

Abstract
The dystonias are comprised of a group of disorders that share common neurological abnormalities of involuntary twisting or repetitive movements and postures. The most common inherited primary dystonia is DYT1 dystonia, which is due to loss of a GAG codon in the TOR1A gene that encodes torsinA. Autopsy studies of brains from patients with DYT1 dystonia have revealed few abnormalities, although recent neuroimaging studies have implied the existence of microstructural defects that might not be detectable with traditional histopathological methods. The current studies took advantage of a knock-in mouse model for DYT1 dystonia to search for subtle anatomical abnormalities in the striatum, a region often implicated in studies of dystonia. Multiple abnormalities were identified using a combination of quantitative stereological measures of immunohistochemical stains for specific neuronal populations, morphometric studies of Golgi-stained neurons, and immuno-electron microscopy of synaptic connectivity. In keeping with other studies, there was no obvious loss of striatal neurons in the DYT1 mutant mice. However, interneurons immunoreactive for choline acetyltransferase or parvalbumin were larger in the mutants than in control mice. In contrast, interneurons immunoreactive for neuronal nitric oxide synthase were smaller in the mutants than in controls. Golgi histochemical studies of medium spiny projection neurons in the mutant mice revealed slightly fewer and thinner dendrites, and a corresponding loss of dendritic spines. Electron microscopic studies showed a reduction in the ratio of axo-spinous to axo-dendritic synaptic inputs from glutamatergic and dopaminergic sources in mutant mice compared with controls. These results suggest specific anatomical substrates for altered signaling in the striatum and potential correlates of the abnormalities implied by human imaging studies of DYT1 dystonia.
Keywords: Mouse mutant, anatomy, Golgi histochemistry, stereology, striatum, electron microscopy
Introduction
The dystonias are a group of disorders that share the common abnormality of involuntary twisting or repetitive movements (Fahn, 1988; Tarsy and Simon, 2006). There are many different clinical manifestations and many different causes. The different types of dystonia traditionally are classified according to three independent axes that involve the region of the body affected, age at onset, and etiology. The etiological axis relies on a fundamental distinction between “primary” and “secondary” dystonias. The primary dystonias are defined as disorders where dystonia is the chief manifestation, while the secondary dystonias more often include other neurological features.
Although many investigators have suggested the brain is structurally normal in primary dystonia (Breakefield et al., 2008; de Carvalho Aguiar and Ozelius, 2002; Nemeth, 2002; Schwarz and Bressman, 2009; Tanabe et al., 2009), neuroimaging studies repeatedly have implied the existence of microstructural defects in the basal ganglia, cerebellum, thalamus, or cerebral cortex (for reviews see: (Neychev et al., 2011; Zoons et al., 2011). However, imaging studies provide only indirect measures of microstructure, and some of the methods used may be detecting functional rather than structural changes. Unfortunately, postmortem studies of brains from patients with dystonia are sparse (Standaert, 2011). The main reason for the paucity of histopathological data is that the dystonias are rare, making it difficult to collect the necessary specimens. A second reason is that the methods needed to reveal potential microstructural defects require special tissue preparation and processing methods that cannot be applied readily in postmortem studies of human brains. The third reason for the limited information is that the types of neuropathological changes suspected for primary dystonias may not involve overt degeneration or emergence of unique pathological hallmarks, but rather quantitative changes in soma or neurite morphology, complexity or connectivity. Revealing these types of changes requires precise quantitative morphometric and ultrastructural comparisons with controls. These three limitations of human autopsy studies have hampered efforts to delineate potential microstructural defects in the human DYT1 brain.
In this situation, animal models can provide a valuable resource to reveal abnormalities that subsequently may be confirmed in more targeted human studies (Jinnah et al., 2005). Several genetically engineered mouse models have been created for DYT1 dystonia, the most common inherited primary dystonia that is caused by loss of a GAG codon (ΔE) in the TOR1A gene that encodes torsinA. Although overt signs of dystonia are lacking in the mouse models, several studies have shown poor motor skills, abnormalities in the physiology and neurochemistry of basal ganglia pathways, and subtle changes in the structure of the membranes surrounding nuclei of neurons of some brain regions (for review see (Song et al., 2012). Nissl stains have revealed no obvious anatomical brain abnormalities, but histological methods capable of identifying microstructural defects in the basal ganglia have not been conducted.
In the current studies, the fine structure of neurons in the striatum and their connectivity was examined in a knock-in mouse model of DYT1 dystonia using a combination of quantitative stereological measures of immunohistochemical stains for specific neuronal populations, morphometric studies of Golgi-stained striatal neurons, and immuno-electron microscopy of synaptic connectivity.
Materials & Methods
Animals
Animals used in these studies were heterozygous DYT1(ΔE) mutant knock-in mice (Goodchild et al., 2005) maintained congenically with C57BL/6J mice from the Jackson Laboratories (Bar Harbor ME). Animals were housed on a 12 hr light/dark cycle, with ad libitum access to food and water. Animals were genotyped by DNA obtained from tail clips using a primer pair for the 34 base pair loxP site in the DYT1 mutant (forward primer, AGTCTGTGGCTGGCTCTCCC; reverse primer, CCTCAGGCTGCTCACAACCAC). The studies were approved by the Institutional Animal Care and Use Committee at Emory University.
Immunohistochemistry
Because anatomical abnormalities may vary according to sex or age, a multivariate statistical strategy was used that enabled simultaneous exploration of these variables in mutant and normal mice. Specifically, immunohistochemical stains were conducted in parallel on a cohort of 24 animals that included 12 mutants and 12 littermate controls. Half of the mutants were male and half female. Half were 3 months of age and half were 6 months of age. Mice were anesthetized with 2,2,2-tribromoethanol and perfused through the heart with a rinse solution of 50 mL of 137 mM NaCl, 22.2 mM dextrose, 23.4 mM sucrose, 2 mM CaCl2 and 1.6 mM sodium cacodylate at pH 7.2. They then were perfused with 4% paraformaldehyde containing 117 mM sucrose and 67 mM sodium cacodylate (pH 7.2) and immersion post-fixed for 16 hrs.
To ensure uniformity of tissue sections, all 24 brains were embedded in a block of gelatin and cut simultaneously in the coronal plane at 40 μm from the frontal pole through the caudal brainstem and cerebellum (Neuroscience Associates, Knoxville TN). To ensure uniformity of staining, each stain was conducted in parallel with an entire series of sections spaced at 240 μm intervals. One series of sections was thionin stained to demonstrate Nissl substance, and the remaining 5 series were used for immunohistochemical stains of specific neuronal populations. Immunostains targeted choline acetyltransferase (ChAT; Milipore, Billerica MA), calbindin (Swant, Bellinzona, Switzerland), parvalbumin (PV; Swant, Bellinzona, Switzerland), neuronal nitric oxide synthase (nNOS; Immunostar, Hudson WI), or tyrosine hydroxylase (TH; Pelfreez, Rogers AR).
Stereological quantifications
For stereological analyses, sections were examined under an Olympus BX51 light microscope (Melville NY), with a motorized stage (MAC5000, Ludl Electronic Products, Hawthorne NY) controlled by a computer with StereoInvestigator software (MicroBrightField, Williston VT). In the rodent basal ganglia, the caudate nucleus and putamen are combined as the caudoputamen, here referred to as the dorsal striatum (excluding the accumbens and olfactory tubercle). To determine the volume of the dorsal striatum using the Cavalieri method (Oorschot, 1996), the entire region was outlined at 4X objective in every 6th Nissl-stained section extending approximately from +1.7 to −2.2 mm from bregma in the anterior-posterior (AP) direction according to the mouse brain atlas (Paxinos and Franklin, 2004).
The total numbers of neurons were counted at 100X in Nissl-stained material or at 60X in immunostained material using the optical fractionator with a 12 μm depth and 1 μm top guard zone, as previously described (Egami et al., 2007). Because the numbers of target neurons for counting varied according to the stain applied, stereological counting frames and grid sizes were adjusted to achieve a Gunderson coefficient of error below 0.10 for each stain. For Nissl-stained neurons, the counting frame was 20 × 20 μm in a sampling grid of 600 × 600 μm. For ChAT and PV+ cells, the frame was 100 × 100 μm in a grid of 250 × 250 μm. For nNOS+ cells, the frame was 120 × 120 μm in a grid of 330 × 330 μm. Cell volumes were determined using the optical rotator with a 3 μm focal plane separation and a 4 grid line separation of 4 μm for ChAT+ cells, or with a 2 μm focal plane separation and a 3 grid line separation of 4 μm for PV+ and nNOS+ cells.
The statistical approach for stereological measures involved Analysis of Variance (ANOVA) with genotype, sex, and age as potential explanatory variables. Since identifying abnormalities related to the mutant Tor1A gene was the primary focus, results were combined across ages and sex for final graphic summary of the data. For all analyses, statistical significance was defined as p<0.05. Because this study was exploratory and effect sizes sometimes were small, any result with 0.05<p<0.10 was noted as being a trend of borderline statistical significance, to acknowledge places where Type II errors could occur.
Spatial distribution of immunostained neurons
To explore potential differences in the spatial distributions in the numbers or sizes of immunostained neurons, stained neurons also were examined according to rostro-caudal, dorso-ventral, and medio-lateral orientations. Because stereological methods are not suitable for delineating 3-dimensional distributions, differences in the rostro-caudal orientation were explored by subdividing results from total stereological counts into 4 rostro-caudal subgroups to provide sufficient power for statistical analyses. For these analyses, stereological counts were grouped from the first 3 sections for AP +1.5~+0.9 mm, second 3 sections for AP +0.7~+0.1 mm, third 3 sections for AP −0.1~−0.7 mm and remaining 6 sections for AP −0.9~−2.0 mm. For dorso-ventral and medio-lateral gradients, total cell counts were taken at the level of the anterior commissure (AP +0.14 mm) and subdivided into four quadrants as previously described (Luk and Sadikot, 2001). For this analysis, all available neurons were re-counted, since stereological sampling yielded insufficient data for statistical analyses. Data regarding neuronal distributions were examined by ANOVA with genotype and subregion as explanatory variables.
Golgi histochemistry
Golgi stains were conducted simultaneously in 6 controls and 6 mutants of both sexes at 3 months of age using the FD Rapid Golgi Stain Kit (FD NeuroTechnologies, Baltimore MD). Briefly, mice were decapitated and the brain was removed immediately. The unfixed brain was immersed in solution A containing potassium dichromate and mercuric chloride and solution B containing potassium chromate for 2 weeks in the dark at room temperature. The brains then were placed in sucrose for 48 hrs at 4°C, and sectioned frozen in the coronal plane at 100 μm using a Microm HM440E sliding microtome (Microm, Waldorf, Germany). Cut sections were stained with a mixture consisting of solutions D and E in distilled water for 15 minutes, and rinsed with distilled water 3 times. Sections were then mounted on glass slides, dehydrated through graded alcohols, and cover-slipped with Permount.
Two medium spiny neurons were chosen from each hemisphere of each mouse (total of 4 neurons per mouse, or 24 neurons per genotype) by a microscopist blinded to genotype at the level of anterior commissure. The selection of neurons was based on previous guidelines for analyzing Golgi-stained material, with the soma in the middle third of the section and complete staining of all dendrites (Mikolaenko et al., 2005). Each neuron was traced with NeuroLucida software (MicroBrightField, Williston VT) and a Wacom Intuos 2 digitizing tablet in conjunction with an Olympus BX51 microscope and a 60X objective. Soma sizes and perimeters were determined by NeuroExplorer software version 10.21 as previously described (Mikolaenko et al., 2005). Spines were defined as any discrete visible protrusion emanating from a dendrite. Because spine counts depend on the total numbers and lengths of dendrites measured, spine density also was calculated as the number of spines per μm of total dendritic lengths.
The approach for statistical examination of Golgi-stained neurons involved two complementary methods to digitally reconstructed neurons available within NeuroExplorer as previously described (Mikolaenko et al., 2005). The first method involved using branch order, where dendrites arising directly from the soma are defined as the first branch order (Figure 1A). Each point where the dendrite branches marks a successively higher branch order. Target variables of interest (dendrite numbers, dendrite thicknesses, dendrite lengths, and spine numbers) are expressed according to branch order. The second method involved using a modified Sholl approach (Sholl, 1953). Here, concentric circles are drawn with an incremental radius of 10 μm around the geometric center of the soma (Figure 1B). Each variable is expressed according to radius level, rather than branch order. Results were examined by ANOVA with genotype and sex as explanatory variables. Branch order or radius from soma center was treated as repeated measures.
Figure 1.

Schematic for analysis of Golgi-stained materials. Dendritic analyses based on branching order (A) are based on consecutive numbering of dendrites determined by each branch point, beginning with the first dendrite coming directly from the soma. Results are presented at each branch level for total dendrite numbers, total dendrite lengths, and total spines. Dendritic analyses based on the Sholl method (B) are based on drawing virtual concentric rings around the geometric center of the soma. Results are presented at each radius (r) for total dendrite numbers, total dendrite lengths, and total spines.
Electron microscopy
Three littermate control mice and four mutants of both sexes at 3 months of age were used for quantitative electron microscopic (EM) studies of the striatal neuropil. Mice were anesthetized with 2,2,2-tribromoethanol and perfused with paraformaldehyde (4%) and glutaraldehyde (0.1%) in 0.1 M phosphate buffer. Sections were cut at 60 μm with a vibrating microtome, and processed to localize TH (Millipore, Temecula CA) and vesicular glutamate transporters 1 and 2 (vGluT1 and vGluT2, Mab Technologies, Atlanta GA) in the striatum using specific antibodies and the immunoperoxidase avidin-biotin complex method (Raju et al., 2008; Villalba and Smith, 2011). Next, the tissue was dehydrated, post-fixed in 1% osmium and embedded in resin on microscope slides. After resin polymerization, 1–2 blocks of striatal tissue from each animal were cut in ultrathin sections and collected on single slot copper grids. After lead citrate staining, sections from the surface of the blocks were scanned at 30,000X with a JEOL electron microscope (Jeol USA, Peabody MA).
Immunoreactive terminals forming synaptic contacts were randomly collected from the blocks of striatal tissue immunostained for TH, vGluT1 or vGluT2. The postsynaptic targets in contact with each labeled terminal were identified based on ultrastructural features (Peters et al., 1991). The percent of labeled boutons in contact with specific postsynaptic targets was then calculated for each terminal population, and statistically compared between normal and DYT1 mice via independent t-test.
Results
Nissl stains
No obvious structural abnormalities were evident in Nissl stains of the striatum in the DYT1 mutant mice (Figure 2A–D). The total volumes of striatal parenchyma and total numbers of striatal neurons were measured stereologically, and the data were analyzed by ANOVA with genotype, sex and age as explanatory variables. For striatal volumes, there were no main effects for genotype (F=0.9; p>0.10), sex (F=0.03; p>0.10), or age (F=1.2; p>0.10). There were no interactions between genotype and sex (F=0.9; p>0.10), or genotype and age (F=1.7; p>0.10). Combining results across sex and age, the total striatal volumes were 9.17 ± 0.08 mm3 in controls and 9.29 ± 0.10 mm3 in mutants.
Figure 2.

Nissl stains of striatum. There were no obvious abnormalities in structure of the striatum between control (A) and DYT1 mutant mice (B). Similarly, there were no obvious abnormalities in the morphology of neurons between control (C) and DYT1 mutant mice (D).
For the number of total Nissl-stained neurons, there again were no main effects for genotype (F=2.0; p>0.10), sex (F=3.7; p=0.07), or age (F=1.3; p>0.10). There were no interactions between genotype and sex (F=0.3; p>0.10), or genotype and age (F=0.5; p>0.10). Combining results across sex and age, there were 1,261,309 ± 28,219 total striatal neurons in controls and 1,221,049 ± 18,877 in mutants. The lack of any obvious changes in Nissl-stained sections of the striatum is consistent with prior studies of these DYT1 mutant mice (Song et al., 2012).
Striatal total neurons consist of 90–95% of medium spiny projection neurons and a small population of interneurons that include large aspiny cholinergic neurons, and smaller GABAergic neurons that express PV, nNOS or calretinin (Kawaguchi et al., 1995; Tepper and Bolam, 2004). Because it is not possible to discriminate among these neuronal subtypes in Nissl stains, immunostains were used to explore potential changes among the sub-populations.
Cholinergic interneurons
The large aspiny cholinergic interneurons identifiable with immunostains for ChAT were examined, because of prior evidence linking human dystonia with dysfunction of striatal cholinergic pathways (Pisani et al., 2007), and animal studies showing abnormal electrophysiological properties of these neurons (Martella et al., 2009; Pisani et al., 2006; Sciamanna et al., 2012b; Sciamanna et al., 2011).
The ChAT+ neurons exhibited a characteristic morphology with a few aspiny dendrites emanating from a soma that was rounded, oval or fusiform (Figure 3A–D). For total numbers of ChAT+ neurons, ANOVA revealed no main effects for genotype (F=0.1; p>0.10). However, there were significant effects for sex with 9% more neurons in females compared to males (F=4.7; p<0.05). The effect of age showed a borderline trend, with 9% more neurons in 6 month old animals compared to those aged 3 months (F=4.4; p=0.05). There were no interactions between genotype and sex (F=0.07; p>0.10), or genotype and age (F=1.5; p>0.10). Combining results across sex and age, control mice had a total of 5,081 ± 159 ChAT+ neurons while mutants had 5,200 ± 214 (Table 1).
Figure 3.

Striatal interneurons expressing choline acetyltransferase (ChAT). There were only subtle differences in the distribution of ChAT+ neurons between normal controls (A) and DYT1 mutant mice (B). There were no obvious differences in the morphology of these neurons between control (C) and mutant mice (D), although those of mutants were slightly larger. Stereological measures for the cell size (E) and regional distribution (F–G) represent average values ± SEM in controls (black bar) and mutants (white bar). A histogram showing the size distributions in bins of 250 μm3 as percent of total cells suggests a shift of the entire mutant cell population to larger sizes compared to controls, rather than a small subpopulation of very large cells. For the dorso-ventral and medio-lateral distributions, all neurons were counted at the level of the anterior commissure split into 4 quadrants: dorso-lateral (DL), dorso-medial (DM), ventro-lateral (VL) and ventro-medial (VM). ChAT+ neuron densities were 22% higher in the DM of mutants compared to normal mice (F). For rostro-caudal distributions (RC), stereological counts were analyzed according to anterior-posterior levels from bregma at +1.5~+0.9 mm (RC1), +0.7~+0.1 mm (RC2), −0.1~−0.7 mm (RC3) and −0.9~−2.0 mm (RC4). The distribution of ChAT+ neurons was normal in mutants (G). Asterisks denote statistical significance at p<0.05.
Table 1.
Numbers and sizes of striatal interneurons.
| Control | Mutant | % control | |
|---|---|---|---|
| ChAT+ cells | |||
| number | 5081 ± 159 | 5200 ± 214 | 102 |
| size (μm3) | 1695 ± 39 | 1977 ± 48 | 117** |
| PV+ cells | |||
| number | 5609 ± 157 | 6013 ± 191 | 107 |
| size (μm3) | 496 ± 9 | 552 ± 13 | 111** |
| nNOS+ cells | |||
| number | 7384 ± 363 | 6785 ± 237 | 92 |
| size (μm3) | 649 ± 17 | 589 ± 17 | 91* |
Data are average numbers or volumes ± SEM of ChAT+, PV+ and nNOS+ cells in the caudoputamen of 12 mutant and 12 control mice with equal numbers of mice from both sexes and two age groups (3 and 6 months of age). Abbreviations; ChAT (cholinergic acetyltransferase), PV (parvalbumin) and nNOS (neuronal nitric oxide synthase). Single asterisks indicate statistical significance at p<0.05, while double asterisks indicate p<0.01.
Although there was no obvious change in ChAT+ neuron numbers, stereological measurements of cell sizes suggested that the somata of these neurons were ~17% larger in the mutant mice (Table 1). ANOVA revealed a significant main effect of cell size for genotype (F=25.6; p<0.01), but no significant effect for sex (F=1.0; p>0.10), while the effect of age showed only a borderline trend (F=3.4; p=0.09). There were no interactions between genotype and sex (F=0.5; p>0.10) or genotype and age (F=0.3; p>0.10). A frequency histogram of cell volumes suggested enlargement of the entire population of ChAT+ cells rather than a smaller subpopulation of particularly large neurons (Figure 3E).
As previously described (Graybiel et al., 1986; Kreitzer, 2009), the ChAT+ interneurons were sparsely distributed throughout the striatum, with a subtle density gradient that increased in the ventral to dorsal, medial to lateral, and caudal to rostral directions. There was a subjective impression that ChAT+ neurons seemed to be anatomically redistributed in the DYT1 mutants, with more neurons in the dorso-lateral regions (Figure 3A–B). To address this possibility, striatal neurons were subgrouped according to quadrants at the level of the anterior commissure, and evaluated by ANOVA with genotype and quadrant as explanatory variables. This analysis showed significant main effects for genotype (F=4.7; p<0.05) and quadrant (F=63.2; p<0.01), as well as a significant interaction between genotype and quadrant (F=3.0; p<0.05). Post-hoc analyses revealed a 22% higher density of ChAT+ neurons in the dorso-lateral striatum of mutants compared to normal mice (p<0.01) (Figure 3F). In the rostro-caudal dimension, ChAT+ neurons appeared distributed similarly in mutants and controls (Figure 3G). Although the effect of rostro-caudal distribution was statistically significant by ANOVA (F=13.1; p<0.01), there were no main effects for genotype (F=0.1; p>0.10), and there was no interaction between genotype and rostro-caudal level (F=0.2; p>0.10). These regional analyses suggest variations in ChAT+ neuron densities of both normal and mutant mice, with slightly higher densities in the dorso-lateral regions of the DYT1 mutant mice.
PV+ interneurons
In the striatum, PV+ neurons represent the so-called “fast spiking” subtype of striatal GABAergic interneurons (Kawaguchi et al., 1995). They were examined because of evidence that striatal injections of GABAergic drugs can induce dystonia in animals (Marrone et al., 2006; Yamada et al., 1995; Yoshida et al., 1991), and because of prior reports showing that these neurons are abnormal in a hamster model of paroxysmal dystonia (Gernert et al., 2000). PV+ neurons had characteristic rounded or oval soma, with a few proximal dendrites without spines in both normal and DYT1 mutant mice (Figure 4A–D). For total numbers of striatal PV+ neurons, ANOVA revealed a borderline statistical trend for genotype (F=3.3; p=0.09) with a 7% increase in mutants. There were no effects for sex (F=0.3; p>0.10) or age (F=2.3; p>0.10), and no interactions between genotype and sex (F=0.2; p>0.10) or genotype and age (F=1.5; p>0.10).
Figure 4.

Striatal interneurons expressing parvalbumin (PV). There were no obvious differences in the distribution of PV+ neurons between normal controls (A) and DYT1 mutant mice (B). There were no obvious differences in the morphology of these neurons between control (C) and mutant mice (D), although those of mutants were slightly larger. Stereological measures for the cell size (E) and regional distribution (F–G) represent average values ± SEM in controls (black bar) and mutants (white bar). A histogram showing the size distributions in bins of 100 μm3 as percent of total cells suggests a shift of the entire mutant cell population to larger sizes compared to controls, rather than a small subpopulation of very large cells. For the dorso-ventral and medio-lateral distributions, all neurons were counted at the level of the anterior commissure split into 4 quadrants: dorso-lateral (DL), dorso-medial (DM), ventro-lateral (VL) and ventro-medial (VM). For rostro-caudal distributions (RC), stereological counts were analyzed according to anterior-posterior levels from bregma at +1.5~+0.9 mm (RC1), +0.7~+0.1 mm (RC2), −0.1~−0.7 mm (RC3) and −0.9~−2.0 mm (RC4). The regional densities of PV+ neurons appeared normal in mutants (F–G).
For PV+ neuron sizes, stereological measures revealed these neurons to be ~11% larger in the mutants (Table 1), with ANOVA confirming a significant main effect for genotype (F=12.8; p<0.01). There were no significant effects for sex (F=0.8; p>0.10) or age (F=1.3; p>1.0), and no interactions between genotype and sex (F=0.08; p>0.10) or genotype and age (F=0.7; p>0.10). A frequency histogram of cell volumes again suggested enlargement of the entire population of PV+ cells rather than an abnormally large subpopulation of cells (Figure 4E).
The PV+ cells were sparsely distributed throughout the striatum in both normal and mutant mice, with density gradients that were enriched in the dorso-lateral and caudal striatum (Figure 4A–B). ANOVA revealed significant differences according to quadrant at the anterior commissure (F=173.1; p<0.01). However, there were no differences between normal and mutant mice (F=1.3; p>0.10), and there was no interaction between genotype and the region (F=1.7; p>0.10) (Figure 4F). ANOVA also revealed a significant main effect for rostro-caudal level (F=23.5; p<0.01), but not for genotype (F=1.3; p>0.10) or interaction between genotype and rostro-caudal level (F=0.5; p>0.10) (Figure 4G). These results suggest that PV+ neurons are distributed similarly in normal and mutant mice.
nNOS+ interneurons
The nNOS+ neurons correspond to the “low threshold spike” subtype of striatal GABAergic interneurons (Kawaguchi et al., 1995), and are another subtype of GABAergic interneuron that have been reported to be abnormal in a hamster model of paroxysmal dystonia (Sander et al., 2006). In both normal and DYT1 mutant animals, nNOS+ neurons had a characteristic fusiform shape, with 2–3 proximal aspiny dendrites (Figure 5A–D). For total numbers of nNOS+ striatal neurons, stereological measures revealed a statistically borderline trend of 9% reduction in the mutant mice (F=4.3; p=0.06). The effect of sex was significant (F=13.9; p<0.01), with 16% fewer neurons in females compared to males. The effect of age was not significant (F=2.3; p>0.10). There were no interactions between genotype and sex (F=2.7; p>0.10) or genotype and age (F=2.0; p>0.10).
Figure 5.

Striatal interneurons expressing neuronal nitric oxide synthase (nNOS). There were no obvious differences in the distribution of nNOS+ neurons between normal controls (A) and DYT1 mutant mice (B). There were no obvious differences in the morphology of these neurons between control (C) and mutant mice (D), although those of mutants were slightly smaller. Stereological measures for the cell size (E) and regional distribution (F–G) represent average values ± SEM in controls (black bar) and mutants (white bar). A histogram showing the size distributions in bins of 100 μm3 as percent of total cells suggests a shift of the entire mutant cell population to smaller sizes compared to controls, rather than a small subpopulation of very smaller cells. For the dorso-ventral and medio-lateral distributions, all neurons were counted at the level of the anterior commissure split into 4 quadrants: dorso-lateral (DL), dorso-medial (DM), ventro-lateral (VL) and ventro-medial (VM). For rostro-caudal distributions (RC), stereological counts were analyzed according to anterior-posterior levels from bregma at +1.5~+0.9 mm (RC1), +0.7~+0.1 mm (RC2), −0.1~−0.7 mm (RC3) and −0.9~−2.0 mm (RC4). The regional densities of nNOS+ neurons appeared normal in mutants (F–G).
Unlike ChAT+ and PV+ neurons that were larger in the mutant mice, the somata of nNOS+ neurons were smaller in the mutants. Stereological measures revealed a 9% reduction in cell body volume (Table 1), an effect that was statistically significant by ANOVA (F=7.1; p<0.05). The effect of sex showed a borderline trend (F=3.9; p=0.07), while the effect of age was not significant (F=0.8; p>0.10). There were no interactions between genotype and sex (F=0.02; p>0.10) or genotype and age (F=0.7; p>0.10). A frequency histogram showed the whole cell population to be shifted towards smaller cells in the mutants, rather than a sub-population of particularly small cells (Figure 5E).
The nNOS+ neurons were sparsely distributed throughout the striatum in both normal and mutant animals, with higher densities in the medial and ventral striatum (Figure 5A–B). ANOVA revealed significant effects for quadrant at the level of the anterior commissure (F=29.5; p<0.01), but not for genotype (F=0.1; p>0.10) or interaction between genotype and the region (F=1.9; p>0.10) (Figure 5F). When density was examined in the rostro-caudal dimension (Figure 5G), ANOVA showed significant effects of genotype (F=5.2; p<0.05) and rostro-caudal level (F=4.0; p<0.05), but no interaction between genotype and rostro-caudal level (F=0.6; p>0.10). These results suggest that nNOS+ neurons are distributed normally in mutant mice compared to controls.
Medium spiny neurons
Medium spiny neurons are the major output neurons of the striatum, comprising 90–95% of all striatal neurons. The fine structure of these neurons was evaluated via quantitative analysis of Golgi-stained sections rather than immunohistochemistry for two reasons. First, these neurons can be unequivocally identified without immunostains because of their highly characteristic morphology (McNeill et al., 1990; Mikolaenko et al., 2005). Second, Golgi histochemistry provides an opportunity for more detailed assessment of microstructural changes than immunostains (Figure 6). A total of 24 control and 24 mutant digitally reconstructed neurons were examined from the brains of 6 control and 6 mutant mice of both sexes at 3 months of age.
Figure 6.

Striatal medium spiny neurons. Panel (A) shows a representative Golgi-stained medium spiny neuron. Spines appeared longer and more complex in controls (B) compared to mutants (C). Representative digital reconstructions of typical medium spiny neurons are shown for a control (D) and mutant mice (E).
Morphometric analyses of Golgi-stained neurons revealed the soma sizes of medium spiny neurons to be normal in the mutant animals. ANOVA focusing on soma cross-sectional areas with genotype and sex as explanatory variables revealed no significant effects of genotype (F=0.2; p>0.10) or sex (F=3.1; p>0.10), and no interaction between genotype and sex (F=0.1; p>0.10). Average soma cross-sectional area was 146.5 ± 10.8 μm2 for controls and 144.4 ± 5.6 μm2 for mutants. Similar analyses focusing on soma perimeter provided results consistent with soma cross-sectional area. Average soma perimeters were 46.8 ± 1.8 μm for controls and 46.2 ± 1.1 μm for mutants.
Although soma sizes were normal, dendrites appeared thinner with fewer branches (Figure 6D–E). Dendrite thickness at each branch order was examined by ANOVA with genotype and sex as explanatory variables, and branch order as a repeated measure. There were significant effects for both genotype (F=5.8; p<0.05) and branch order (F=131.5; p<0.01), but no effect for sex (F=0.3; p>0.10). The interaction between branch order and genotype was not significant (F=1.3; p>0.10). These results imply thinner dendrites in mutant animals at all branch levels (Figure 7A).
Figure 7.
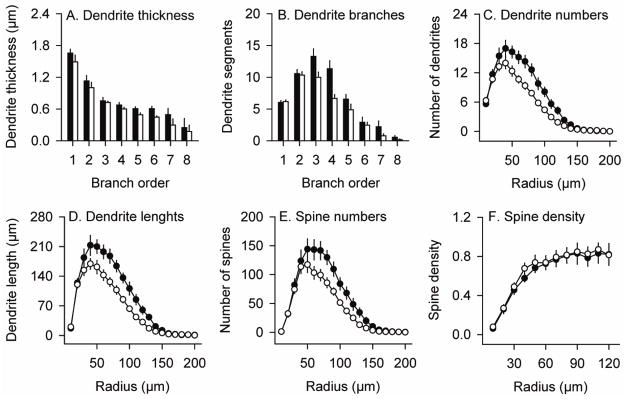
Quantitative morphologic features of striatal medium spiny neurons. Results depict average values ± SEM for a total of 24 control neurons and 24 mutant neurons from the brains of 6 control mice and 6 DYT1 mutants. Dendrite thickness (A) and number of dendrite segments (B) at each branch order were higher in normal (black bar) than mutant (white bar) neurons at some levels. A modified Sholl analysis was used to quantify numbers of dendrites (C), dendrite lengths (D), spine numbers (E) and spine densities (F) according to incremental radii of 10 μm from the soma center in controls (filled circles) and mutants (open circles). The results from branch order and Sholl analyses were concordant, suggesting fewer dendrites with less total dendritic length at certain levels. The reduction in total spines in mutants (E) may reflect dendrite loss, since spine density (spines/μm dendrite) was normal (F).
The number of dendritic segments also was evaluated according to branch order. ANOVA revealed a borderline main effect for genotype (F=4.7; p=0.06), and no effect of sex (F=0.1; p>0.10). There was also a significant effect for branch order (F=157.7; p<0.01) and a significant interaction between genotype and branch order (F=5.1; p<0.01). Post-hoc t-tests revealed reduced numbers of dendrite segments at branch orders 3 and 4 in the DYT1 mutants (p<0.05) (Figure 7B).
A second method that involved a modified Sholl analysis also was used to quantify numbers of dendrites, total dendritic lengths, and spine densities. In this analysis, each of these parameters was quantified according to a specific radius from the soma center rather than branching order. For dendrite numbers, there were significant main effects for both genotype (F=7.7; p<0.05) and radius from cell body (F=190.0; p<0.01), as well as an interaction between the genotype and radius (F=5.3; p<0.05). Post-hoc t-tests revealed reduced numbers of dendrites in mutant animals at distances of 50–120 μm from the soma center (p<0.05, Figure 7C). For total dendrite lengths, there were significant main effects again for genotype (F=8.6; p<0.05) and radius (F=178.3; p<0.01), as well as a significant interaction between the genotype and radius (F=4.7; p<0.05). Post-hoc tests revealed reduced dendritic lengths in the DYT1 mutants with a 50–130 μm radius from the soma center (p<0.05, Figure 7D). However, ANOVA addressing the percentage of neurons with at least one dendrite reaching each radius revealed no significant effect for genotype (F=1.3; p>0.10; data not shown), suggesting that dendrites were fewer, but not generally shorter.
Dendritic spines were readily visualized in both normal and DYT1 mutant mice (Figure 6B–C). In vivo, spines have dynamically changing morphologies that may appear short and stubby, long and thin, or with distal swellings or bifurcations (Bonhoeffer and Yuste, 2002; Fiala et al., 2002; Harris and Kater, 1994). In the DYT1 mutants, spines appeared relatively shorter and less complex in the DYT1 mutants (Figure 6B, C). For spine numbers, there were significant main effects for genotype (F=6.0; p<0.05) and radius (F=123.2; p<0.01), with a borderline trend for significant interaction between the radius and genotype (F=3.4; p=0.06). Post-hoc tests revealed reduced spine numbers in the DYT1 mutants at distances of 70–130 μm from soma center (Figure 7E). The reduction in spine numbers paralleled the reduction in dendrite segments and lengths, and calculating spine density (spines/dendrite length) on remaining dendrites revealed no significant differences between normal and DYT1 mutants (Figure 7F).
Electron microscopy
The multiple subtle microstructural changes at the light microscopic level suggest the possibility of associated abnormalities in synaptic connectivity. Specifically, the reduction in the number of dendrites and spines seen in Golgi-stained sections may be associated with changes in afferent connections from other regions. In order to further address this issue, immuno-electron microscopy was used to characterize the synaptic connections of dopaminergic and glutamatergic terminals in normal and DYT1 mice. Medium spiny neurons receive glutamatergic inputs from the cerebral cortex or thalamus predominantly on spines (Kemp and Powell, 1971; Raju et al., 2008), and dopaminergic inputs from the substantia nigra on dendrites and spine necks (Bolam and Smith, 1990; Smith et al., 1994). Corticostriatal and thalamostriatal synapses onto medium spiny neurons were identified with antibodies to the vesicular glutamate transporters, vGluT1 and vGluT2, respectively (Raju et al., 2008), while nigrostriatal dopaminergic synapses were identified with an antibody to TH (Figure 8A, C, E).
Figure 8.

Electron microscopy for synapses of medium spiny neurons with glutamatergic or dopaminergic inputs. Cortical glutamatergic synapses were labeled by immunostains for vGluT1 (A) while thalamic glutamatergic synapses were labeled by immunostains vGluT2 (C). Dopaminergic synapses were observed by immunostains for TH (E). The electron micrographs depict examples of axo-spinous (Sp) and axo-dendritic (Den) synapses of medium spiny neurons with the glutamatergic and dopaminergic terminals (arrows). Terminals labeled by vGluT1 and vGluT2 immunostains formed asymmetric synapses with unlabeled spines or dendrites, while terminals labeled by TH formed dual symmetric synapses with a small spine and a dendrite. Corresponding quantifications for axo-dendritic (black bars) and axo-spinous synapses (white bars) as percent of total synapses are shown in panels B, D, and F. The numbers above each bar indicate the total numbers of synapses evaluated. All data represent average values ± SEM, and asterisks denote statistical significance at p<0.05.
Compared to control mice, the DYT1 mutant mice had significantly more axo-dendritic synapses for vGluT1-positive and TH-positive terminals, and significantly fewer axo-spinous synapses (p<0.05; Figure 8B, D, F). Additionally, the pattern of synaptic connectivity of each terminal subtype showed decreases in the ratio of axo-spinous over axo-dendritic synapses in mutants compared with controls (TH: F=18.1; p<0.05; vGluT1: F=12.2; p<0.05; vGluT2: F=13.8; p<0.05). The ratios of axo-spinous to axo-dendritic synapses were 23.0 ± 0.2 for mutants vs. 10.9 ± 0.2 for controls for vGluT1, 6.1 ± 1.1 vs. 3.1 ± 0.6 for vGluT2, and 1.6 ± 0.2 vs. 0.7 ± 0.2 for TH. The changes in ratios of these types of synapses labeled for TH were notably for inversion of the usual predominance of axo-spinous synapses over axo-dendritic synapses.
Discussion
These studies reveal multiple microstructural alterations in the dorsal striatum of DYT1 mutant mice. Although there was no obvious neuronal loss in the mutant mice, there were alterations in neuronal sizes, including a 17% increase in the size of ChAT+ interneurons, an 11% increase in the size of PV+ interneurons, and a 9% decrease in the size of nNOS+ interneurons. There also appeared to be a spatial redistribution of ChAT+ neurons, with slightly higher densities in the dosolateral regions. In addition, medium spiny projection neurons had fewer and thinner dendrites in the DYT1 mutant mice, with associated reduction in spines. The mutant mice also had a reduction in the ratio of axo-spinous to axo-dendritic synaptic inputs from both glutamatergic and dopaminergic sources. These findings suggest specific anatomical substrates for altered signaling in the basal ganglia and potential correlates of the abnormalities implied by human imaging studies in patients with DYT1 dystonia.
Comparison with studies of other DYT1 mouse models
We chose to focus on the striatum of a knock-in mouse model of DYT1 dystonia, because of converging evidence from previous studies that the basal ganglia are involved in different types of dystonia (Wichmann, 2008; Wichmann and Dostrovsky, 2011). Although our study is the most comprehensive evaluation of microstructural abnormalities in this region, several prior studies have revealed subtle anatomical defects in other brain regions of DYT1 mutant mice. For example, some studies have shown DYT1 mouse models to have ubiquitin-positive peri-nuclear inclusions in the pedunculopontine nucleus, pons, and peri-aqueductal gray (Dang et al., 2005; Grundmann et al., 2007; Shashidharan et al., 2005). These inclusions are not seen in all DYT1 mouse models (Sharma et al., 2005; Zhao et al., 2008), and their relevance to dystonia is unclear because they occur in regions not typically considered in the pathogenesis of dystonia. However, these inclusions reveal the existence of another type of microstructural abnormality in the DYT1 brain.
Several other studies also have pointed towards abnormal electrophysiological signaling in the basal ganglia of DYT1 mouse models. Our studies showing enlarged ChAT+ neurons are consistent with electrophysiological data that have implied excessive cholinergic tone in these mutants (Martella et al., 2009; Pisani et al., 2007; Pisani et al., 2006; Sciamanna et al., 2012b; Sciamanna et al., 2011). In contrast to our results, a prior study of a mouse model with selective knock-out of the Tor1A gene in cholinergic neurons showed no structural abnormalities among striatal cholinergic neurons (Sciamanna et al., 2012a). Possible reasons for the differences include the use of a different mouse model, and limited statistical power to detect small differences because of the very small number of animals evaluated (only 3 mutants and 3 controls total). In any event, it is possible that the structural and functional studies observed in the mouse model are relevant to human studies indicating that anti-cholinergic medications have partial therapeutic effects (Pisani et al., 2007).
Another significant structural change seen in the striatum of the DYT1 mutant mice was a decreased number of dendrites and spines on medium spiny striatal projection neurons, accompanied by a decreased ratio of axo-spinous to axo-dendritic synapses from terminals immunoreactive for vGluT1, vGluT2 and TH. The Golgi stains suggested spines were smaller and less complex in the mutant mice, although three-dimensional reconstructions from serialized EM sections would be needed to verify this observation. Although the functional significance of changes in spine morphology and synaptic connectivity remains unknown, these data suggest alterations in the transmission, integration and processing of cortical, thalamic and nigral inputs to the striatum of DYT1 mice. This suggestion is supported by several in vitro slice electrophysiology studies in other DYT1 mice models of dystonia (Dang et al., 2012; Martella et al., 2009; Sciamanna et al., 2011).
We previously conducted a stereological study of DYT1 knock-in mice showing that substantia nigra neurons immunopositive for TH are slightly reduced in number and have a larger soma (Song et al., 2012). Finally, there is one prior study showing abnormal dendrites among cerebellar Purkinje neurons in Golgi stains for a different line of DYT1 knock-in mice (Zhang et al., 2011). This study focused only on proximal dendrites of Purkinje neurons, so it remains possible that additional microstructural defects may exist in cerebellar volumes or laminar structure, cerebellar Purkinje neuron numbers or sizes, or other neuronal populations. Indeed, a recent imaging study of DYT1 knock-in mice implied microstructural defects in cerebello-thalamic connectivity (Ulug et al., 2011), although histological studies delineating the nature of these defects have not yet been reported. Together, these studies suggest a variety of microstructural changes that are not limited to the basal ganglia in mouse models of DYT1 dystonia. Further studies of these and other regions are warranted to determine the anatomical specificity and physiological relevance of the microstructural changes.
Relevance to human DYT1 dystonia
Comprehensive autopsy studies involving a relatively small numbers of DYT1 brains have revealed few obvious defects (Standaert, 2011). The only abnormalities reported have so far been subtle and subjective. One autopsy study of 3 DYT1 brains revealed substantia nigra neurons to be enlarged (Rostasy et al., 2003), while another study of 4 DYT1 brains showed peri-nuclear inclusions in the brainstem (McNaught et al., 2004). Neither study has been independently confirmed. These limited findings have contributed to the belief that the brain is structurally normal in DYT1 dystonia. In fact, some investigators have suggested that distinctions between primary and secondary dystonias should include the absence of structural abnormalities (Breakefield et al., 2008; de Carvalho Aguiar and Ozelius, 2002; Nemeth, 2002; Schwarz and Bressman, 2009; Tanabe et al., 2009). However, imaging studies repeatedly have implied that structural abnormalities may exist in certain regions. For example, a voxel based morphometry study revealed a reduced volume of the putamen (Draganski et al., 2009), and diffusion-tensor imaging studies have revealed abnormal connectivity of cerebello-thalamo-cortical pathways (Argyelan et al., 2009; Carbon et al., 2010a; Carbon et al., 2010b). These imaging studies suggest that more detailed studies of histopathology of the human DYT1 brain are needed.
The current results provide some potential histological correlates of the microstructural abnormalities implied by human imaging studies. The types of defects uncovered in the mouse model, such as changes in the size or distribution of neurons or the numbers and thicknesses of dendrites, would not be detectable with the histological methods previously applied to human DYT1 brains.
The current results therefore may provide a useful guide for the types of methods that may need to be considered in future autopsy studies of human DYT1 brains. The current results also suggest specific anatomical mechanisms that may alter signaling in the basal ganglia. A reduction in dendrites and spines among medium spiny neurons implies reduced capacity for synaptic inputs to the basal ganglia. It also is tempting to speculate that the enlarged ChAT+ neurons reflect an anatomical correlate of excess cholinergic tone implied by prior human and animal studies (Pisani et al., 2006). Changes in the size of PV+ and nNOS+ neurons similarly may reflect changes in GABAergic inhibitory tone in the basal ganglia implied by prior studies (Gernert et al., 2000; Sander et al., 2006; Sciamanna et al., 2011). However, the functional significance of the microstructural abnormalities found in the current studies remains to be established. Since torsinA is ubiquitously expressed in the brain, mutations may disrupt the cellular processes of many different types of neurons in different ways, leading to many subtle changes in neuronal morphology. It is difficult to know which of these morphological changes might be involved in the pathogenesis of dystonia, which may reflect compensations from defects arising elsewhere, and which are functionally unimportant.
Relevance for classification of the dystonias
The current results also have implications for using histopathological features in the classification of the dystonias. Although autopsy studies of primary dystonia have shown no obvious degenerative changes, the number of brains examined and the techniques of evaluation have not been adequate to detect subtle changes in neuron numbers, neuron sizes or morphology, or regional distributions. The methods used also have been inadequate to evaluate changes in numbers or sizes of axons, dendrites and spines, and in their synaptic relationships with major striatal afferents. While histopathological abnormalities in primary dystonia may not involve overt degeneration, they may involve dystrophic cells, axon or dendrite loss or reconfiguration, synaptic abnormalities, pathological inclusions, or other subtle changes that may have profound functional consequences. Indeed, a recent study of primary cervical dystonia revealed such subtle quantitative changes in the cerebellar Purkinje neurons (Prudente et al., 2012).
Studies of other disorders have made it clear that subtle microstructural defects may be indicative of significant functional abnormalities. For example, it is widely believed that the loss of synapses and axonal arborizations are among the earliest degenerative changes in Parkinson’s disease or Alzheimer’s disease (Luo and O’Leary, 2005). Simplification of dendrite and spine structure is a pathological feature commonly found in numerous neurological and psychiatric diseases (Fiala et al., 2002), and is believed to underlie profound intellectual disability in fragile-X syndrome and other mental retardation syndromes where overt degeneration is lacking (Irwin et al., 2000; von Bohlen Und Halbach, 2010). These observations raise obvious questions regarding the definition of criteria used to define specific histopathological features as “degenerative” or “developmental” and they highlight the arbitrary nature of using histopathological features to classify different types of dystonia. In view of these issues, classifying the different types of dystonia according to strict histopathological criteria should be discouraged.
Highlights.
Striatal cholinergic neurons were enlarged in a mouse model of DYT1 dystonia.
Striatal parvalbumin+ neurons were enlarged in the mutant mice.
Striatal nNOS+ neurons were smaller in the mutant mice.
Striatal medium spiny neurons had fewer dendrites and smaller spines in the mutants.
The ratio of axo-spinous to axo-dendritic synapses was reduced in the mutant mice.
Acknowledgments
We thank Dr. William Dauer for making the DYT1 knock-in mice available for these studies. Thanks are also due to Susan Jenkins and Jean-Francois Pare for their technical help with the processing of tissue for electron microscopy studies. This work was supported in part by The Dystonia Medical Research Foundation, NIH grants NS040470 and NS033592, and the Yerkes Primate Centre base grant from NCRR (RR00165).
Abbreviations
- ΔE
deletion of a single GAG codon in Tor1A gene
- ChAT
choline acetyltransferase
- PV
parvalbumin
- nNOS
neuronal nitric oxide synthase
- TH
tyrosine hydroxylase
- AP
region from bregma in the anterior-posterior direction
- EM
electron microscopic
- vGluT
vesicular glutamate transporters
- ANOVA
analysis of variance
- DL
dorso-lateral
- DM
dorso-medial
- VL
ventro-lateral
- VM
ventro-medial
- RC
rostro-caudal distributions
- SP
axo-spinous
- Den
axo-dendritic
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- Argyelan M, et al. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–7. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Smith Y. The GABA and substance P input to dopaminergic neurones in the substantia nigra of the rat. Brain Res. 1990;529:57–78. doi: 10.1016/0006-8993(90)90811-o. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T, Yuste R. Spine motility. Phenomenology, mechanisms, and function. Neuron. 2002;35:1019–27. doi: 10.1016/s0896-6273(02)00906-6. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, et al. The pathophysiological basis of dystonias. Nat Rev Neurosci. 2008;9:222–34. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- Carbon M, et al. Functional imaging in hereditary dystonia. Eur J Neurol. 2010a;17(Suppl 1):58–64. doi: 10.1111/j.1468-1331.2010.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, et al. Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain. 2010b;133:690–700. doi: 10.1093/brain/awq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, et al. An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 DeltaGAG knock-in mice. Behav Brain Res. 2012;226:465–72. doi: 10.1016/j.bbr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang MT, et al. Generation and characterization of Dyt1 DeltaGAG knock-in mouse as a model for early-onset dystonia. Exp Neurol. 2005;196:452–63. doi: 10.1016/j.expneurol.2005.08.025. [DOI] [PubMed] [Google Scholar]
- de Carvalho Aguiar PM, Ozelius LJ. Classification and genetics of dystonia. Lancet Neurol. 2002;1:316–25. doi: 10.1016/s1474-4422(02)00137-0. [DOI] [PubMed] [Google Scholar]
- Draganski B, et al. Genotype-phenotype interactions in primary dystonias revealed by differential changes in brain structure. Neuroimage. 2009;47:1141–7. doi: 10.1016/j.neuroimage.2009.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egami K, et al. Basal ganglia dopamine loss due to defect in purine recycling. Neurobiol Dis. 2007;26:396–407. doi: 10.1016/j.nbd.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S. Concept and classification of dystonia. Adv Neurol. 1988 Jan 8;50 [PubMed] [Google Scholar]
- Fiala JC, et al. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Gernert M, et al. Deficit of striatal parvalbumin-reactive GABAergic interneurons and decreased basal ganglia output in a genetic rodent model of idiopathic paroxysmal dystonia. J Neurosci. 2000;20:7052–8. doi: 10.1523/JNEUROSCI.20-18-07052.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, et al. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, et al. Cholinergic neuropil of the striatum observes striosomal boundaries. Nature. 1986;323:625–7. doi: 10.1038/323625a0. [DOI] [PubMed] [Google Scholar]
- Grundmann K, et al. Overexpression of human wildtype torsinA and human DeltaGAG torsinA in a transgenic mouse model causes phenotypic abnormalities. Neurobiol Dis. 2007;27:190–206. doi: 10.1016/j.nbd.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–71. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Irwin SA, et al. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10:1038–44. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, et al. Rodent models for dystonia research: characteristics, evaluation, and utility. Mov Disord. 2005;20:283–292. doi: 10.1002/mds.20364. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, et al. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–35. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kemp JM, Powell TP. The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: a study with the Golgi method. Philos Trans R Soc Lond B Biol Sci. 1971;262:429–39. doi: 10.1098/rstb.1971.0105. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC. Physiology and pharmacology of striatal neurons. Annu Rev Neurosci. 2009;32:127–47. doi: 10.1146/annurev.neuro.051508.135422. [DOI] [PubMed] [Google Scholar]
- Luk KC, Sadikot AF. GABA promotes survival but not proliferation of parvalbumin-immunoreactive interneurons in rodent neostriatum: an in vivo study with stereology. Neuroscience. 2001;104:93–103. doi: 10.1016/s0306-4522(01)00038-0. [DOI] [PubMed] [Google Scholar]
- Luo L, O’Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–56. doi: 10.1146/annurev.neuro.28.061604.135632. [DOI] [PubMed] [Google Scholar]
- Marrone MC, et al. Altered cortico-striatal synaptic plasticity and related behavioural impairments in reeler mice. Eur J Neurosci. 2006;24:2061–70. doi: 10.1111/j.1460-9568.2006.05083.x. [DOI] [PubMed] [Google Scholar]
- Martella G, et al. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–49. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KS, et al. Brainstem pathology in DYT1 primary torsion dystonia. Ann Neurol. 2004;56:540–7. doi: 10.1002/ana.20225. [DOI] [PubMed] [Google Scholar]
- McNeill TH, et al. Quantitative analysis of age-related dendritic changes in medium spine I (MSI) striatal neurons of C57BL/6N mice. Neurobiol Aging. 1990;11:537–550. doi: 10.1016/0197-4580(90)90115-g. [DOI] [PubMed] [Google Scholar]
- Mikolaenko I, et al. A Golgi study of neuronal architecture in a genetic mouse model for Lesch-Nyhan disease. Neurobiol Dis. 2005;20:479–490. doi: 10.1016/j.nbd.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Nemeth AH. The genetics of primary dystonias and related disorders. Brain. 2002;125:695–721. doi: 10.1093/brain/awf090. [DOI] [PubMed] [Google Scholar]
- Neychev VK, et al. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the cavalieri and optical disector methods. J Comp Neurol. 1996;366:580–99. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Elsevier Academic Press; Amsterdam; Boston: 2004. [Google Scholar]
- Peters A, et al. The fine structure of the nervous system: neurons and their supporting cells. Oxford University Press; New York: 1991. [Google Scholar]
- Pisani A, et al. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–53. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Pisani A, et al. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–25. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Prudente CN, et al. Neuropathology of cervical dystonia. Exp Neurol. 2012 doi: 10.1016/j.expneurol.2012.11.019. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju DV, et al. Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur J Neurosci. 2008;27:1647–58. doi: 10.1111/j.1460-9568.2008.06136.x. [DOI] [PubMed] [Google Scholar]
- Rostasy K, et al. TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol Dis. 2003;12:11–24. doi: 10.1016/s0969-9961(02)00010-4. [DOI] [PubMed] [Google Scholar]
- Sander SE, et al. Age-related changes in striatal nitric oxide synthase-immunoreactive interneurones in the dystonic dtsz mutant hamster. Neuropathol Appl Neurobiol. 2006;32:74–82. doi: 10.1111/j.1365-2990.2005.00703.x. [DOI] [PubMed] [Google Scholar]
- Schwarz CS, Bressman SB. Genetics and treatment of dystonia. Neurol Clin. 2009;27:697–718. vi. doi: 10.1016/j.ncl.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Sciamanna G, et al. Cholinergic dysregulation produced by selective inactivation of the dystonia-associated protein torsinA. Neurobiol Dis. 2012a;47:416–27. doi: 10.1016/j.nbd.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, et al. Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J Neurosci. 2012b;32:11991–2004. doi: 10.1523/JNEUROSCI.0041-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, et al. Developmental profile of the aberrant dopamine D2 receptor response in striatal cholinergic interneurons in DYT1 dystonia. PLoS One. 2011;6:e24261. doi: 10.1371/journal.pone.0024261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, et al. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci. 2005;25:5351–5. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shashidharan P, et al. Transgenic mouse model of early-onset DYT1 dystonia. Hum Mol Genet. 2005;14:125–33. doi: 10.1093/hmg/ddi012. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–407. [PMC free article] [PubMed] [Google Scholar]
- Smith Y, et al. Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J Comp Neurol. 1994;344:1–19. doi: 10.1002/cne.903440102. [DOI] [PubMed] [Google Scholar]
- Song CH, et al. Functional analysis of dopaminergic systems in a DYT1 knock-in mouse model of dystonia. Neurobiol Dis. 2012;48:66–78. doi: 10.1016/j.nbd.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standaert DG. Update on the pathology of dystonia. Neurobiol Dis. 2011;42:148–51. doi: 10.1016/j.nbd.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe LM, et al. Primary dystonia: molecules and mechanisms. Nat Rev Neurol. 2009;5:598–609. doi: 10.1038/nrneurol.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsy D, Simon DK. Dystonia. N Engl J Med. 2006;355:818–829. doi: 10.1056/NEJMra055549. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Bolam JP. Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol. 2004;14:685–92. doi: 10.1016/j.conb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Ulug AM, et al. Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc Natl Acad Sci U S A. 2011;108:6638–43. doi: 10.1073/pnas.1016445108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba RM, Smith Y. Differential structural plasticity of corticostriatal and thalamostriatal axo-spinous synapses in MPTP-treated Parkinsonian monkeys. J Comp Neurol. 2011;519:989–1005. doi: 10.1002/cne.22563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen Und Halbach O. Dendritic spine abnormalities in mental retardation. Cell Tissue Res. 2010;342:317–23. doi: 10.1007/s00441-010-1070-9. [DOI] [PubMed] [Google Scholar]
- Wichmann T. Commentary: Dopaminergic dysfunction in DYT1 dystonia. Exp Neurol. 2008;212:242–6. doi: 10.1016/j.expneurol.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichmann T, Dostrovsky JO. Pathological basal ganglia activity in movement disorders. Neuroscience. 2011;198:232–44. doi: 10.1016/j.neuroscience.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, et al. Neuronal mechanism underlying dystonia induced by bicuculline injection into the putamen of the cat. Brain Res. 1995;677:333–6. doi: 10.1016/0006-8993(95)00190-2. [DOI] [PubMed] [Google Scholar]
- Yoshida M, et al. Differential roles of the caudate nucleus and putamen in motor behavior of the cat as investigated by local injection of GABA antagonists. Neurosci Res. 1991;10:34–51. doi: 10.1016/0168-0102(91)90018-t. [DOI] [PubMed] [Google Scholar]
- Zhang L, et al. Altered dendritic morphology of Purkinje cells in Dyt1 DeltaGAG knock-in and purkinje cell-specific Dyt1 conditional knockout mice. PLoS One. 2011;6:e18357. doi: 10.1371/journal.pone.0018357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. Abnormal motor function and dopamine neurotransmission in DYT1 DeltaGAG transgenic mice. Exp Neurol. 2008;210:719–30. doi: 10.1016/j.expneurol.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoons E, et al. Structural, functional and molecular imaging of the brain in primary focal dystonia--a review. Neuroimage. 2011;56:1011–20. doi: 10.1016/j.neuroimage.2011.02.045. [DOI] [PubMed] [Google Scholar]