

Abstract
Post-traumatic stress disorder (PTSD) is a common and chronic anxiety disorder that can result after exposure to a traumatic event. Though our understanding of the aetiology of PTSD is incomplete, several neurobiological systems have been implicated in the pathophysiology and vulnerability towards developing PTSD after trauma exposure. We aimed to provide a concise review of benchmark findings in important neurobiological systems related to the aetiology and maintenance of PTSD symptomology. Specifically, we discuss functional aetiologies in the noradrenergic, serotonergic, endogenous cannabinoid and opioid systems as well as the hypothalamic-pituitary adrenal (HPA) axis. This article provides a succinct framework to appreciate the current understanding of neurobiological mechanisms related to the pathophysiology of PTSD and how these findings may impact the development of future, targeted pharmacological treatments for this debilitating disorder.
1 Introduction
Post-traumatic stress disorder (PTSD) is a common and chronic anxiety disorder that can develop following exposure to a traumatic life event such as military combat, a natural disaster, and/or physical or sexual assault. PTSD occurs in approximately 8–14 % of the population in the USA [1-3], and rates of PTSD among women in the USA (12—18 %) are approximately twice than in men [3, 4].
PTSD presents a significant burden not only to individuals but society at-large. The majority of people with PTSD meet the diagnostic criteria for other psychiatric disorders [4, 5], including major depression [6, 7] and anxiety disorders [8]. Individuals with a PTSD diagnosis are more likely than the general population to use drugs and experience impairments in psychosocial functioning [9] and to engage in suicidal behaviours [1]. Not only does this population have increased psychiatric treatment needs, but when compared with the general population, people with PTSD also require greater healthcare utilization, have more costs attributed to co-morbid medical conditions such as heart problems, diabetes and peptic ulcers [10], and gastrointestinal problems [11], and have increased rates of surgery and visits to the physician [12, 13].
Despite the deleterious impact of PTSD, our current understanding about the human pathophysiology governing the divergent paths associated with extreme stress response is lacking [14, 15], and these models have failed to provide effective therapeutic targets. US-based practice guidelines for PTSD have recommended cognitive behavioural therapy (CBT) and selective serotonin reuptake inhibitors (SSRIs) or selective noradrenaline (norepinephrine) reuptake inhibitors (SNRIs) as first-line treatments [16, 17]. However, PTSD response rates to pharmacological treatments such as the two US FDA-approved SSRIs paroxetine and sertraline rarely exceed 60 %, and even fewer patients (20–30 %) achieve clinical remission [18]. Several placebo-controlled trials of other medications in PTSD have failed, and even recent studies of approved medications (e.g. sertraline) have failed to show efficacy in specific subgroups of PTSD patients such as combat veterans [19]. The recent review of treatments for PTSD by the United States Institute of Medicine, located in Washington, DC, concluded there was not sufficient evidence for any drug or class of drug for the treatment of PTSD [20]. To date, drug development in PTSD has been opportunistic, building almost solely on empirical observations with drugs approved for other indications, and not surprisingly, treatment options for the often chronically symptomatic PTSD patients remain limited.
This paper reviews our current understanding of the pathophysiology underlying PTSD with evidence suggesting functional aetiologies in the noradrenergic, serotonergic, endogenous cannabinoid, and opioid systems as well as the hypothalamic-pituitary adrenal (HPA) axis. Other systems, for example glutamate, are also relevant and important, but have been reviewed extensively elsewhere [21] [22]. By revealing the neurobiological mechanisms that play a role in the aetiology of PTSD, we aim to identify novel targets that offer potential therapeutic value in developing future evidence-based PTSD pharmacological interventions.
2 Noradrenergic System
The adrenoreceptors (ARs) are a group of G protein-coupled receptors consisting of three major classifications: α1, α2 and β with associated subtypes [23]. The AR system stimulates CNS activity and sympathetic autonomic responses through cell bodies located in the locus coeruleus and projects to the prefrontal cortex and limbic system structures (e.g., amygdala, hypothalamus) [24], which implicate it in selective attention to rewarding and aversive stimuli [25] and stress and fear-related responses [26] [27]. Through dysregulation of physiological mechanisms, hyperadrenergic activity has been linked to psychiatric conditions such as major depression [28] [29], traumatic brain injury [30] and anxiety disorders [31-33] [23, 34].
The AR system has held a preeminent role in PTSD research, as it influences amygdala functioning and associated fear signalling [27, 35, 36]. Table 1 lists evidence of altered peripheral and central AR functioning in PTSD populations suggesting state and trait alterations in AR functions.
Table 1.
Evidence for altered noradrenergic function in PTSDa
| Physiological observations | Study results |
|---|---|
| Baseline/resting state measures | |
| Increased resting heart rate and blood pressure | +/− |
| Increased resting urinary noradrenaline (norepinephrine) and adrenaline (epinephrine) |
+ |
| Decrease in basal and stimulated activity of cAMP | +/− |
| Decreased binding to platelet α2 receptors | + |
| Decrease in platelet MAO activity | + |
| Increased resting plasma noradrenaline or MHPG | +/− |
| Challenge test markers | |
| Increased plasma noradrenaline with traumatic reminders/panic attacks |
+ |
| Increased heart rate and blood pressure response to traumatic reminders/panic attacks |
+++ |
| Increased orthostatic heart rate response to exercise | + |
| Increased symptoms, heart rate and plasma MHPG with yohimbine noradrenergic challenge |
++ |
| Differential brain metabolic response to yohimbine | + |
The evidence for altered noradrenergic functioning in PTSD is stronger in challenge test designs (i.e., incorporating threat responses, trauma reminders) in comparison to baseline/resting state studies
cAMP cyclic adenosine 3′5′-monophosphate, MAO monoamine oxidase, MHPG 3-methosy-4-hydroxyphenylglycol, PTSD post-traumatic stress disorder, +/− indicates an equal number of studies support this finding and do not support this finding, + indicates at least one study supports this finding and no studies do not support the finding, or the majority of studies support the finding, ++ indicates two or more studies support this finding, and no studies do not support the finding, +++ indicates three or more studies support this finding, and no studies do not support the finding
Early studies using the α2 selective antagonist yohimbine, which acts on both pre- and post-synaptic receptors and increases noradrenaline activity, helped form a model suggesting that increased noradrenaline activity leads to impaired medial prefrontal cortex functioning [27, 37] and fear extinction [38, 39], explaining increases on behavioural measures of anxiety and PTSD symptom severity [40, 41]. β-AR antagonists (i.e., propranolol) have produced mixed results in animal models, with some evidence for inhibiting fear memory reconsolidation and responding [35]. However, post-trauma administration of propranolol has not yielded significant effects on preventing fear conditioning [42] or inhibitory avoidance reconsolidation [43]. Clinical evidence has shown a similar trend regarding propranolol as a PTSD prophylactic [44, 45] with several contrary findings [46, 47]. However, clinical trials to date have not yet provided conclusive results given small sample sizes, difficulties recruiting for these trials and heterogeneous patient cohorts.
The noradrenaline transporter (NET) is a potential noradrenaline target for studying the pathophysiology of PTSD and may emerge as a target for treatment development in the future. The NET is a part of the Na+/Cl− neurotransmitter transporters [48] that has high concentrations in the locus coeruleus and moderate levels within cortical regions including the frontal cortex, hippocampus, amygdala, thalamus and cerebellar cortex [49]. Besides acting as a noradrenaline plasma membrane monoamine transporter and maintaining presynaptic noradrenaline storage [50], the NET has been connected to the regulation of dopamine reuptake, and its dysregulation may be involved in mood and stress disorders [37, 51-53].
There is ongoing work suggesting NET involvement in PTSD. Preclinical evidence has demonstrated that chronic stress exposure may lead to AR system dysregulation and can decrease NET availability in the locus coeruleus and increase noradrenaline synaptic availability in cortical areas [54], while the selective noradrenaline reuptake inhibitor (NRI) reboxetine can antagonize noradrenaline synaptic availability and decrease foot shock stress reactivity [55]. These preclincial findings have been recently substantiated in a human positron emission tomography (PET) study in humans showing decreased NET availability in patients with PTSD [194]. This alteration in homeostatic stress responding could trigger anxious and depressive phenotypes [56], which characterize PTSD [23, 29, 31-34, 57]. Recent clinical research [58] used a combination of naltrexone and either the SSRI, paroxetine, or the NRI, desipramine, in treating veterans with PTSD and alcohol dependence. The desipramine group performed equivalently in PTSD symptom reduction compared with the standard SSRI treatment and was more effective in reducing alcohol consumption and study attrition and therefore improving overall clinical presentation.
The NET may inform the development of future PTSD treatments. Trauma exposure can cause impaired impulse control and emotional dysregulation [59-61]. Early clinical evidence suggests that atomoxetine, which has a high affinity for the NET, increases inhibitory response control [62] and reduces attention-deficit hyperactivity disorder (ADHD) symptoms with co-morbid anxiety [63] and PTSD [64] diagnoses. This drug may be effective in treating phenotypes exhibiting pronounced hyperarousal and impulsive behaviours. Venlafaxine, an SNRI, has shown clinical utility in reducing re-experiencing and avoidance/numbing symptoms [65, 66] and can improve fear extinction in rats, thus suggesting its utility for supplement PTSD exposure therapy [67]. As the NET interacts with other stress systems such as dopaminergic [37, 51-53] and serotonergic [65, 66] systems, combination drugs (i.e., SNRIs) and combination treatments that affect NET functioning [58] may lead to improved PTSD treatment modalities.
3 Serotonergic Receptors: 5-HT1A, 5-HT1B
The serotonergic (5-HT) receptors are a group of G protein-coupled receptors and one identified ligand-gated ion channel (5-HT3) that encompass seven classifications, ranging from 5-HT1 through 5-HT7 with associated subtypes [68]. The 5-HT system is involved in cognition, emotional processing and behavioural regulation [69]. Animal and human models have demonstrated that 5-HT receptor systems are implicated in the pathophysiology of several psychiatric disorders, including depression [70] [71], alcoholism [72, 73] and PTSD [74, 75].
Fear regulation and threat responsiveness have been linked to 5-HT signalling in the amygdala [76], a brain region integral to understanding the fear response and PTSD aetiology [77-80]. 5-HT agonists can selectively induce anxiety attacks and trauma-related flashbacks in PTSD populations [41, 81, 82]. The 5-HT1A and 5-HT1B receptors have been identified in the study of stress disorders [74, 83, 84] and could provide new clinical understanding of PTSD phenotypes. The 5-HT1A receptors are located presynaptically on 5-HT cell bodies in the raphe and postsynaptically in other brain regions [85]. The density of 5-HT1A receptors in humans and monkeys appears highest in the raphe, hippocampal formation, hypothalamus, and insula, temporal, cingulate and ventral prefrontal cortices [86], [87, 88]. Postsynaptic 5-HT1A receptors are expressed mainly in the astroglia of the frontal and limbic cortices [85], [89] and stimulate the release of trophic-factor S-100β, which promotes serotonergic system development [85], [90] and cytoskeletal maintenance [91]. Therefore, the neuropathological abnormalities exhibited in limbic and paralimbic cortical areas in mood disorders (i.e., reduced cortex volume, reduced synaptic proteins, increased neuronal density, reduced glial counts) may be attributed to impaired 5-HT1A receptor functioning [92, 93]. Preclinical evidence has shown 5-HT1A receptor knockout mice display increased anxiety and fear responses [94]. Relevant to stress regulation and PTSD, mice administered the 5-HT1A agonist 8-OH-DPAT demonstrated anxiogenic responses to the elevated plus maze (EPM) test, which could modulate glutamatergic and GABAergic systems located in the medial prefrontal cortex [95].
The 5-HT1B receptors have the highest expression in the striatum, pallidum, nucleus acumbens, substantia nigra and the ventral tegmental area and to a lesser extent in the dorsal raphe nucleus, the amygdala, hippocampus and cortical areas [96]. These receptors operate presynaptically as autoreceptors on the axon terminals of 5-HT-containing neurons and as heteroreceptors on non-5-HT-containing neurons [71, 96]. As a heteroreceptor, the 5-HT1B receptor is involved in regulating the activity of multiple neurobiological systems in the brain including gamma-aminobutyric acid (GABA) [96, 97], noradrenaline [98], dopamine [99], acetylcholine [100] and glutamate [97, 101] in areas of a cortico-striato-pallido-thalamic-limbic circuit that have been implicated in the pathogenesis of PTSD. Animal studies have yielded behavioural pharmacological results indicating 5-HT1B receptor involvement in locomotor activity, reinforcement learning, appetitive behaviours (i.e., sexual, feeding), sleep and aggression [96, 102]. PTSD patients and healthy trauma-exposed individuals have demonstrated reduced 5-HT1B binding potentials in the caudate, amygdala and anterior cingulate cortex with positive correlations observed according to trauma history and symptom severity [74]. Recently, alterations in 5-HT1B receptor density have been shown to be linked to specific PTSD symptomology suggesting that these changes may be responsible for certain features of the clinical phenotype of PTSD [193] (Fig. 1). This evidence suggests that trauma exposure can have long-term effects on 5-HT1B receptor functioning and could explain some of the enduring behavioural, neurological and psychological changes observed in PTSD phenotypes.
Fig. 1.

Post-traumatic stress disorder (PTSD) patients, overall, have lower serotonin 5-HT1B receptor density in the anterior cingulate cortex (ACC), hippocampus (HC) and pallidum as well as other regions implicated in PTSD [70]. Specific alterations in 5-HT1B receptor density in PTSD have been shown, using the 5-HT1B selective radioligand [11C]P943, to be linked to specific domains of PTSD symptomology. Decreased 5-HT1B receptor binding in the ACC has been shown to increase re-experiencing symptoms. Decreased 5-HT1B receptor binding in the HC has been shown to increase numbing symptoms. Decreased 5-HT1B receptor binding in the pallidum has been shown to increase anxious arousal, re-experiencing, and numbing symptoms [193]
5-HT1A and 5-HT1B receptor modulators may help in future treatment development. Animal [95] and human [103, 104] models suggest that further inquiry into the influence of 5-HT1A agonists on anxiety regulation could inform new forms of PTSD treatments. Drugs targeting 5-HT1B receptors may have clinical utility in treating PTSD, which are possible efficacious treatments for co-morbid mood disorders [70, 84]. A recent clinical trial utilizing coadministration of the now discontinued 5-HT1B antagonist, elazsonan (CP-448187), with sertraline yielded 14.9 % and 15.7 % treatment response rates for reducing depressive symptoms by at least half during weeks 2 and 3, respectively, compared with 3.3 % and 7 % for sertraline alone [105]. These symptom reductions were sustained throughout the 8-week trial, which suggests that 5-HT1B antagonists may have expediting therapeutic properties in PTSD.
4 Endocannabinoids and the Cannabinoid CB1 Receptor
Various lines of evidence suggest that the endogenous cannabinoids (eCB), anandamide (AEA) and 2-arachidonolyflycerol (2-AG) [Fig. 2], which exert much of their actions through the two known cannabinoid (CB) receptors (CB1, CB2), play an important role in the development [106] and function of the PTSD circuit, specifically in stress responses [107-112].
Fig. 2.
Chemical structures of endogenous compounds that bind to cannabinoid receptors
First, animal studies [113] show that chronic stress is associated with significantly decreased AEA levels in all brain areas studied. Furthermore, inhibiting the metabolism of AEA leads to a reduction of anxiety in rodents [114]. In our own work in PTSD we found decreased plasma AEA levels in PTSD patients relative to healthy control subjects (0.72 ± 0.12 vs. 2.74 ± 0.85 pmol/mL, t = 2.47, degrees of freedom [df] = 17, p = 0.024; Fig. 3, personal unpublished observations), providing additional evidence for an important role of altered eCB signalling in PTSD. Notably, these findings agree with results of human studies in depression [112] reporting that female outpatients with major depressive disorder (MDD) have lower serum 2-AG levels than controls, and the magnitude of the decrease was associated with the length and severity of the depressive episode. While AEA was not associated with major depression per se, an inverse relationship was found between serum AEA content and Hamilton Anxiety Rating Scale scores, suggesting that AEA content may relate to the anxiety dimension of the depression phenotype. In addition, earlier age at first trauma was correlated with lower AEA levels in PTSD and the magnitude of the decrease was associated with the length of illness, providing additional evidence for an important role of dysfunctional eCB signalling in PTSD.
Fig. 3.
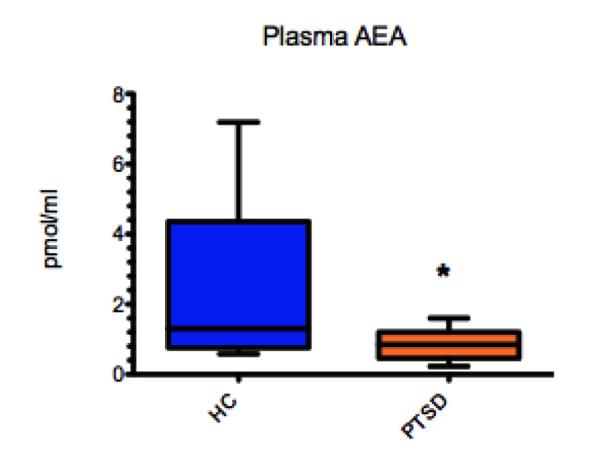
Plasma anandamide (AEA) levels are decreased in post-traumatic stress disorder (PTSD) patients (0.72 ± 0.12 pmol/mL) relative to healthy control subjects without trauma history (hippocampus [HC]; 2.74 ± 0.85 pmol/mL, t = 2.47, degrees of freedom [df] = 17, p = 0.024)
The CB1 receptors are of particular interest as recent studies demonstrate a specific and primary [115] role for the CB1 receptor in mediating the neurobiological underpinnings and behavioural consequences of stress exposure. The first CB1 receptor was discovered relatively recently by Allyn Howlett’s laboratory in 1988 [116, 117], and since then there has been substantial attention devoted to this neuromodulatory system.
CB1 receptors are found in moderate to high levels throughout forebrain limbic structures and have been shown to modulate a variety of behaviours, including mood, stress, anxiety, learning, memory [118-120] and notably the extinction of fear [121-123]. They are the most abundant G protein-coupled receptors in the CNS, [124, 125] and are found in high concentrations in a fear circuit of cortical and subcortical brain regions that are consistently implicated in PTSD. Moreover, genetic or pharmacological disruption of CB1 receptor signalling, which is required for normal fear extinction [121-123], results in an anxious PTSD phenotype [126, 127], and brain CB1 receptor signalling controls extinction of aversive memories [121-123]. Mice lacking CB1 receptors exhibit heightened indices of anxiety and depression, reinforcing the concept that eCBs are required to maintain emotional homeostasis [117, 119, 126]. While the eCB system is highly reactive in response to acute stress, there is evidence to suggest that chronic stress causes a breakdown in eCB signalling, thus compromising an endogenous buffer system to stress in the brain [113, 117]. Therefore, CB1 receptor function is a mechanism that seems to play an important role in the neurobiology of emotional behaviour with specific implications in PTSD.
Additionally, animal studies have shown gender disparities in the stress-induced regulation of CB1 receptors in male and female animals [109, 113, 128, 129] with elevated CB1 receptor expression in female but not male animals. Additionally, impaired CB1 receptor-mediated eCB signalling [130] was identified as a key mechanism explaining the increased vulnerability of female animals to develop an anxious phenotype in chronic stress models (for a review see Palanza [131]). This evidence may help explain why women are at higher risk for developing PTSD symptoms following exposure to various types of trauma [132-140] (odds ratio approximately 5), even when sexual trauma, which predominates in women, was excluded (odds ratio approximately 3) [141].
To further understand the neuromodulatory role of the eCBs and their corresponding receptors, we look to PET imaging as it is the most direct, sensitive and straightforward means of probing the functional neurochemistry of humans and assessing molecular targets in the brain in vivo, provided the proper tracer and modelling approaches are available. Due to the recent development of CB1 receptor-selective radiotracers, it is now possible for the first time to conduct an in vivo assessment of CB1 receptor density using PET.
Given the overwhelming evidence that the eCBs and their attending receptors play a key role in the aetiology of PTSD, pharmacological interventions specifically designed to target eCB signalling could emerge as a novel, potentially breakthrough treatment for PTSD. Conversely, the development of CB1 receptor antagonists, such as SR-141716 (rimonabant), has been associated with the emergence of negative mood symptoms and an increased risk of suicidality. Therefore, direct modulation of the CB1 receptor may not be the most appropriate point of intervention for therapeutic drug development. Fatty-acid amide hydrolase (FAAH) is an integral membrane enzyme primarily responsible for the degradation of the eCBs in the brain[142] and may yield a safer alternative treatment. FAAH activity has been linked with aversive memory processing [143] and arousability [142]. In addition to these findings, a single nucleotide polymorphism of FAAH was significantly associated with PTSD diagnosis in subjects without lesions in the ventromedial prefrontal cortex [142]. Therefore, FAAH inhibition may point to a novel treatment mechanism with unique therapeutic potential in the treatment of PTSD.
5 Hypothalamic-Pituitary-Adrenal Axis and Corticotropin-Releasing Factor
The eCB system is believed to be strongly tied to the HPA axis, another neurocircuit implicated in stress responding [144]. The HPA axis is a stress-responsive neuroendocrine system that ties the CNS to the endocrine system. The HPA axis assists with the adaptation to stress and the maintenance of homeostasis after challenge, yet is also vital in supporting baseline functioning [145]. A dysfunctional HPA axis is associated with numerous psychosomatic and psychiatric disorders [146-154].
Corticotropin-releasing factor (CRF) is a neuronal signalling molecule produced by cells in the hypothalamus in response to physical or psychological stress. Increased levels of CRF in the hypothalamus in response to stress results in the activation of the HPA axis and increased release of cortisol. CRF acts at the G protein-coupled receptors CRF-1 and CRF-2. It is believed that high CRF levels at the time of trauma may facilitate encoding of traumatic memory and enduring anxiety effects via direct action at CRF-1 receptors [155-158].
Animal models provide further evidence for the role of CRF in the development and maintenance of stress-induced behaviours. Mice deficient in the G protein-coupled receptor CRF-1 display an impaired stress response and decreased anxious behaviours [159, 160], while mice deficient in the G protein-coupled CRF-2 receptor display increased anxious phenotypes and hypersensitivity to stress [159, 161]. Regulation of the relative contributions of the two CRF receptor subtypes may be essential to regulating physiological and psychological responses to stress [160]. In a mouse-predator stress model of PTSD, CFR-1 receptor antagonism prevented the initiation of stress effects [157, 158], suggesting a potential therapeutic target for the primary prevention of stress-induced symptoms.
PTSD patients exhibit increased cerebrospinal fluid levels of CRF [162, 163] and abnormalities in other HPA axis systems [164], indicating the utility of compounds that dampen the CRF system or other HPA axis hormones in the treatment of PTSD [158, 165]. CRF-1 receptor antagonism is a treatment modality that has been explored for the treatment of MDD [166], While an initial open-label study suggested promising efficacy data, these results were not confirmed in follow-up placebo-controlled trials [167]. Testing of CRF-1 receptor antagonists in the treatment of chronic PTSD is currently underway, but the potential role for CRF-1 antagonists as a method of primary prevention has not yet been explored.
Recently, much attention has been devoted to furthering our understanding of the relationship between the eCB system and the HPA axis in response to stress. Animal models showed that (1) in vivo exposure to stress was capable of modulating eCB content in the hypothalamus, and (2) eCB signalling in vivo exerted negative regulation over the HPA axis [117, 168]. It is believed that a functional crosstalk exists between eCB signalling and the HPA axis, and changes in one system have a ripple effect on the other [117]. This relationship poses the opportunity for further development and understanding of unique therapeutic targets in the treatment of PTSD.
6 Opioid System
Multiple lines of evidence connect opioid systems in the pathophysiology of PTSD. Opioid receptors (ORs) belong to the superfamily of G protein-coupled receptors and are generally classified into at least three subtypes: δ (encephalin preferring), κ (dynorphin preferring) and μ (morphine preferring) [169]. The dynorphin/κ opioid receptor (κ-OR) is of particular interest, as it has been implicated in several brain disorders including substance (particularly psychostimulant) abuse [170], epilepsy [171], Tourette’s syndrome [172] and Alzheimer’s disease [173]. Moreover, recent evidence suggests a role for the dynorphin/κ-OR in the expression of stress-induced behaviours [174].
Relevant to the aetiology of PTSD, is the expression pattern of the κ-OR with high receptor levels in a ventral medial, prefrontal, cortex-hippocampal-limbic circuit (see Fig. 4) [175, 176] where they mediate anxiety-like behaviours [177]. These brain regions are also implicated in the aetiology of PTSD [27].
Fig. 4.

MRI scans and positron emission tomography (PET) images of kappa opiate receptors in the brain of a healthy individual. For both left and right panels, the left column shows the axial slice, the middle column shows the coronal slice and the right column shows the sagittal slice. The PET images of the receptors confirm their known distribution in the human brain, with high levels in a circuit implicated in post-traumatic stress disorder, which includes the amygdala, hippocampus and ventromedial prefrontal cortex. Left panel: Top row: MRI images. Middle row: 0–10 min summed PET image. Bottom row: 60–120 min summed PET image. Right panel: Top row: MRI images. Middle row: corresponding binding potential (BPND images estimated using a simplified reference tissue model [SRTM]). Bottom row: corresponding BPND images estimated using the SRTM2; it is slightly less noisy than the SRTM, as would be expected.
There is a growing body of basic science data [178] showing that although κ-OR signalling during acute stress may create the physical ability and motivation to escape a threatening situation, κ-OR signalling in response to chronic and inescapable stress can lead to persistent depressive and anxious behaviours [179], which resemble important components of the phenotype of PTSD [180].
Recent preclinical evidence suggests that κ-ORs may emerge as targets for treatment development for patients with anxious-depressive phenotypes [179]. This would be an important step forward in the treatment of PTSD because current FDA-approved medication treatments (SSRIs) offer relatively little benefit to most PTSD patients [181, 182]. A recent study showed that κ-OR antagonists, in contrast to the SSRI fluoxetine, possess unique antidepressant-anxiolytic properties in models of unlearned and learned fear [183], which are informative models to our further understanding of trauma-related psychopathology leading to the development of new treatment approaches [184].
Clinical evidence implicating opioid systems in the aetiology of PTSD are three-fold. First, survivors of intimate partner violence can exhibit persistent pain syndromes [185]. Second, there is an association between childhood sexual trauma and exaggerated rate of opioid use later in life [186]. Lastly, the use of morphine during trauma care may reduce the risk of subsequent development of PTSD after traumatic injuries in military personnel [187], burn victims [188], and children [189] and adult [190] patients.
The ability to image the κ-OR would be important in allowing us to further understand the circuitry of the κ-OR and characterize the involvement of κ-OR transmission in PTSD. A greater understanding of the neuromodulatory role the κ-OR plays in the aetiology and course of PTSD may provide a basis and justification for the development of experimental drugs targeting the κ-OR. In vivo functional imaging using PET is currently the sole method for providing a quantitative measurement of κ-OR-mediated signalling in the brain [191], and novel imaging approaches are currently under development.
To date, data suggest direct involvement of the κ-OR system in the maladaptive stress response resulting in the phenotype of PTSD. The implications of these lines of evidence are that the κ-OR system could emerge as a system of interest for targeted evidence-based treatment development for this patient population, either alone or as combined pharmacotherapy with behavioural exposure, a therapeutic strategy that has recently been validated to enhance extinction of conditioned fear in PTSD patients [192]. Pharmacological therapies targeting the κ-OR might be particularly effective in the management of depression and co-morbid anxiety, the typical phenotype of PTSD. Such developments would be critical steps forward in the clinical care of this severely ill patient population.
7 Conclusion
In a sharp distinction from other medical disorders such as cancer, coronary artery disease and diabetes, which have objective biological tests for diagnosis, severity of illness and response to treatment, biological markers cannot yet independently confirm the assessment of PTSD. Current procedures for diagnosing PTSD rely on self-report screening measures and clinical interviews. Treatment has been limited to symptom management rather than targeting the biological aetiology. To date, drug development in PTSD has been opportunistic, building almost solely on empirical observations with drugs approved for other conditions. As of today, not a single pharmacological treatment has been developed specifically for PTSD. Utilizing our existing knowledge of the circuits and substrates underlying the development and maintenance of PTSD, and the critical importance of forthcoming research findings, we are now able to guide future PTSD treatments altering brain mechanisms with proven relevance to PTSD.
Acknowledgements
This project was supported by the National Institutes of Health through the following awards: R21 MH081103 (ARRA), R21 MH096105-01A1, R21 MH085627 and R01MH096876-01A1.
Footnotes
The authors have no conflicts of interest that are directly relevant to the content of this article.
References
- 1.Davidson JR, Hughes D, Blazer DG, et al. Post-traumatic stress disorder in the community: an epidemiological study. Psychol Med. 1991;21(3):713–21. doi: 10.1017/s0033291700022352. [DOI] [PubMed] [Google Scholar]
- 2.Kessler RC, Sonnega A, Bromet E, et al. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52(12):1048–60. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- 3.Breslau N, Kessler RC, Chilcoat HD, et al. Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch Gen Psychiatry. 1998;55(7):626–32. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- 4.Resnick HS, Kilpatrick DG, Dansky BS, et al. Prevalence of civilian trauma and posttraumatic stress disorder in a representative national sample of women. J Consult Clin Psychol. 1993;61(6):984–91. doi: 10.1037//0022-006x.61.6.984. [DOI] [PubMed] [Google Scholar]
- 5.Breslau N, Davis GC, Andreski P, et al. Traumatic events and posttraumatic stress disorder in an urban population of young adults. Arch Gen Psychiatry. 1991;48(3):216–22. doi: 10.1001/archpsyc.1991.01810270028003. [DOI] [PubMed] [Google Scholar]
- 6.Koenen KC, Lyons MJ, Goldberg J, et al. A high risk twin study of combat-related PTSD comorbidity. Twin Res. 2003;6(3):218–26. doi: 10.1375/136905203765693870. [DOI] [PubMed] [Google Scholar]
- 7.Neria Y, Bromet EJ. Comorbidity of PTSD and depression: linked or separate incidence. Biol Psychiatry. 2000;48(9):878–80. doi: 10.1016/s0006-3223(00)01012-x. [DOI] [PubMed] [Google Scholar]
- 8.Gros DF, Frueh BC, Magruder KM. Prevalence and features of panic disorder and comparison to posttraumatic stress disorder in VA primary care. Gen Hosp Psychiatry. 2011;33(5):482–8. doi: 10.1016/j.genhosppsych.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 9.Pietrzak RH, Goldstein RB, Southwick SM, et al. Psychiatric comorbidity of full and partial posttraumatic stress disorder among older adults in the United States: results from wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. Am J Geriatr Psychiatry. 2012;20(5):380–90. doi: 10.1097/JGP.0b013e31820d92e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pietrzak RH, Goldstein RB, Southwick SM, et al. Medical comorbidity of full and partial posttraumatic stress disorder in US adults: results from Wave 2 of the National Epidemiologic Survey on Alcohol and Related Conditions. Psychosom Med. 2011;73(8):697–707. doi: 10.1097/PSY.0b013e3182303775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leserman J, Drossman DA, Li Z, et al. Sexual and physical abuse history in gastroenterology practice: how types of abuse impact health status. Psychosom Med. 1996;58(1):4–15. doi: 10.1097/00006842-199601000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Solomon SD, Davidson JR. Trauma: prevalence, impairment, service use, and cost. J Clin Psychiatry. 1997;58(Suppl. 9):5–11. [PubMed] [Google Scholar]
- 13.Zoellner LA, Foa EB, Brigidi BD. Interpersonal friction and PTSD in female victims of sexual and nonsexual assault. J Trauma Stress. 1999;12(4):689–700. doi: 10.1023/A:1024777303848. [DOI] [PubMed] [Google Scholar]
- 14.Charney DS, Manji HK. Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE. 2004;2004(225):RE5. doi: 10.1126/stke.2252004re5. [DOI] [PubMed] [Google Scholar]
- 15.Krystal JH, Neumeister A. Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Res. 2009;1293:13–23. doi: 10.1016/j.brainres.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benedek DM, Friedman MJ, Zatzick D, et al. Guideline watch (March 2009): Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. American Psychiatric Association; Arlington (VA): 2009. [PubMed] [Google Scholar]
- 17.VA/DOD . VA/DoD clinical practice guideline for management of post-traumatic stress. U.S. Department of Veterans Affairs; Washington, DC: 2010. [Google Scholar]
- 18.Davidson JR. Pharmacologic treatment of acute and chronic stress following trauma: 2006. J Clin Psychiatry. 2006;67(Suppl. 2):34–9. [PubMed] [Google Scholar]
- 19.Friedman MJ, Marmar CR, Baker DG, et al. Randomized, double-blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting. J Clin Psychiatry. 2007;68(5):711–20. doi: 10.4088/jcp.v68n0508. [DOI] [PubMed] [Google Scholar]
- 20.Institute of Medicine . Treatment of PTSD: an assessment of the evidence. National Academies Press; Washington, DC: 2007. [Google Scholar]
- 21.Myers KM, Carlezon WA, Jr., Davis M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology. 2011;36(1):274–93. doi: 10.1038/npp.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nair J, Singh Ajit S. The role of the glutamatergic system in posttraumatic stress disorder. CNS Spectr. 2008;13(7):585–91. doi: 10.1017/s1092852900016862. [DOI] [PubMed] [Google Scholar]
- 23.Strawn JR, Geracioti TD., Jr. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety. 2008;25(3):260–71. doi: 10.1002/da.20292. [DOI] [PubMed] [Google Scholar]
- 24.Morilak DA, Barrera G, Echevarria DJ, et al. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(8):1214–24. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Southwick SM, Bremner JD, Rasmusson A, et al. Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;46(9):1192–204. doi: 10.1016/s0006-3223(99)00219-x. [DOI] [PubMed] [Google Scholar]
- 26.O’Donnell T, Hegadoren KM, Coupland NC. Noradrenergic mechanisms in the pathophysiology of post-traumatic stress disorder. Neuropsychobiology. 2004;50(4):273–83. doi: 10.1159/000080952. [DOI] [PubMed] [Google Scholar]
- 27.Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- 28.Wong EH, Sonders MS, Amara SG, et al. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol Psychiatry. 2000;47(9):818–29. doi: 10.1016/s0006-3223(99)00291-7. [DOI] [PubMed] [Google Scholar]
- 29.Cheok F, Schrader G, Banham D, et al. Identification, course, and treatment of depression after admission for a cardiac condition: rationale and patient characteristics for the Identifying Depression As a Comorbid Condition (IDACC) project. Am Heart J. 2003;146(6):978–84. doi: 10.1016/S0002-8703(03)00481-2. [DOI] [PubMed] [Google Scholar]
- 30.Kobori N, Hu B, Dash PK. Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience. 2011;172:293–302. doi: 10.1016/j.neuroscience.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Charney DS, Woods SW, Goodman WK, et al. Neurobiological mechanisms of panic anxiety: biochemical and behavioral correlates of yohimbine-induced panic attacks. Am J Psychiatry. 1987;144(8):1030–6. doi: 10.1176/ajp.144.8.1030. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka M, Yoshida M, Emoto H, et al. Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: basic studies. Eur J Pharmacol. 2000;405(1-3):397–406. doi: 10.1016/s0014-2999(00)00569-0. [DOI] [PubMed] [Google Scholar]
- 33.Geracioti TD, Jr., Baker DG, Ekhator NN, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry. 2001;158(8):1227–30. doi: 10.1176/appi.ajp.158.8.1227. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y, Ishitobi Y, Maruyama Y, et al. Salivary alpha-amylase and cortisol responsiveness following electrical stimulation stress in panic disorder patients. Neurosci Res. 2012;73(1):80–4. doi: 10.1016/j.neures.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Debiec J, Bush DE, LeDoux JE. Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats: a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety. 2011;28(3):186–93. doi: 10.1002/da.20803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onur OA, Walter H, Schlaepfer TE, et al. Noradrenergic enhancement of amygdala responses to fear. Soc Cogn Affect Neurosci. 2009;4(2):119–26. doi: 10.1093/scan/nsn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miner LH, Jedema HP, Moore FW, et al. Chronic stress increases the plasmalemmal distribution of the norepinephrine transporter and the coexpression of tyrosine hydroxylase in norepinephrine axons in the prefrontal cortex. J Neuroscience. 2006;26(5):1571–8. doi: 10.1523/JNEUROSCI.4450-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420(6911):70–4. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 39.Morgan CA, 3rd, Southwick SM, Grillon C, et al. Yohimbine-facilitated acoustic startle reflex in humans. Psychopharmacology. 1993;110(3):342–6. doi: 10.1007/BF02251291. [DOI] [PubMed] [Google Scholar]
- 40.Bremner JD, Innis RB, Ng CK, et al. Positron emission tomography measurement of cerebral metabolic correlates of yohimbine administration in combat-related posttraumatic stress disorder. Arch Gen Psychiatry. 1997;54(3):246–54. doi: 10.1001/archpsyc.1997.01830150070011. [DOI] [PubMed] [Google Scholar]
- 41.Southwick SM, Krystal JH, Bremner JD, et al. Noradrenergic and serotonergic function in posttraumatic stress disorder. Arch Gen Psychiatry. 1997;54(8):749–58. doi: 10.1001/archpsyc.1997.01830200083012. [DOI] [PubMed] [Google Scholar]
- 42.Cohen H, Kaplan Z, Koresh O, et al. Early post-stressor intervention with propranolol is ineffective in preventing posttraumatic stress responses in an animal model for PTSD. Eur Neuropsychopharmacol. 2011;21(3):230–40. doi: 10.1016/j.euroneuro.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 43.Muravieva EV, Alberini CM. Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem. 2010;17(6):306–13. doi: 10.1101/lm.1794710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pitman RK, Sanders KM, Zusman RM, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry. 2002;51(2):189–92. doi: 10.1016/s0006-3223(01)01279-3. [DOI] [PubMed] [Google Scholar]
- 45.Tarsitani L, De Santis V, Mistretta M, et al. Treatment with beta-blockers and incidence of post-traumatic stress disorder after cardiac surgery: a prospective observational study. J Cardiothorac Vasc Anesth. 2012;26(2):265–9. doi: 10.1053/j.jvca.2011.09.011. [DOI] [PubMed] [Google Scholar]
- 46.Stein MB, Kerridge C, Dimsdale JE, et al. Pharmacotherapy to prevent PTSD: results from a randomized controlled proof-of-concept trial in physically injured patients. J Trauma Stress. 2007;20(6):923–32. doi: 10.1002/jts.20270. [DOI] [PubMed] [Google Scholar]
- 47.Hoge EA, Worthington JJ, Nagurney JT, et al. Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci Ther. 2012;18(1):21–7. doi: 10.1111/j.1755-5949.2010.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- 49.Ordway GA, Stockmeier C, Cason GW, et al. Pharmacology and distribution of norepinephrine transporters in the human locus coeruleus and raphe nuclei. J Neuroscience. 1997;17(21):8451–8458. doi: 10.1523/JNEUROSCI.17-05-01710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat Rev Neurosci. 2003;4(1):13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 51.Moron JA, Brockington A, Wise RA, et al. Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neuroscience. 2002;22(2):389–95. doi: 10.1523/JNEUROSCI.22-02-00389.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liprando LA, Miner LH, Blakely RD, et al. Ultrastructural interactions between terminals expressing the norepinephrine transporter and dopamine neurons in the rat and monkey ventral tegmental area. Synapse. 2004;52(4):233–44. doi: 10.1002/syn.20023. [DOI] [PubMed] [Google Scholar]
- 53.Arnsten AF, Li BM. Neurobiology of executive functions: catecholamine influences on prefrontal cortical functions. Biol Psychiatry. 2005;57(11):1377–84. doi: 10.1016/j.biopsych.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 54.Rusnak M, Kvetnansky R, Jelokova J, et al. Effect of novel stressors on gene expression of tyrosine hydroxylase and monoamine transporters in brainstem noradrenergic neurons of long-term repeatedly immobilized rats. Brain Res. 2001;899(1-2):20–35. doi: 10.1016/s0006-8993(01)02126-6. [DOI] [PubMed] [Google Scholar]
- 55.Dazzi L, Seu E, Cherchi G, et al. Antagonism of the stress-induced increase in cortical norepinephrine output by the selective norepinephrine reuptake inhibitor reboxetine. Eur J Pharmacol. 2003;476(1-2):55–61. doi: 10.1016/s0014-2999(03)02130-7. [DOI] [PubMed] [Google Scholar]
- 56.Goddard AW, Ball SG, Martinez J, et al. Current perspectives of the roles of the central norepinephrine system in anxiety and depression. Depress Anxiety. 2010;27(4):339–50. doi: 10.1002/da.20642. [DOI] [PubMed] [Google Scholar]
- 57.Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proc Natl Acad Sci U S A. 2000;97(1):325–30. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petrakis IL, Ralevski E, Desai N, et al. Noradrenergic vs serotonergic antidepressant with or without naltrexone for veterans with PTSD and comorbid alcohol dependence. Neuropsychopharmacology. 2012;37(4):996–1004. doi: 10.1038/npp.2011.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barrett EL, Mills KL, Teesson M. Hurt people who hurt people: violence amongst individuals with comorbid substance use disorder and post traumatic stress disorder. Addict Behav. 2011;36(7):721–8. doi: 10.1016/j.addbeh.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 60.Marshall-Berenz EC, Vujanovic AA, Macpherson L. Impulsivity and alcohol use coping motives in a trauma-exposed sample: the mediating role of distress tolerance. Pers Individ Dif. 2011;50(5):588–592. doi: 10.1016/j.paid.2010.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss NH, Tull MT, Viana AG, et al. Impulsive behaviors as an emotion regulation strategy: examining associations between PTSD, emotion dysregulation, and impulsive behaviors among substance dependent inpatients. J Anxiety Disord. 2012;26(3):453–8. doi: 10.1016/j.janxdis.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chamberlain SR, Hampshire A, Muller U, et al. Atomoxetine modulates right inferior frontal activation during inhibitory control: a pharmacological functional magnetic resonance imaging study. Biol Psychiatry. 2009;65(7):550–5. doi: 10.1016/j.biopsych.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 63.Gabriel A, Violato C. Adjunctive atomoxetine to SSRIs or SNRIs in the treatment of adult ADHD patients with comorbid partially responsive generalized anxiety (GA): an open-label study. Atten Defic Hyperact Disord. 2011;3(4):319–26. doi: 10.1007/s12402-011-0063-1. [DOI] [PubMed] [Google Scholar]
- 64.Spencer TJ, Faraone SV, Michelson D, et al. Atomoxetine and adult attention-deficit/hyperactivity disorder: the effects of comorbidity. J Clin Psychiatry. 2006;67(3):415–20. doi: 10.4088/jcp.v67n0312. [DOI] [PubMed] [Google Scholar]
- 65.Davidson J, Baldwin D, Stein DJ, et al. Treatment of posttraumatic stress disorder with venlafaxine extended release: a 6-month randomized controlled trial. Arch Gen Psychiatry. 2006;63(10):1158–65. doi: 10.1001/archpsyc.63.10.1158. [DOI] [PubMed] [Google Scholar]
- 66.Davidson J, Rothbaum BO, Tucker P, et al. Venlafaxine extended release in posttraumatic stress disorder: a sertraline- and placebo-controlled study. J Clin Psychopharmacol. 2006;26(3):259–67. doi: 10.1097/01.jcp.0000222514.71390.c1. [DOI] [PubMed] [Google Scholar]
- 67.Yang CH, Shi HS, Zhu WL, et al. Venlafaxine facilitates between-session extinction and prevents reinstatement of auditory-cue conditioned fear. Behav Brain Res. 2012;230(1):268–73. doi: 10.1016/j.bbr.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 68.Hannon J, Hoyer D. Molecular biology of 5-HT receptors. Behav Brain Res. 2008;195(1):198–213. doi: 10.1016/j.bbr.2008.03.020. [DOI] [PubMed] [Google Scholar]
- 69.Cools R, Roberts AC, Robbins TW. Serotoninergic regulation of emotional and behavioural control processes. Trends Cogn Sci. 2008;12(1):31–40. doi: 10.1016/j.tics.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 70.Murrough JW, Neumeister A. The serotonin 1B receptor: a new target for depression therapeutics? Biol Psychiatry. 2011;69(8):714–5. doi: 10.1016/j.biopsych.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 71.Carr GV, Lucki I. The role of serotonin receptor subtypes in treating depression: a review of animal studies. Psychopharmacology (Berl) 2011;213(2-3):265–87. doi: 10.1007/s00213-010-2097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Furay AR, Neumaier JF, Mullenix AT, et al. Overexpression of 5-HT(1B) mRNA in nucleus accumbens shell projection neurons differentially affects microarchitecture of initiation and maintenance of ethanol consumption. Alcohol. 2011;45(1):19–32. doi: 10.1016/j.alcohol.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu J, Henry S, Gallezot JD, et al. Serotonin 1B receptor imaging in alcohol dependence. Biol Psychiatry. 2010;67(9):800–3. doi: 10.1016/j.biopsych.2009.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murrough JW, Czermak C, Henry S, et al. The effect of early trauma exposure on serotonin type 1B receptor expression revealed by reduced selective radioligand binding. Arch Gen Psychiatry. 2011;68(9):892–900. doi: 10.1001/archgenpsychiatry.2011.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie P, Kranzler HR, Poling J, et al. Interactive effect of stressful life events and the serotonin transporter 5-HTTLPR genotype on posttraumatic stress disorder diagnosis in 2 independent populations. Arch Gen Psychiatry. 2009;66(11):1201–9. doi: 10.1001/archgenpsychiatry.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zanoveli JM, Carvalho MC, Cunha JM, et al. Extracellular serotonin level in the basolateral nucleus of the amygdala and dorsal periaqueductal gray under unconditioned and conditioned fear states: an in vivo microdialysis study. Brain Res. 2009;1294:106–15. doi: 10.1016/j.brainres.2009.07.074. [DOI] [PubMed] [Google Scholar]
- 77.Muller JM, Morelli E, Ansorge M, et al. Serotonin transporter deficient mice are vulnerable to escape deficits following inescapable shocks. Genes Brain Behav. 2011;10(2):166–75. doi: 10.1111/j.1601-183X.2010.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390(6660):604–7. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 79.Rodrigues SM, Schafe GE, LeDoux JE. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron. 2004;44(1):75–91. doi: 10.1016/j.neuron.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 80.Wellman CL, Izquierdo A, Garrett JE, et al. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neuroscience. 2007;27(3):684–91. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krystal JH, Webb E, Cooney NL, et al. Serotonergic and noradrenergic dysregulation in alcoholism: m-chlorophenylpiperazine and yohimbine effects in recently detoxified alcoholics and healthy comparison subjects. Am J Psychiatry. 1996;153(1):83–92. doi: 10.1176/ajp.153.1.83. [DOI] [PubMed] [Google Scholar]
- 82.Price LH, Malison RT, McDougle CJ, et al. Neurobiology of tryptophan depletion in depression: effects of m-chlorophenylpiperazine (mCPP) Neuropsychopharmacology. 1997;17(5):342–50. doi: 10.1016/S0893-133X(97)00084-5. [DOI] [PubMed] [Google Scholar]
- 83.Spinelli S, Chefer S, Carson RE, et al. Effects of early-life stress on serotonin(1A) receptors in juvenile Rhesus monkeys measured by positron emission tomography. Biol Psychiatry. 2010;67(12):1146–53. doi: 10.1016/j.biopsych.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McDevitt RA, Hiroi R, Mackenzie SM, et al. Serotonin 1B autoreceptors originating in the caudal dorsal raphe nucleus reduce expression of fear and depression-like behavior. Biol Psychiatry. 2011;69(8):780–7. doi: 10.1016/j.biopsych.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Azmitia EC, Whitaker-Azmitia PM. Awakening the sleeping giant: anatomy and plasticity of the brain serotonergic system. J Clin Psychiatry. 1991;52(Suppl.):4–16. [PubMed] [Google Scholar]
- 86.Frazer A, Hensler JG. 5-HT1A receptors and 5-HT1A-mediated responses: effect on treatments that modify serotonergic neurotransmission. In: Whitaker-Azmitia PM, Peroutka SJ, editors. The neuropharmacology of serotonin. The New York Academy of Sciences; New York: 1990. pp. 460–475. [DOI] [PubMed] [Google Scholar]
- 87.Palacios J, Pazos A, Hoyer D. Characterisation and mapping of 5-HT1A receptors in animals and in man. In: Dourish C, Ahlenius S, Hutson P, editors. Brain 5-HT1A receptors: behavioural and neurochemical pharmacology. Ellis Horwood; Chichester: 1987. Chapter 6. [Google Scholar]
- 88.Pazos A, Probst A, Palacios JM. Serotonin receptors in the human brain: III. Autoradiographic mapping of serotonin-1 receptors. Neuroscience. 1987;21(1):97–122. doi: 10.1016/0306-4522(87)90326-5. [DOI] [PubMed] [Google Scholar]
- 89.Whitaker-Azmitia PM, Clarke C, Azmitia EC. Localization of 5-HT1A receptors to astroglial cells in adult rats: implications for neuronal-glial interactions and psychoactive drug mechanism of action. Synapse. 1993;14(3):201–5. doi: 10.1002/syn.890140303. [DOI] [PubMed] [Google Scholar]
- 90.Whitaker-Azmitia PM, Azmitia EC. Stimulation of astroglial serotonin receptors produces culture media which regulates growth of serotonergic neurons. Brain Res. 1989;497(1):80–5. doi: 10.1016/0006-8993(89)90972-4. [DOI] [PubMed] [Google Scholar]
- 91.Azmitia EC. Serotonin neurons, neuroplasticity, and homeostasis of neural tissue. Neuropsychopharmacology. 1999;21(2 Suppl.):33S–45S. doi: 10.1016/S0893-133X(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 92.McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–22. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 93.Drevets WC, Frank E, Price JC, et al. Serotonin type-1A receptor imaging in depression. Nucl Med Biol. 2000;27(5):499–507. doi: 10.1016/s0969-8051(00)00119-0. [DOI] [PubMed] [Google Scholar]
- 94.Ramboz S, Oosting R, Amara DA, et al. Serotonin receptor 1A knockout: an animal model of anxiety-related disorder. Proc Nat Acad Sci. 1998;95:14476–14481. doi: 10.1073/pnas.95.24.14476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Solati J, Salari AA, Bakhtiari A. 5HT(1A) and 5HT(1B) receptors of medial prefrontal cortex modulate anxiogenic-like behaviors in rats. Neurosci Lett. 2011;504(3):325–9. doi: 10.1016/j.neulet.2011.09.058. [DOI] [PubMed] [Google Scholar]
- 96.Sari Y. Serotonin1B receptors: from protein to physiological function and behavior. Neurosci Biobehav Rev. 2004;28(6):565–82. doi: 10.1016/j.neubiorev.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 97.Bramley JR, Sollars PJ, Pickard GE, et al. HT1B receptor-mediated presynaptic inhibition of GABA release in the suprachiasmatic nucleus. J Neurophysiol. 2005;93(6):3157–64. doi: 10.1152/jn.00770.2004. [DOI] [PubMed] [Google Scholar]
- 98.Haddjeri N, de Montigny C, Blier P. Modulation of the firing activity of noradrenergic neurones in the rat locus coeruleus by the 5-hydroxtryptamine system. Br J Pharmacol. 1997;120(5):865–75. doi: 10.1038/sj.bjp.0700968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yan QS, Zheng SZ, Feng MJ, et al. Involvement of 5-HT1B receptors within the ventral tegmental area in ethanol-induced increases in mesolimbic dopaminergic transmission. Brain Res. 2005;1060(1-2):126–37. doi: 10.1016/j.brainres.2005.08.051. [DOI] [PubMed] [Google Scholar]
- 100.File SE, Kenny PJ, Cheeta S. The role of the dorsal hippocampal serotonergic and cholinergic systems in the modulation of anxiety. Pharmacol Biochem Behav. 2000;66(1):65–72. doi: 10.1016/s0091-3057(00)00198-2. [DOI] [PubMed] [Google Scholar]
- 101.Bonaventure P, Langlois X, Leysen JE. Co-localization of 5-HT1B- and 5-HT1D receptor mRNA in serotonergic cell bodies in guinea pig dorsal raphe nucleus: a double labeling in situ hybridization histochemistry study. Neurosci Lett. 1998;254(2):113–6. doi: 10.1016/s0304-3940(98)00680-6. [DOI] [PubMed] [Google Scholar]
- 102.Ruf BM, Bhagwagar Z. The 5-HT1B receptor: a novel target for the pathophysiology of depression. Curr Drug Targets. 2009;10(11):1118–38. doi: 10.2174/138945009789735192. [DOI] [PubMed] [Google Scholar]
- 103.Broocks A, Meyer T, Opitz M, et al. HT1A responsivity in patients with panic disorder before and after treatment with aerobic exercise, clomipramine or placebo. Eur Neuropsychopharmacol. 2003;13(3):153–64. doi: 10.1016/s0924-977x(02)00177-3. [DOI] [PubMed] [Google Scholar]
- 104.McAllister-Williams RH, Massey AE, Fairchild G. Repeated cortisol administration attenuates the EEG response to buspirone in healthy volunteers: evidence for desensitization of the 5-HT1A autoreceptor. J Psychopharmacol. 2007;21(8):826–32. doi: 10.1177/0269881107078292. [DOI] [PubMed] [Google Scholar]
- 105.Ramey T, Stiger T, Banerjee A, et al. Methodology of time to onset of response characterization in the proof of concept trial for a combination drug in MDD. 9th Annual Scientific Meeting plus Research-To-Policy Forum, International Society for CNS Clinical Trials and Methodology; San Diego (CA). 2009 Oct 5-6. [Google Scholar]
- 106.Krebs-Kraft DL, Hill MN, Hillard CJ, et al. Sex difference in cell proliferation in developing rat amygdala mediated by endocannabinoids has implications for social behavior. Proc Natl Acad Sci U S A. 107(47):20535–40. doi: 10.1073/pnas.1005003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hill MN, McLaughlin RJ, Bingham B, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 107(20):9406–11. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rademacher DJ, Meier SE, Shi L, et al. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54(1):108–16. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 109.Reich CG, Taylor ME, McCarthy MM. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav Brain Res. 2009;203(2):264–9. doi: 10.1016/j.bbr.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gorzalka BB, Hill MN, Hillard CJ. Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci Biobehav Rev. 2008;32(6):1152–60. doi: 10.1016/j.neubiorev.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 111.Hill MN, Miller GE, Carrier EJ, et al. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology. 2009;34(8):1257–62. doi: 10.1016/j.psyneuen.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hill MN, Miller GE, Ho WS, et al. Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry. 2008;41(2):48–53. doi: 10.1055/s-2007-993211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hill MN, Patel S, Carrier EJ, et al. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30(3):508–15. doi: 10.1038/sj.npp.1300601. [DOI] [PubMed] [Google Scholar]
- 114.Kathuria S, Gaetani S, Fegley D, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9(1):76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 115.Hill MN, McLaughlin RJ, Morrish AC, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34(13):2733–45. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Devane WA, Dysarz FA, 3rd, Johnson MR, et al. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharm. 1988;34(5):605–13. [PubMed] [Google Scholar]
- 117.Hill MN. Introduction to the special issue on stress, emotional behavior, and the endocannabinoid system: a decade of research. Neuroscience. 2012;204:1–4. doi: 10.1016/j.neuroscience.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 118.Viveros MP, Marco EM, File SE. Endocannabinoid system and stress and anxiety responses. Pharmacol Biochem Behav. 2005;81(2):331–42. doi: 10.1016/j.pbb.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 119.Martin M, Ledent C, Parmentier M, et al. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berl) 2002;159(4):379–87. doi: 10.1007/s00213-001-0946-5. [DOI] [PubMed] [Google Scholar]
- 120.Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58(4):315–48. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 121.Marsicano G, Wotjak CT, Azad SC, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418(6897):530–4. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 122.Varvel SA, Wise LE, Niyuhire F, et al. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32(5):1032–41. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- 123.Chhatwal JP, Davis M, Maguschak KA, et al. Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology. 2005;30(3):516–24. doi: 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 124.Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77(2):299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- 125.Herkenham M, Lynn AB, Little MD, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87(5):1932–6. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haller J, Bakos N, Szirmay M, et al. The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neuroscience. 2002;16(7):1395–8. doi: 10.1046/j.1460-9568.2002.02192.x. [DOI] [PubMed] [Google Scholar]
- 127.Haller J, Varga B, Ledent C, et al. CB1 cannabinoid receptors mediate anxiolytic effects: convergent genetic and pharmacological evidence with CB1-specific agents. Behav Pharmacol. 2004;15(4):299–304. doi: 10.1097/01.fbp.0000135704.56422.40. [DOI] [PubMed] [Google Scholar]
- 128.Viveros MP, Llorente R, Lopez-Gallardo M, et al. Sex-dependent alterations in response to maternal deprivation in rats. Psychoneuroendocrinology. 2009;34(Suppl. 1):S217–26. doi: 10.1016/j.psyneuen.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 129.Hill MN, Carrier EJ, McLaughlin RJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008;106(6):2322–36. doi: 10.1111/j.1471-4159.2008.05567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Suarez J, Llorente R, Romero-Zerbo SY, et al. Early maternal deprivation induces gender-dependent changes on the expression of hippocampal CB(1) and CB(2) cannabinoid receptors of neonatal rats. Hippocampus. 2009;19(7):623–32. doi: 10.1002/hipo.20537. [DOI] [PubMed] [Google Scholar]
- 131.Palanza P. Animal models of anxiety and depression: how are females different? Neurosci Biobehav Rev. 2001;25(3):219–33. doi: 10.1016/s0149-7634(01)00010-0. [DOI] [PubMed] [Google Scholar]
- 132.Dell’osso L, Carmassi C, Massimetti G, et al. Full and partial PTSD among young adult survivors 10 months after the L’Aquila 2009 earthquake: gender differences. J Affect Disord. 2011;131(1-3):79–83. doi: 10.1016/j.jad.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 133.Freedy JR, Magruder KM, Mainous AG, et al. Gender differences in traumatic event exposure and mental health among veteran primary care patients. Mil Med. 2010;175(10):750–8. doi: 10.7205/milmed-d-10-00123. [DOI] [PubMed] [Google Scholar]
- 134.Irish LA, Fischer B, Fallon W, et al. Gender differences in PTSD symptoms: an exploration of peritraumatic mechanisms. J Anxiety Disord. 2011;25(2):209–16. doi: 10.1016/j.janxdis.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Luxton DD, Skopp NA, Maguen S. Gender differences in depression and PTSD symptoms following combat exposure. Depress Anxiety. 2010;27(11):1027–33. doi: 10.1002/da.20730. [DOI] [PubMed] [Google Scholar]
- 136.Ditlevsen DN, Elklit A. The combined effect of gender and age on post traumatic stress disorder: do men and women show differences in the lifespan distribution of the disorder? Ann Gen Psychiatry. 2010;9:32. doi: 10.1186/1744-859X-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bowler RM, Han H, Gocheva V, et al. Gender differences in probable posttraumatic stress disorder among police responders to the 2001 World Trade Center terrorist attack. Am J Ind Med. 2010;53(12):1186–96. doi: 10.1002/ajim.20876. [DOI] [PubMed] [Google Scholar]
- 138.Galovski TE, Mott J, Young-Xu Y, et al. Gender differences in the clinical presentation of PTSD and its concomitants in survivors of interpersonal assault. J Interpers Violence. 2011;26(4):789–806. doi: 10.1177/0886260510365865. [DOI] [PubMed] [Google Scholar]
- 139.Breslau N, Anthony JC. Gender differences in the sensitivity to posttraumatic stress disorder: an epidemiological study of urban young adults. J Abnorm Psychol. 2007;116(3):607–11. doi: 10.1037/0021-843X.116.3.607. [DOI] [PubMed] [Google Scholar]
- 140.Breslau N. Gender differences in trauma and posttraumatic stress disorder. J Gend Specif Med. 2002;5(1):34–40. [PubMed] [Google Scholar]
- 141.Stein MB, Walker JR, Forde DR. Gender differences in susceptibility to posttraumatic stress disorder. Behav Res Ther. 2000;38(6):619–28. doi: 10.1016/s0005-7967(99)00098-4. [DOI] [PubMed] [Google Scholar]
- 142.Pardini M, Krueger F, Koenigs M, et al. Fatty-acid amide hydrolase polymorphisms and post-traumatic stress disorder after penetrating brain injury. Translational Psychiatry. 2012;2:e75. doi: 10.1038/tp.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lutz B. The endocannabinoid system and extinction learning. Mol Neurobiol. 2007;36:92–101. doi: 10.1007/s12035-007-8004-x. [DOI] [PubMed] [Google Scholar]
- 144.Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16. doi: 10.1016/j.neuroscience.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychol. 2005;69(1):113–32. doi: 10.1016/j.biopsycho.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 146.Chrousos GP, Gold PW. The concepts of stress and stress system disorders: overview of physical and behavioral homeostasis. JAMA. 1992;267(9):1244–52. [PubMed] [Google Scholar]
- 147.Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25(1):1–35. doi: 10.1016/s0306-4530(99)00035-9. [DOI] [PubMed] [Google Scholar]
- 148.Heim C, Newport DJ, Heit S, et al. Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA. 2000;284(5):592–7. doi: 10.1001/jama.284.5.592. [DOI] [PubMed] [Google Scholar]
- 149.Holsboer F. Psychiatric implications of altered limbic-hypothalamic-pituitary-adrenocortical activity. Eur Arch Psychiatry Neurol Sci. 1989;238(5-6):302–22. doi: 10.1007/BF00449812. [DOI] [PubMed] [Google Scholar]
- 150.Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554–65. doi: 10.1176/appi.ajp.160.9.1554. [DOI] [PubMed] [Google Scholar]
- 151.Stratakis CA, Chrousos GP. Neuroendocrinology and pathophysiology of the stress system. Ann New York Acad Sci. 1995;771:1–18. doi: 10.1111/j.1749-6632.1995.tb44666.x. [DOI] [PubMed] [Google Scholar]
- 152.Tsigos C, Chrousos GP. Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulation in psychiatric and autoimmune disorders. Endocrinol Metab Clin North Am. 1994;23(3):451–66. [PubMed] [Google Scholar]
- 153.Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53(4):865–71. doi: 10.1016/s0022-3999(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 154.Young EA. Sex differences and the HPA axis: implications for psychiatric disease. JGSM. 1998;1(1):21–7. [PubMed] [Google Scholar]
- 155.Hubbard DT, Nakashima BR, Lee I, et al. Activation of basolateral amygdala corticotropin-releasing factor 1 receptors modulates the consolidation of contextual fear. Neuroscience. 2007;150(4):818–28. doi: 10.1016/j.neuroscience.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Roozendaal B, Schelling G, McGaugh JL. Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the beta-adrenoceptor-cAMP pathway: dependence on glucocorticoid receptor activation. J Neuroscience. 2008;28(26):6642–51. doi: 10.1523/JNEUROSCI.1336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adamec R, Fougere D, Risbrough V. CRF receptor blockade prevents initiation and consolidation of stress effects on affect in the predator stress model of PTSD. Int J Neuropsychopharmacol. 2010;13(6):747–57. doi: 10.1017/S1461145709990496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Steckler T, Risbrough V. Pharmacological treatment of PTSD:-established and new approaches. Neuropharmacology. 2012;62(2):617–27. doi: 10.1016/j.neuropharm.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bale TL, Contarino A, Smith GW, et al. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nature Gen. 2000;24(4):410–4. doi: 10.1038/74263. [DOI] [PubMed] [Google Scholar]
- 160.Bale TL, Picetti R, Contarino A, et al. Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior. J Neuroscience. 2002;22(1):193–9. doi: 10.1523/JNEUROSCI.22-01-00193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Coste SC, Kesterson RA, Heldwein KA, et al. Abnormal adaptations to stress and impaired cardiovascular function in mice lacking corticotropin-releasing hormone receptor-2. Nature Gen. 2000;24(4):403–9. doi: 10.1038/74255. [DOI] [PubMed] [Google Scholar]
- 162.Baker DG, West SA, Nicholson WE, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am J Psychiatry. 1999;156(4):585–8. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- 163.Bremner JD, Licinio J, Darnell A, et al. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am J Psychiatry. 1997;154(5):624–9. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ressler KJ, Mercer KB, Bradley B, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–7. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Baker DG, Nievergelt CM, Risbrough VB. Post-traumatic stress disorder: emerging concepts of pharmacotherapy. Expert Opin Emerg Drugs. 2009;14(2):251–72. doi: 10.1517/14728210902972494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zobel AW, Nickel T, Kunzel HE, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34(3):171–81. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 167.Binneman B, Feltner D, Kolluri S, et al. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. 2008;165(5):617–20. doi: 10.1176/appi.ajp.2008.07071199. [DOI] [PubMed] [Google Scholar]
- 168.Patel S, Roelke CT, Rademacher DJ, et al. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145(12):5431–8. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- 169.Dhawan BN, Cesselin F, Raghubir R, et al. International Union of Pharmacology: XII. Classification of opioid receptors. Pharmacol Rev. 1996;48(4):567–92. [PubMed] [Google Scholar]
- 170.Mello NK, Negus SS. Interactions between kappa opioid agonists and cocaine: preclinical studies. Ann N Y Acad Sci. 2000;909:104–32. doi: 10.1111/j.1749-6632.2000.tb06678.x. [DOI] [PubMed] [Google Scholar]
- 171.de Lanerolle NC, Williamson A, Meredith C, et al. Dynorphin and the kappa 1 ligand [3H]U69,593 binding in the human epileptogenic hippocampus. Epilepsy Res. 1997;28(3):189–205. doi: 10.1016/s0920-1211(97)00044-2. [DOI] [PubMed] [Google Scholar]
- 172.Chappell PB, Leckman JF, Scahill LD, et al. Neuroendocrine and behavioral effects of the selective kappa agonist spiradoline in Tourette’s syndrome: a pilot study. Psychiatry Res. 1993;47(3):267–80. doi: 10.1016/0165-1781(93)90084-t. [DOI] [PubMed] [Google Scholar]
- 173.Mathieu-Kia AM, Fan LQ, Kreek MJ, et al. Mu-, delta- and kappa-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer’s disease patients. Brain Res. 2001;893(1-2):121–34. doi: 10.1016/s0006-8993(00)03302-3. [DOI] [PubMed] [Google Scholar]
- 174.Sauriyal DS, Jaggi AS, Singh N. Extending pharmacological spectrum of opioids beyond analgesia: multifunctional aspects in different pathophysiological states. Neuropeptides. 2011;45(3):175–88. doi: 10.1016/j.npep.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 175.Hiller JM, Fan LQ. Laminar distribution of the multiple opioid receptors in the human cerebral cortex. Neurochem Res. 1996;21(11):1333–45. doi: 10.1007/BF02532374. [DOI] [PubMed] [Google Scholar]
- 176.Simonin F, Gaveriaux-Ruff C, Befort K, et al. kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci U S A. 1995;92(15):7006–10. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bruchas MR, Land BB, Lemos JC, et al. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One. 2009;4(12):e8528. doi: 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Knoll AT, Carlezon WA., Jr. Dynorphin, stress, and depression. Brain Res. 1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Carr GV, Bangasser DA, Bethea T, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in Wistar Kyoto rats. Neuropsychopharmacology. 2010;35(3):752–63. doi: 10.1038/npp.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Friedman MJ, Resick PA, Bryant RA, et al. Considering PTSD for DSM-5. Depress Anxiety. 2011;28(9):750–69. doi: 10.1002/da.20767. [DOI] [PubMed] [Google Scholar]
- 181.Berger W, Mendlowicz MV, Marques-Portella C, et al. Pharmacologic alternatives to antidepressants in posttraumatic stress disorder: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33(2):169–80. doi: 10.1016/j.pnpbp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for post traumatic stress disorder (PTSD) Cochrane Database Syst Rev. 2006;(1):CD002795. doi: 10.1002/14651858.CD002795.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Knoll AT, Meloni EG, Thomas JB, et al. Anxiolytic-like effects of kappa-opioid receptor antagonists in models of unlearned and learned fear in rats. J Pharmacol Exp Ther. 2007;323(3):838–45. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- 184.Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167(6):648–62. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wuest J, Merritt-Gray M, Ford-Gilboe M, et al. Chronic pain in women survivors of intimate partner violence. J Pain. 2008;9(11):1049–57. doi: 10.1016/j.jpain.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 186.Schiff M, Levit S, Cohen-Moreno R. Childhood sexual abuse, post-traumatic stress disorder, and use of heroin among female clients in Israeli methadone maintenance treatment programs (MMTPS) Soc Work Health Care. 2010;49(9):799–813. doi: 10.1080/00981381003745103. [DOI] [PubMed] [Google Scholar]
- 187.Holbrook TL, Galarneau MR, Dye JL, et al. Morphine use after combat injury in Iraq and post-traumatic stress disorder. N Engl J Med. 2010;362(2):110–7. doi: 10.1056/NEJMoa0903326. [DOI] [PubMed] [Google Scholar]
- 188.Stoddard FJ, Jr., Sorrentino EA, Ceranoglu TA, et al. Preliminary evidence for the effects of morphine on posttraumatic stress disorder symptoms in one- to four-year-olds with burns. J Burn Care Res. 2009;30(5):836–43. doi: 10.1097/BCR.0b013e3181b48102. [DOI] [PubMed] [Google Scholar]
- 189.Nixon RD, Nehmy TJ, Ellis AA, et al. Predictors of posttraumatic stress in children following injury: the influence of appraisals, heart rate, and morphine use. Behav Res Ther. 2010;48(8):810–5. doi: 10.1016/j.brat.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 190.Bryant RA, Creamer M, O’Donnell M, et al. A study of the protective function of acute morphine administration on subsequent posttraumatic stress disorder. Biol Psychiatry. 2009;65(5):438–40. doi: 10.1016/j.biopsych.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 191.Henriksen G, Willoch F. Imaging of opioid receptors in the central nervous system. Brain. 2008;131(Pt 5):1171–96. doi: 10.1093/brain/awm255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cukor J, Spitalnick J, Difede J, et al. Emerging treatments for PTSD. Clin Psychol Rev. 2009;29(8):715–26. doi: 10.1016/j.cpr.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 193.Pietrzak RH, Henry S, Southwick SM, et al. Linking in vivo brain serotonin type 1B receptor density to phenotypic heterogeneity of posttraumatic stress symptomatology. Mol Psychiatry. doi: 10.1038/mp.2012.60. Epub 2012 May 15. [DOI] [PubMed] [Google Scholar]
- 194.Pietrzak R, Gallezot J-D, Ding Y-S, et al. Posttraumatic stress disorder is associated with reduced in vivo norepinephrine transporter availability in the locus coeruleus. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2013.399. In press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]