis:acrylamide).
For the duplex substrate S5 under physiological salt conditions (140 mM potassium glutamate, 200 μM MgSO4, 10 mM Na-HEPES, pH 7.5) the uracil containing strand was first labeled with 32P at the 5′ and 3′ ends as described for S6Mss above. The labeled strand was then hybridized to the complementary strand and the unincorporated radiolabel was removed using P30 resin (Biorad™). Forty nanomolar of the duplex substrate was then reacted with hUNG and quenched at various time points using UGI as described above. To each aliquot, 3 μl of 0.25 M ethylenediamine pH 8.0 was added and the reaction was immediately heated to 95 °C for 5 minutes to cleave the DNA at the abasic sites. Formamide gel loading buffer was then added to 65% final concentration and the samples were heated at 95 °C for an additional 3 minutes. The samples were immediately loaded onto a pre-heated 10% (19:1 bis:acrylamide) denaturing gel in order to fully denature any residual structure.
Analysis of the Site Transfer Data
All gels were exposed to a storage phosphor screen and digitized using a phosphorimager. For each reaction time course, product band densities were quantified in QuantityOne™ using the box method. More details concerning the data analysis are presented in the Results. All errors presented in the text are standard deviations derived from at least three independent measurements.
Results
Approach
A method was recently described that allows measurement of the probability (Ptrans) that hUNG will successfully transfer between two uracil sites embedded in a single DNA chain separated by a known distance using a sliding or hopping pathway (Fig. 1a) (9). This is the first approach that allows dissection of the total transfer probability into the individual contributions from hopping (Phop) and sliding (Pslide), where Ptrans = Phop + Pslide (Fig. 1).
Figure 1.

a) The `molecular clock' approach uses a small molecule inhibitor of hUNG (uracil) to trap enzyme molecules that have hopped off the DNA chain during transfer between two uracil target sites, while leaving sliding enzymes unperturbed (9). Thus, this method allows quantitative determination of the individual contributions of hopping and sliding transfers (where the total transfer probability is Ptrans = Phop + Pslide). b) Simulations showing the dependence of facilitated transfer (Ptrans) on the concentration of the trap [based on the mechanism in (a)]. As previously noted (9), the probability of locating a site by hopping includes the probability that the enzyme initially falls off the DNA (Poff) as well as the probability that it returns to the same DNA chain (Preturn) without getting lost to bulk solution (Phop = PoffPreturn). The trap allows selective disruption of the hopping pathway because the probability that a hopping enzyme returns to the DNA chain decreases according to Preturn = kreturn/(kreurn + kTrap[Trap]). The utility of this approach and the numerous control experiments that confirm its utility have been previously published (9).
The method requires quantitative site transfer probability measurements (see below) in the absence and presence of a small molecule trap of the enzyme. Inclusion of the trap (the free uracil base) serves to capture all enzyme molecules that have dissociated from the DNA during the process of transferring between the two uracil sites (i.e. the enzyme molecules that have hopped off the DNA). The trap has no effect on enzyme transfers that follow the sliding pathway because the binding site for the trap is blocked when hUNG is bound to nonspecific DNA. Separation of the two pathways (Phop and Pslide) is possible because at zero concentration of trap transfer can occur by both hopping and sliding, but as the trap concentration increases, the hopping contribution diminishes in a hyperbolic fashion, ultimately approaching a limiting asymptote equal to Pslide (long dashed line, Fig. 1b). If the site spacing is large enough, no enzyme molecules will reach the second site without departing the DNA at least once, and transfer will be entirely ablated at high concentrations of trap (short dashed line, Fig. 1b). Conversely, at short site spacings all transfers may occur by sliding and therefore will be impervious to the trap (solid line, Fig. 1b).
The reader is referred to reference (9) for a detailed description of the method including control experiments that establish its utility for hUNG. The experimental observations that support the conclusion that uracil serves as a trap of a dissociated state of hUNG without disrupting DNA sliding are: (i) Two pathways for transfer between substrate sites are observed (uracil insensitive and sensitive). (ii) The uracil concentration dependence of Ptrans follows the expected hyperbolic kinetic behavior (Fig. 1b), including the non-zero plateau value at short site spacings and high uracil concentrations, as would be expected for concurrent hopping and sliding. (iii) The site spacing dependences of Pslide and Phop are consistent with those expected for hopping and sliding pathways. That is, the probability for hopping (uracil sensitive) follows a 1/r dependence on site spacing, while the probability for sliding (uracil insensitive) shows a bp2 dependence on site spacing. (iii) Transfer was completely eliminated at high uracil concentrations when the substrate sites were positioned on opposite DNA strands where an obligate dissociation/reassociation step is required for transfer. This was observed even though the opposite strand sites were closer in space than when positioned on the same strand. Additionally, at site spacings exceeding the sliding length, identical values of Ptrans were previously observed for sites positioned on the same and opposite strands, consistent with hopping (11). (iv) The hopping pathway was highly sensitive to increases in ionic strength while the sliding pathway was not (9). (v) High concentrations of uracil have no effect on the dissociation constant for nonspecific binding of UNG to DNA, but uracil blocks the catalytic activity of the enzyme (9). These observations are consistent with the trap having no effect on sliding and acting solely by trapping the active site of the dissociated enzyme.
Calculating Site Transfer Probabilities
To determine the probabilities for facilitated site transfer by hUNG, we use an initial-rate, steady-state assay that quantifies the fraction of enzyme molecules that excise one uracil site (primary excision events) and then successfully transfer and excise the other uracil in the same DNA molecule (secondary excision events) (Fig. 2a) (9, 24, 25). Primary or secondary uracil excision events will lead to discrete fragments of the double end-labeled DNA which may be resolved by polyacrylamide gel electrophoresis after post-reaction sample processing (see Materials and Methods) (Fig. 2b). If only primary excision events occur at site 1 or 2, the DNA fragments A + BC or AB + C will be produced in equal amounts with apparent velocities v1 = v2 if each site reacts identically. However, if intramolecular transfer occurs, the larger AB and BC fragments will be efficiently converted into the smaller fragments A and C (as well as the unobserved B fragment) with velocities v2→1 (reflecting 2→1 transfers) and v1→2 (reflecting 1→2 transfers). It is worth noting that the initial rates for formation of fragments A and C depend on both primary and secondary events, and therefore, v2→1 and v1→2 are not necessarily equivalent to the initial rates of appearance of fragments A and C. In general, the qualitative hallmark of intramolecular transfer is the production of greater amounts of the secondary excision products A and C at the expense of the single excision products AB and BC (20, 24).
Figure 2.

Site transfer assay. a) Schematic of the site transfer assay where a single strand of a duplex DNA substrate contains two uracil sites is reacted with hUNG ([hUNG] << [DNA]). After quenching, cleavage of the product abasic sites results in single site (AB and BC) and double site (A and C) product fragments (produced from intramolecular translocation events). Qualitatively, intramolecular translocation of hUNG is indicated by an excess of the A and C fragments under initial rate conditions (see text). b) Schematic of the reaction products as analyzed by gel electrophoresis. Bands are quantified by imaging and the intramolecular transfer efficiency is calculated from eq 1 (see text).
The overall transfer probability (Ptrans) may be precisely calculated from the time dependent fragment concentrations. These concentrations are inserted into eq 1, which requires extrapolation to zero time to obtain the true transfer probability (24). The basis for this equation can be easily understood: the denominator counts all excision events and the numerator counts only secondary excision events.
| (1) |
Thus, the ratio reveals the fraction of all excision events that lead to successful transfers to the second site. (The term −[AB] − [BC] in the numerator corrects for the fact that fragments A and C can result from both primary excision events ABC → A + BC and ABC → AB + C, or secondary excision events AB → A + B and BC → B + C.) We find that this is a useful and straightforward analytical approach when there is no site preference for excision of the individual sites or no directional bias to transfer (9, 11). In the following paper we use a modified analytical approach that is useful when a site excision or transfer bias is present (insert reference upon publication).
Effects of Neutral Methylphosphonate (M) Substitutions on Nonspecific DNA Binding
Previous work suggested that a continuous DNA phosphate backbone was necessary and sufficient for DNA sliding because (i) hUNG sliding only occurred between uracil sites that were positioned on the same strand in duplex DNA, and (ii) sliding between uracil sites was observed on ssDNA substrates (reference (9) and accompanying paper) (insert reference upon publication). To begin to explore the role of phosphate backbone charge on DNA sliding, the effects of neutral M substitutions on nonspecific DNA binding were first determined using fluorescence anisotropy measurements (Fig. 3). Three 5′ fluorescein-labeled DNA constructs were investigated that contained mixed diastereomer M linkages at one or more positions, which were then compared with the corresponding all phosphodiester versions (Fig. 3a). The M-DNA constructs shown in Fig. 3a were chosen to match the intervening DNA strand segments used in the site transfer measurements described below (NS5M, NS6M), and also to evaluate the effect of removing as many as four phosphate charges (NS10M). Using single DNA strands as models for the intervening sequences in duplex DNA is justified because (i) structural studies indicate that hUNG primarily interacts with the phosphate backbone on the single strand of DNA that connects the two target bases (26), (ii) hUNG is known to slide along a single strand in duplex DNA (reference (9) and accompanying paper) (insert reference upon publication), and (iii) M substitutions in a single strand of duplex DNA still allow binding to the other strand and would complicate the interpretation of equilibrium dissociation constants.
Figure 3.
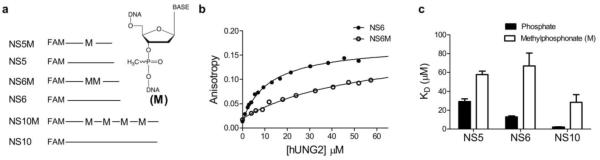
Substrate design and effects of methylphosphonate (M) substitutions on nonspecific DNA binding as measured using fluorescence anisotropy. (a) Design of 5′ fluorescein labeled oligonucleotides containing all phosphodiester or methylphosphonate (M) linkages. NS5 and NS6 and their corresponding M containing oligonucleotide sequences were chosen to match the intervening sequences in the uracil containing substrate used in the site transfer assays. (b) Equilibrium hUNG binding to oligonucleotides NS6 and NS6M. (c) Summary of determined dissociation constants for hUNG and non-specific DNA. Error bars represent mean ± 1 s.d. of at least three independent trials.
Binding measurements revealed that single or multiple M substitutions in single stranded DNA decreased the binding affinity (Kd) of hUNG compared to matched all phosphodiester controls (Fig. 3b & c). The 2 fold-effect of a single M substitution in the center of a 5 mer strand (NS5M and NS5, ΔΔG = 0.42 kcal/mol) was increased 5-fold in the 6 mer strand containing two M substitutions (NS6M, ΔΔG = 1.01 kcal/mol, or ~0.5 kcal/mol per M linkage). Similarly, the 10mer strand containing four methylphosphonates (NS10M) showed a 14-fold deficit in binding (ΔΔG = 1.52 kcal/mol, or ~0.4 kcal/mol per M linkage). Qualitatively, these results demonstrate that the removal of phosphate charge has a damaging effect on nonspecific DNA binding, and raise the expectation that if site transfer by hUNG requires interaction with the phosphate backbone, the removal of these interactions should diminish successful sliding between two uracil sites. We defer to the Discussion possible further physical interpretations of the effects of M substitution on nonspecific DNA binding.
Sliding of hUNG Does Not Require a Continuous Polyanion DNA Strand
To address the question of whether a continuous backbone charge is a requirement for sliding along duplex DNA, we synthesized two 90mer M-substituted DNA substrates containing M linkages on the DNA strand connecting the two uracils (Fig. 4a). The intervening nonspecific DNA strand that connects the two uracil sites in these substrates corresponds exactly to the sequences of NS5M or NS6M described above. Importantly, the substrates in Fig. 4a were constructed with uracil spacings of five and six bp (S5M, S6M) because it has been previously shown that 40% and 20% of the hUNG site transfers occur by a sliding pathway at these spacings when uninterrupted phosphodiester linkages are present (9). In addition, we took care to position the M linkages far enough away from the uracil sites such that the footprint of the specific hUNG catalytic complex does not overlap these positions. This aspect of the substrate design is critical because it has been shown that specific M substitutions within two nucleotides of the uracil site can have a large damaging effect on catalysis (ΔΔG up to 10 kcal/mole) (22, 23).
Figure 4.

hUNG sliding and hopping is unaffected on double stranded DNA substrates containing intervening neutral methylphosphonate (M) linkages. a) Schematic of the substrates used (S5M, S6M). Methylphosphonate positioning was chosen so that the catalytic excision of uracil by hUNG is unaffected (22, 23). b) Gel images of the site transfer products derived from S5M in the presence and absence of uracil. c) Determination of Ptrans, where the observed site transfer (Ptransobs, eq 1) is calculated at each time point and linearly extrapolated to zero time to determine the true site transfer value (Ptrans). Values at 10 and 15 mM uracil were identical indicating measurements were made within the plateau region depicted in Fig. 1b. d) Summary of the site transfer properties of hUNG for double stranded methylphosphonate containing DNA substrates compared to the all phosphodiester versions. Data for S5 and S6 were reported previously and are shown for comparison (9). Values are equal to the mean ± 1 s.d. of at least 3 trials at 0 mM Uracil and 6 at high uracil (3 each at 10 and 15 mM uracil).
We measured Ptrans, Pslide, and Phop for S5M and S6M, and compared these values to those previously measured for the analogous phosphodiester substrates S5 and S6 (Fig. 4) (9). As described above, measurement of Ptrans was obtained in the absence of the uracil trap, and measurements of Pslide were obtained in the presence of 10 and 15 mM trap (the two values were identical, confirming that the transfer measurements were in the plateau region depicted in Fig. 1b). For all of the data, Pslide is reported as the average value obtained at 10 and 15 mM uracil concentration (n = 3 for each concentration) and Phop is the difference between Ptrans and Pslide. Representative transfer data for S5M in the absence and presence of the uracil trap shows a significant degree of intramolecular transfer as revealed by excess A and C fragments (Fig. 4b). Addition of 10 or 15 mM uracil trap leads to a reduction in the successful transfer events, but transfer is not entirely ablated indicating that a sliding pathway is present (Fig. 4b). Extrapolation to zero time using eq 1 shows that Ptrans = 0.61 ± 0.08 and Pslide = 0.35 ± 0.07 for S5M, which are values indistinguishable from those previously reported for S5 (Fig. 4c and d). In addition, there was no difference between the transfer parameters of S6M containing two intervening M linkages and the all phosphodiester analog S6 (Fig. 4d). The transfer parameters for these phosphodiester and M-substituted substrates are summarized in Fig. 4d from which we conclude that ablating as many as one-third of the intervening negative charges connecting the two uracil sites has no measurable effect on Ptrans, Pslide, or Phop.
We next examined M linkages in the context of transfer of hUNG on single stranded DNA using a ssDNA substrate that contained two M linkages analogous to the duplex S6M (S6Mss). S6Mss was designed to have minimal secondary structure and to have no more than two adjacent adjacent Watson-Crick pairings to eliminate potential secondary structure (Supplementary Fig. S2). The data for S6Mss is summarized in Fig. 5 and show that M linkages have no effect on site transfer compared to the all phosphodiester ssDNA substrate S5ss. Comparing S6ss to S5ss is justified because Pslide for ssDNA has a flat dependence with site spacing between 5 and 10 ntds (9), [see also accompanying paper (insert reference upon publication)].
Figure 5.

hUNG sliding on ssDNA is unaffected by methylphosphonate substitutions. (a) Gel images of the site transfer assay in the presence and absence of uracil for S6Mss which contains two intervening M substitutions. We note the presence of a small amount of cleavage in the zero time lane (<1%). This was found to be the result of the commercially available terminal deoxynucleotidyl transferase used in the 3′ end labeling having a very small amount of uracil DNA glycosylase activity over the >2 hr incubation at 37 °C, likely from copurification. These background bands were determined to have no effect on the site transfer calculations. (b) Linear extrapolation of Ptransobs for S6Mss in the presence and absence of uracil to determine Ptrans. (c) Comparison of the site transfer measurements of S6Mss to that of the all phosphodiester substrate S5ss. Comparison with S5ss is reasonable because site transfer by sliding on ssDNA is flat for site spacings between 5 and 10 ntds (see text).
The absence of a requirement for a continuous phosphate charge in sliding or hopping between two closely spaced sites in dsDNA or ssDNA is striking in comparison with the damaging effect of M substitutions on the Km for a uracil-containing substrate (ΔΔG = ~1–4 kcal/mole depending on position) (22), the large and highly stereospecific 5–10 kcal/mol effects of single M substitutions on the activation barriers for uracil excision in single strand or dsDNA (22, 23), and the significant effects of M substitution on nonspecific DNA binding reported above. These differences suggest that the rapid kinetic process of nonspecific sliding does not involve the same phosphate backbone interactions observed in crystal structures of nonspecific and specific complexes between hUNG and DNA (26, 27).
hUNG site transfer using physiological ion concentrations
Site transfer measurements published previously have been studied under conditions where [NaCl] ranged from 22 to 72 mM. In this range, sliding was found to be insensitive to salt, but hopping was fully ablated at salt concentrations exceeding 42 mM (9). However, it is desirable to evaluate these parameters under conditions that more closely mimic the intracellular ion concentrations. For this purpose we use a buffer consisting of 140 mM potassium glutamate, 10 mM Na-HEPES pH 7.5 and 200 μM MgSO4.
Measurements of equilibrium nonspecific DNA binding and hUNG catalytic activity under these conditions were first made. Compared to low ionic strength conditions, the equilibrium dissociation constant for nonspecific binding was increased ~100 fold (0.82 ± 0.26 μM to 85 ± 19 μM) (Supplemental Fig. S3a). Similarly, the catalytic activity of hUNG for a 90mer DNA duplex substrate containing a single uracil site was reduced ~300 fold under these conditions with a measured kcat/Km = 1 × 106 M−1s−1 (Supplemental Fig. S3b). This value may be compared with the previously measured kcat/Km = 3.4 × 108 M−1s−1 for the identical substrate at low ionic strength. We note that accurate determinations of kcat and Km were not possible under physiological salt conditions, but a good estimate of kcat/Km could be obtained from the linear increase in rate using 0 – 4 μM substrate (Supplemental Fig. S3b). Most of the effect on activity is assignable to Km because the maximal observed rates under physiological salt concentrations approached the kcat value of 5 s−1 under low salt conditions (9).
Site transfer measurements were then made using the physiological buffer with duplex substrate S5, which contains two uracils positioned 5 bp apart. At this spacing, intramolecular site transfer was still observed (Ptrans = 0.20 ± 0.04). The transfer probability was similar at high uracil (Pslide = 0.16 ± 0.06), indicating that within error of the measurements all transfers occur by sliding, although Pslide was reduced compared to the value at low ionic strength (Pslide = 0.37 ± 0.06). This result matches the previous finding that the hopping pathway is eliminated with a salt concentration of 42 mM while sliding persists unabated at the same concentration (9). Thus despite the 100-fold decrease in nonspecific binding affinity under mock physiological conditions, short-range sliding of hUNG on DNA can still occur. These findings imply that the binding interface of the sliding form of hUNG is immune to invasion by salt ions. This observation is consistent with the inability of the uracil trap to access the active site of hUNG during the process of sliding.
Discussion
Different Effects of M Substitution on Nonspecific DNA Binding, DNA Translocation and Uracil Excision
Comparison of the divergent effects of methylphosphonate (M) substitutions on nonspecific DNA binding, translocation between uracil sites, and catalysis by UNG leads to the conclusion that the requirements for a charged phosphate group in these processes are very different. Previous studies where stereospecific M substitutions were made in single stranded and duplex substrates of UNG have revealed that substitutions at the +1, −1, and −2 phosphates surrounding the uracil site (5′p+1Up−1Np−2N3′) result in stereospecific 101−108 fold damaging effects on catalysis (22, 23). Most of these large effects were attributed to the beneficial energetic effects of the anionic phosphate groups towards stabilization of the glycosyl cation transition state. The previously measured damaging effects of single M substitutions on the ground state Michaelis complex were not stereospecific and were less than the effects on the activation barrier (i.e. Km effects were in the range 10–100 fold) (22, 23). The even smaller damaging effect of a single M substitution on nonspecific DNA binding (~2-fold, Fig. 3c), would suggest that the nonspecific complex differs in its interactions with the phosphate backbone as compared to the Michaelis complex. Despite the apparent differences between these complexes revealed by M substitution, the high resolution crystal structures of the specific and nonspecific hUNG-DNA complexes show that the same phosphate groups form hydrogen bonds with neutral serine or histidine side chains, or backbone amide groups, and that there are few cationic groups ≤ 3.3 Å from phosphate oxygens (Fig. 7) (26). Thus, taken together, these energetic measurements suggest that as the enzyme moves forward along the reaction coordinate it forms increasingly important electrostatic interactions with the phosphate backbone. The interesting exception, as shown in this work, is the transient state for DNA sliding which apparently has no requirement for an uninterrupted charged phosphate chain.
Figure 7.
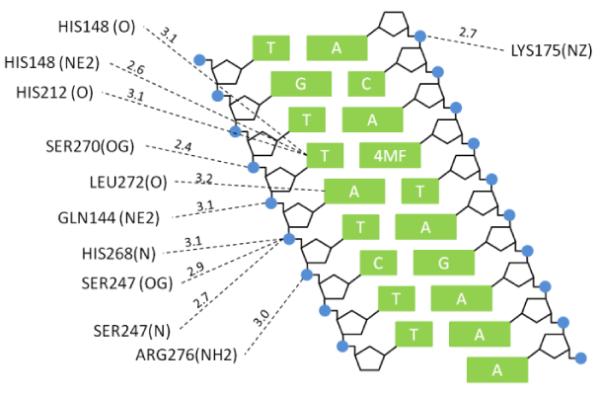
DNA interactions of hUNG non-specifically bound to a destabilized thymine basepair (PDB ID: 2OXM (27), 4MF = 4-methylindole). Residues shown have a nitrogen or oxygen atom < 3.3 Å of a nitrogen atom of a DNA base or a phosphate oxygen of the DNA backbone.
What is the physical basis for the different effects of M substitution on nonspecific binding and DNA translocation? Although M substitution has only a minor effect on B DNA structural parameters (28–30), this substitution can change duplex hydration patterns (29, 31) and quite possibly reduce the ion count in the cloud loosely associated with the DNA (32, 33). Thus, these indirect outcomes of M substitution can make unique mechanistic interpretations of the observed effects elusive. In the present case, the small damaging effects of M substitution on nonspecific DNA binding (0.5 kcal/mol per substitution) could reflect direct disruption of the backbone hydrogen bonding in the complex (Fig. 7), or a reduction in minor groove hydration waters or ions around the neutral patch (29, 31). If these indirect effects prevail, then the reduction in binding affinity upon M substitution could arise from a smaller favorable entropy change resulting from fewer water molecules or ions being released to bulk solution upon complexation (34). Although such indirect effects might provide viable explanations for the reduced binding affinity of hUNG for M substituted DNA, they do not reasonably account for the absence of an effect of M substitution on DNA sliding because sliding occurs in a kinetic event after the ion cloud has been dispersed.
The absence of a functional requirement for a continuous negatively charged backbone in site translocation strongly suggests that the sliding form of hUNG cannot simply involve translocation of the crystallographic conformation of hUNG along DNA (26, 27, 35, 36). Rather, the data would suggest that the sliding conformation of hUNG is an open state that interacts loosely with the DNA backbone, with perhaps intervening water molecules (but not solute ions) that would serve to shield charge. This view of a loose, transiently bound conformation is consistent with CPMG NMR dynamic measurements indicating that UNG oscillates between an open and closed form on the millisecond time scale when bound to nonspecific DNA (37). The open form was proposed to function in stochastic sliding along the DNA chain, and the closed form resembles the crystallographic conformation, allowing hUNG to interrogate the integrity of base pairs. Indeed, a two state conformational change has been postulated as a general mechanism for site specific DNA binding proteins to overcome the “search-speed/stability” paradox (3, 4, 38), and recent structural evidence obtained with other DNA glycosylases suggests evidence for more than one conformation involved in search and recognition by these enzymes (12, 13). The findings reported here provide a first glimpse at the electrostatic properties of this transient state of hUNG.
Boundary Estimates for 1D Translocation on Duplex DNA
Employing the measured values for the average lifetime of hUNG on nonspecific DNA (τbind = 3 ms), and its mean sliding length (Lslide = 4.2 bp), we previously used eq 2 to estimate the 1-dimensional diffusion constant (D1) of hUNG on nonspecific duplex DNA (D1 = 6 × 103 bp2 s−1 = 7 × 10−4 μm2 s−1) (9). This value was several orders of magnitude below the theoretical upper limit (~107 bp2 s−1 or ~1 μm2 s−1) (5, 39, 40).
| (2) |
This calculation assumes that the entire bound lifetime of hUNG is available for DNA sliding. However the current data, which requires the presence of at least two nonspecific states of hUNG, also requires that only a fraction of the bound lifetime is available for sliding (i.e the time spent in the open state). An estimated lower limit for the population of the transient sliding state may be estimated based on the sensitivity of the NMR-relaxation dispersion dynamic measurements previously performed on the hUNG-nonspecific DNA complex (37). This methodology would not be able to detect a transient sliding state with a population of less than ~5% of the total, setting a lower limit for the time spent sliding of 0.05 × 3 ms ≥ 0.15 ms. It is difficult to set an upper boundary, but it must be considerably less than τbind = 3 ms. Using eq 2 and this lower limit for the sliding time, we calculate an upper limit for D1 ≤ 105 bp2 s−1. Thus, the previous and current estimates place D1 in the range ~104 to 105 bp2 s−1. Given this refined two-state view of sliding by hUNG, we suggest that the sliding state resembles the transition state for DNA dissociation. However, instead of falling off the DNA chain the enzyme closes on the DNA and completes a sliding transfer. This viewpoint of short range sliding as an aborted transition state for DNA dissociation differs considerably from other characterizations of protein sliding whereby the protein moves isoenergetically along the surface of the DNA (41–43). These aspects of the hUNG search mechanism are depicted in the model presented in Fig. 8.
Figure 8.

Two-state model for hUNG sliding on nonspecific DNA. In this model hUNG exists in a highly populated “closed” recognition conformation that makes multiple interactions with the phosphate backbone as observed in crystallographic studies, and also a transient mobile “open” sliding conformation that makes little or no interactions with the phosphate backbone (this work). It is reasonable to view the open state as the aborted transition state preceding DNA dissociation. The open state, which must be present at least 5% of the total bound lifetime (see text), allows for fast movement on the DNA while also allowing time for recognition of uracil bases when they are encountered.
Search and Recognition in the Cell Nucleus
Under optimal low salt reaction conditions hUNG is an evolutionarily optimized enzyme with a catalytic power that vastly exceeds any other DNA glycosylase (1). However, under conditions that more closely mimic the intracellular ionic environment, its ability to bind nonspecific DNA is severely hampered by a factor of around 100-fold, which exerts a profound effect on the mechanism of site location. One major ramification of the ionic strength effect on nonspecific DNA binding is that hopping becomes a less productive pathway. Each time hUNG dissociates from the DNA chain under high salt conditions, there will be a reduced probability that a reassociation attempt will result in a productive binding event. Thus many more attempts will have to be made, which will result in an increase in the search time contributed by hopping. In contrast, DNA sliding is largely refractory to increases in ionic strength, and the search time resulting from sliding will remain largely unchanged. This important property of sliding, even over the short ranges traveled by hUNG, is essential for increasing coverage of the genome and for the ultimate detection of damage (Fig. 8). An additional consideration within the nuclear environment is the effect of crowding, as well as excluded volume effects (44, 45). Such factors could favor compact sliding states and also increase the contribution of hopping because of the high local concentration of DNA chains. Consideration of such effects requires improved experimental models for search and recognition.
Supplementary Material
Acknowledgements
We thank Dr. David Draper for valuable discussions on this work. This work was supported by US National Institute of Health grant GM056834.
Abbreviations
- hUNG
catalytic domain of human uracil DNA glycosylase
- ntds
nucleotides
- bps
base pairs
Footnotes
This work was supported by NIH grant GM056834 (J.T.S).
Supporting Information Available. Supplemental methods and three supporting figures: (1) Schematic description of splint ligation strategy used to synthesize 90mer DNA substrates containing methylphosphonate linkages. (2) mFold output showing the lack of secondary structure for the single strand substrate S6Mss. (3) Steady-state kinetic parameters of hUNG and non-specific DNA binding under physiological salt concentrations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Stivers JT, Jiang YL. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 2.Friedman JI, Stivers JT. Detection of Damaged DNA Bases by DNA Glycosylase Enzymes. Biochemistry. 2010;49:4957–4967. doi: 10.1021/bi100593a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirny L, Slutsky M, Wunderlich Z, Tafvizi A, Leith J, Kosmrlj A. How a protein searches for its site on DNA: the mechanism of facilitated diffusion. J. Phys. A: Math. Theor. 2009;42:434013. [Google Scholar]
- 4.Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys J. 2004;87:4021–4035. doi: 10.1529/biophysj.104.050765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berg OG, Winter RB, Hippel, Von PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 6.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15883–15888. doi: 10.1073/pnas.0505378102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hedglin M, O'Brien PJ. Hopping Enables a DNA Repair Glycosylase To Search Both Strands and Bypass a Bound Protein. ACS Chemical Biology. 2010;5:427–436. doi: 10.1021/cb1000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schonhoft JD, Stivers JT. Timing facilitated site transfer of an enzyme on DNA. Nat. Chem. Biol. 2012;8:205–210. doi: 10.1038/nchembio.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blainey PC, van Oijen AM, Banerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qi Y, Nam K, Spong MC, Banerjee A, Sung R-J, Zhang M, Karplus M, Verdine GL. Strandwise translocation of a DNA glycosylase on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012;109:1086–1091. doi: 10.1073/pnas.1111237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Setser JW, Lingaraju GM, Davis CA, Samson LD, Drennan CL. Searching for DNA lesions: structural evidence for lower- and higher-affinity DNA binding conformations of human alkyladenine DNA glycosylase. Biochemistry. 2012;51:382–390. doi: 10.1021/bi201484k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunn AR, Kad NM, Nelson SR, Warshaw DM, Wallace SS. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terakawa T, Kenzaki H, Takada S. p53 Searches on DNA by Rotation-Uncoupled Sliding at C-Terminal Tails and Restricted Hopping of Core Domains. J. Am. Chem. Soc. 2012;134:14555–14562. doi: 10.1021/ja305369u. [DOI] [PubMed] [Google Scholar]
- 16.Leith JS, Tafvizi A, Huang F, Uspal WE, Doyle PS, Fersht AR, Mirny LA, van Oijen AM. Sequence-dependent sliding kinetics of p53. Proc. Natl. Acad. Sci. U.S.A. 2012 doi: 10.1073/pnas.1120452109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.li AG, Yeykal CC, Robertson RB, Greene EC. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1221–1226. doi: 10.1073/pnas.0508366103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Senavirathne G, Jaszczur M, Auerbach PA, Upton TG, Chelico L, Goodman MF, Rueda D. Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J. Biol. Chem. 2012;287:15826–15835. doi: 10.1074/jbc.M112.342790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammar P, Leroy P, Mahmutovic A, Marklund EG, al E. The lac repressor displays facilitated diffusion in living cells. Science. 2012;336:1595–1598. doi: 10.1126/science.1221648. [DOI] [PubMed] [Google Scholar]
- 20.Stanford NP, Szczelkun MD, Marko JF, Halford SE. One- and three-dimensional pathways for proteins to reach specific DNA sites. EMBO J. 2000;19:6546–6557. doi: 10.1093/emboj/19.23.6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Werner RM, Jiang YL, Gordley RG, Jagadeesh GJ, Ladner JE, Xiao G, Tordova M, Gilliland GL, Stivers JT. Stressing-Out DNA? The Contribution of Serine–Phosphodiester Interactions in Catalysis by Uracil DNA Glycosylase. Biochemistry. 2000;39:12585–12594. doi: 10.1021/bi001532v. [DOI] [PubMed] [Google Scholar]
- 22.Jiang YL, Ichikawa Y, Song F, Stivers JT. Powering DNA Repair through Substrate Electrostatic Interactions. Biochemistry. 2003;42:1922–1929. doi: 10.1021/bi027014x. [DOI] [PubMed] [Google Scholar]
- 23.Parker JB, Stivers JT. Uracil DNA glycosylase: revisiting substrate-assisted catalysis by DNA phosphate anions. Biochemistry. 2008;47:8614–8622. doi: 10.1021/bi800854g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terry BJ, Jack WE, Modrich P. Facilitated diffusion during catalysis by EcoRI endonuclease. Nonspecific interactions in EcoRI catalysis. J. Biol. Chem. 1985;260:13130–13137. [PubMed] [Google Scholar]
- 25.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl. Acad. Sci. U.S.A. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 27.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thiviyanathan V, Vyazovkina KV, Gozansky EK, Bichenchova E, Abramova TV, Luxon BA, Lebedev AV, Gorenstein DG. Structure of Hybrid Backbone Methylphosphonate DNA Heteroduplexes: Effect of R and S Stereochemistry †,‡. Biochemistry. 2002;41:827–838. doi: 10.1021/bi011551k. [DOI] [PubMed] [Google Scholar]
- 29.Hamelberg D, Williams LD, Wilson WD. Effect of a neutralized phosphate backbone on the minor groove of B-DNA: molecular dynamics simulation studies. Nucleic Acids Res. 2002;30:3615–3623. doi: 10.1093/nar/gkf472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maher LDWALJ, III, Williams LD, Maher LJ., III Electrostatic mechanisms of DNA deformation. Annu Rev Biophys Biomol Struct. 2013;29:497–521. doi: 10.1146/annurev.biophys.29.1.497. [DOI] [PubMed] [Google Scholar]
- 31.Hausheer FH, Rao BG, Saxe JD, Singh UC. Physicochemical properties of (R)- vs (S)-methylphosphonate substitution on antisense DNA hybridization determined by free energy perturbation and molecular dynamics. J. Am. Chem. Soc. 1992;114:3201–3206. [Google Scholar]
- 32.Bai Y, Chu VB, Lipfert J, Pande VS, Herschlag D, Doniach S. Critical Assessment of Nucleic Acid Electrostatics via Experimental and Computational Investigation of an Unfolded State Ensemble. J. Am. Chem. Soc. 2008;130:12334–12341. doi: 10.1021/ja800854u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das R, Mills T, Kwok L, Maskel G, Millett I, Doniach S, Finkelstein K, Herschlag D, Pollack L. Counterion Distribution around DNA Probed by Solution X-Ray Scattering. Physical Review Letters. 2003;90:188103. doi: 10.1103/PhysRevLett.90.188103. [DOI] [PubMed] [Google Scholar]
- 34.Privalov PL, Dragan AI, Crane-Robinson C. Interpreting protein/DNA interactions: distinguishing specific from non-specific and electrostatic from nonelectrostatic components. Nucleic Acids Res. 2011;39:2483–2491. doi: 10.1093/nar/gkq984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis. Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 36.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman JI, Majumdar A, Stivers JT. Nontarget DNA binding shapes the dynamic landscape for enzymatic recognition of DNA damage. Nucleic Acids Res. 2009;37:3493–3500. doi: 10.1093/nar/gkp161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou H-X. Rapid search for specific sites on DNA through conformational switch of nonspecifically bound proteins. Proc. Natl. Acad. Sci. U.S.A. 2011;108:8651–8656. doi: 10.1073/pnas.1101555108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schurr JM. The one-dimensional diffusion coefficient of proteins absorbed on DNA: Hydrodynamic considerations. Biophysical chemistry. 1979;9:413–414. [PubMed] [Google Scholar]
- 40.Bagchi B, Blainey PC, Xie XS. Diffusion Constant of a Nonspecifically Bound Protein Undergoing Curvilinear Motion along DNA. J. Phys. Chem. B. 2008;112:6282–6284. doi: 10.1021/jp077568f. [DOI] [PubMed] [Google Scholar]
- 41.Berg OG, Hippel, Von PH. Diffusion-controlled macromolecular interactions. Annu Rev Biophys Biophys Chem. 1985;14:131–160. doi: 10.1146/annurev.bb.14.060185.001023. [DOI] [PubMed] [Google Scholar]
- 42.Winter RB, Berg OG, Hippel, Von PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor-operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 43.Winter RB, Hippel, Von PH. How do genome-regulatory proteins locate their DNA target sites? Trends Biochem. Sci. 1982;7:52–55. [Google Scholar]
- 44.Zhou H-X, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein-protein association kinetics. Chem. Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.