

Abstract
Xanthurenic acid (XA), an endogenous kynurenine, is a known vesicular glutamate transport (VGLUT) inhibitor and has also been proposed as an mGlu2/3 receptor agonist. Changes in these systems have been implicated in the pathophysiology of schizophrenia and other psychiatric disorders; however, little is known of how XA affects synaptic transmission. We therefore investigated the effects of XA on synaptic transmission at two hippocampal glutamatergic pathways and evaluated the ability of XA to bind to mGlu2/3 receptors. Field excitatory postsynaptic potentials (fEPSPs) were recorded from either the dentate gyrus (DG) or CA1 region of mouse hippocampal slices in vitro. Addition of XA to the bathing medium (1–10 mM) resulted in a dose-related reduction of fEPSP amplitudes (up to 52% reduction) in both hippocampal regions. In the DG, the VGLUT inhibitors Congo Red and Rose Bengal, and the mGlu2/3 agonist LY354740, also reduced fEPSPs (up to 80% reduction). The mGlu2/3 antagonist LY341495 reversed the LY354740 effect, but not the XA effect. LY354740, but not XA, also reduced DG paired-pulse depression. XA had no effect on specific binding of 1 nM [3H]LY341495 to membranes with human mGlu2 receptors. We conclude that XA can modulate synaptic transmission via a mechanism that may involve VGLUT inhibition rather than activation of mGlu2/3 receptors. This could be important in the pathophysiology of nervous system disorders including schizophrenia and might represent a target for developing novel pharmacological therapies.
Keywords: metabotropic glutamate receptors, mGlu2, schizophrenia, synaptic transmission, inhibition, vesicular glutamate transport (VGLUT)
INTRODUCTION
The kynurenine pathway represents a major route of metabolism for the amino acid tryptophan and leads to production of a number of neuromodulatory compounds (Schwarcz et al, 2012; Stone, 1993). Numerous studies have demonstrated modulation of neuronal excitability by the kynurenines, with early evidence indicating that quinolinic acid and related compounds induce convulsions in rodents (Lapin, 1978a, 1978b) and subsequent studies indicating that kynurenines modulate both normal neuronal function and neuropathophysiological mechanisms (Stone, 1993; Schwarcz et al, 2012; Myint, 2012).
Tryptophan metabolism and kynurenine production were suggested to have a role in psychiatric disorders in the 1960s (Lapin and Oxenkrug, 1969) and disruption of these pathways has subsequently been implicated in schizophrenia and other central nervous system (CNS) disorders (Schwarcz et al, 2012; Myint, 2012). It is of interest that several kynurenines modulate glutamate receptor function and synaptic activity (Stone, 1993) and that altered glutamatergic synaptic function is believed to underlie some of the symptoms of schizophrenia: modulation of N-methyl-D-aspartate (NMDA) receptor function by antagonists such as ketamine and phencyclidine exacerbate positive, negative, and cognitive symptoms, and can induce schizophrenia-like symptoms in normal individuals (Coyle et al, 2003; Krystal et al, 1994; Millan, 2005). There is also evidence of abnormal expression of a number of proteins associated with both the release of neurotransmitter, such as the vesicular glutamate transporter (VGLUT) family, and the activity of postsynaptic receptors, such as the NMDA receptor (Hahn et al, 2006; Maher and LoTurco, 2012; Eastwood and Harrison, 2005; Varea et al, 2012; Uezato et al, 2009; Oni-Orisan et al, 2008; Smith et al, 2001; Sodhi et al, 2011). It is therefore tempting to speculate that abnormal activity of the kynurenine pathway reported in schizophrenia may lead to altered glutamatergic synaptic transmission, thereby contributing to the neuropathophysiology of schizophrenia and other CNS disorders (Schwarcz et al, 2012; Myint, 2012).
Xanthurenic acid (XA) is a product of the kynurenine pathway arising from tryptophan via transamination of 3-hydroxykynurenine (Gobaille et al, 2008). A body of evidence suggests that XA may serve a neuromodulatory function, including wide-spread, heterogeneous CNS levels; accumulation in synaptic terminals; release of XA upon electrical stimulation (Gobaille et al, 2008); intracerebrocentricular XA administration inducing seizures (Lapin, 1978b) and intraperitoneal administration in rats being antinociceptive (Heyliger et al, 1998; Fazio et al, 2012). A number of factors suggest that XA might have a role in psychiatric disorders, such as schizophrenia: first, some early reports suggested altered levels of XA and tryptophan metabolism in schizophrenic patients (Price et al, 1959; Benassi et al, 1961); second, we have found that XA modulates sensory inhibition in the thalamus in vivo (Copeland et al, 2013), a process that may be of importance in schizophrenia (Pinault, 2011); third, the reported molecular targets of XA are believed to be VGLUTs (Bartlett et al, 1998; Carrigan et al, 2002) and, recently, preliminary evidence suggested that XA may also modulate Group II metabotropic glutamate (mGlu) receptor function (Fazio et al, 2012; Mauro et al, 2010). Altered activity of both VGLUT and Group II mGlu receptors has been linked to schizophrenia (Fell et al, 2011; Gonzalez-Maeso et al, 2008; Eastwood and Harrison, 2005; Varea et al, 2012).These strands of evidence are all suggestive that maladaptation of XA metabolism may be involved in the pathophysiology of schizophrenia; therefore, we thought it is important to further explore the potential of XA to modulate neuronal function. We tested the effects of XA on synaptic transmission in the in vitro slice preparation of mouse hippocampus, and have also explored the XA mechanism of action via an in vitro radioligand binding assay. In addition, we have tested the effects of two VGLUT inhibitors that are structurally unrelated to XA on glutamatergic transmission. Data from mutant mice lacking VGLUT point to a key role in neurotransmission (Wojcik et al, 2004; Fremeau et al, 2004); however, there is a paucity of data regarding the effect of pharmacological modulation of VGLUTs. We have found that XA modulates synaptic transmission, although this appears to be via a mechanism distinct from direct modulation of Group II mGlu receptors and may be via VGLUT inhibition.
MATERIALS AND METHODS
Electrophysiology
All experiments were conducted on tissue from adult (>P28) C57black/6j mice (Harlan, UK), housed on a 12-h light/dark cycle with unlimited access to food and water. Experimental conditions and procedures were in accordance with the UK Animals (Scientific Procedures) Act 1986 and associated guidelines.
Mice were killed by decapitation and the brain was removed and placed into ice-cold oxygenated sucrose Krebs' medium containing (mM): sucrose 202, KCl 2, KH2PO4 1.25, MgSO4 10, CaCl2 0.5, NaHCO3 26, and glucose 10. The brain was hemisected along the midline and 300 μm parasagittal slices were prepared with an oscillating microtome (Integraslice, Campden Instruments). Slices were then transferred to a recovery chamber at room temperature containing oxygenated Krebs' solution (mM): NaCl 124, KCl 2, KH2PO4 1.25, MgSO4 1, CaCl2 2, NaHCO3 26, and glucose 10.
Following at least 1 h of recovery, individual slices were transferred to an interface recording chamber where they were perfused with Krebs' solution (35.6–36.2 °C). Extracellular field potential recordings were made via a Krebs'-filled glass micropipette (resistance 6–9 M( )) with an Axoprobe 1 A amplifier (Axon Instruments), digitized (5 kHz) via a CED1401 interface and stored on a computer with Spike2 software (Cambridge Electronic Design). Field excitatory postsynaptic potential (fEPSP) responses were evoked (0.1 ms pulses applied every 10 s; 2.4–5.5 V adjusted to ∼70% of the maximal spike-free response) by a bipolar stimulating electrode. For the dentate gyrus (DG), recording and stimulating electrodes were positioned in the lower to middle portion of the molecular layer. CA1 recordings were performed with the stimulating electrode positioned in the stratum radiatum near the CA3-CA1 border and the recording electrode positioned in the stratum radiatum of the CA1.
Responses to stimuli were waveform averaged (six trials). The amplitude of the average fEPSPs was quantified by computing the amplitude over a 0.5- to 0.8-ms time window centered on the peak of the fEPSP. Statistical significance was determined by the appropriate t-test, with P<0.05 taken as statistically significant.
Test compounds were applied by addition to the bathing medium. XA (Tocris or Sigma-Aldrich) was prepared either by dissolving directly into the Krebs' solution, or by preparation of a stock solution in equimolar NaOH and then diluting to the required concentration in Krebs' solution. The preparation method did not appear to impact the characteristics of the XA effect and the results are therefore pooled. Both LY354740 (Tocris) and LY341495 (Tocris) were prepared as 10 mM stock solutions in equimolar NaOH, stored in frozen aliquots, and diluted to the desired concentration in Krebs' solution on the day of the experiment. Congo Red (Sigma-Aldrich) was dissolved directly into Krebs' solution on the day of testing. Rose Bengal (Sigma-Aldrich) was prepared on the day of testing as a 10-mM stock in water and diluted to the desired concentration in the Krebs' solution. To inhibit GABA receptor-mediated effects, CGP55845 (5 μM; Tocris) and either picrotoxin (100 μM; Tocris) or bicuculline methochloride (30 μM; Tocris) were added to the Krebs' solution.
Radioligand Binding
Human mGlu2 receptor membrane preparation (Millipore) was diluted in assay buffer (20 mM HEPES, 100 mM NaCl, 3 mM MgCl2, pH 7.4) to give a final concentration of 6.6 μg/ml. To start the reaction, membrane (0.75 μg/well) was added to deep-well polypropylene microtiter plates containing [3H]LY341495 (1 nM; 40 Ci/mmol; American Radiolabeled Chemicals) and appropriate concentrations of XA (Sigma-Aldrich) in assay buffer. Final assay volume was 0.2 ml. Non-specific binding was defined with 100 μM LY354740. Assay plates were incubated at room temperature (21–23 °C) for 2 h, and bound and free radioligands were separated by rapid filtration with 3 × 1 ml of cold assay buffer using Whatman GF-B uniplates (Brandel) presoaked in 0.3% polyethylenimine (Sigma-Aldrich). The uniplates were dried at 37 °C for 30 min, 50 μl Microscint 20 (Perkin-Elmer) added to each well, and radioactivity determined using a Packard Topcount 2 (Perkin-Elmer). Experiments were performed in duplicates performed four times. Data are expressed as counts per minute (±SEM) and comparisons were made using Wilcoxon matched-pairs test (P<0.05).
RESULTS
XA Modulation of Synaptic Transmission in the DG
In the DG, pairs of stimuli evoked fEPSPs that were abolished in the presence of 10 μM of the AMPA receptor antagonist NBQX (Sheardown et al, 1990). When pairs of stimuli were applied (40 ms interval), fEPSPs exhibited paired-pulse depression, indicative of medial perforant pathway activation (Macek et al, 1996). Application of XA resulted in a reversible and concentration-dependent depression in the amplitude of the fEPSP, with 3 mM depressing the fEPSP by 21±2% (n=11; P<0.001) with a further reduction of 52±9% (n=4; P<0.05) from control observed in the presence of 10 mM XA (Figure 1). Similarly, LY354740, a selective Group II mGlu receptor agonist (Monn et al, 1997), depressed the fEPSP in a concentration-dependent manner, with the top concentration tested (1 μM) reducing the fEPSP by 74±3% (n=18; P<0.001) from control amplitude (Figure 1).
Figure 1.

Actions of XA, LY354740, Congo Red, and Rose Bengal in the dentate gyrus. (a) Representative traces (averaged fEPSPs in response to perforant path stimulation) from a single hippocampal slice showing recordings made under either control conditions or during the application of XA (3 mM) or LY354740 (1 μM). Both XA and LY354740 produced reductions in fEPSP amplitude. For clarity, the stimulus artifacts have been truncated. Average percentage reductions in fEPSP amplitudes (±SEM) resulting from application of various concentrations of XA or LY354740 in several experiments are shown in the bar graph (n values shown in the bars; *P<0.05; ***P<0.001). (b) Similar traces to those in (a), showing recordings made under either control conditions or during the application of Rose Bengal (1–30 μM). Average percentage reductions in fEPSP amplitudes (±SEM) resulting from application of Rose Bengal or Congo Red in several experiments are shown in the bar graph (n values shown in the bars; ns=P>0.05; **P<0.01; ***P<0.001).
To test whether the XA effect was via Group II mGlu receptors, we conducted a number of investigations. First, reduction of the fEPSP amplitude observed in the presence of LY354740 (300 nM) was reversed in the presence of the Group II receptor antagonist LY341495 (Kingston et al, 1998) from 31±6% when LY354740 was applied alone to 3±2% (n=3; P>0.05 compared with control; P<0.05 compared with LY354740 alone) in the presence of LY341495 (300 nM). However, in the same recordings, the depression of fEPSP amplitude in the presence of XA (3 mM; 23±2% n=3) was not reversed when XA was co-applied with LY341495 (300 nM; 26±2% n=3; Figure 2). In the presence of GABA receptor antagonists, XA (3 mM) produced a depression of synaptic transmission of 26±4% (n=3) that was not significantly different to the depression observed in the presence of XA (3 mM) under control conditions (P>0.05; Figure 2). Finally, to test whether XA acts as a modulator of Group II receptor function, XA (3 mM) was co-applied with a submaximal concentration of LY354740 (30 nM). No potentiation of the submaximal effect of LY354740 by XA was seen (Figure 2).
Figure 2.

Actions of the Group II antagonist LY341495 in the dentate gyrus. (a–c) Representative averaged fEPSP traces (as in Figure 1) from a single hippocampal slice experiment taken at different time points (i–viii) as indicated in the time line in (d), showing effects of XA and LY354740 in the presence or absence of the antagonist LY341495. For clarity, the stimulus artifacts have been truncated. (d) Time line from a single representative experiment showing fEPSP amplitude while different compounds are applied as indicated by the bars above the time line. LY341495 (300 nM) did not affect the action of XA (ii, iii), but did reverse the greater (but submaximal) action of 300 nM LY354740 (v, vi). Finally, LY354740 (1 μM) produced a maximal reduction in fEPSP amplitude (viii). (e) Average percentage reductions in fEPSPs upon application of XA or LY354740 in the absence or presence of LY341495. Error bars represent the SEM; values in parentheses represent the number of recordings; ns=no significant difference from control; *P<0.05. Note that LY341495 reversed the action of LY354740 but had no effect on the action of XA. (f) Average percentage reductions in fEPSPs upon application of XA in the presence or absence of GABA receptor antagonists. Error bars represent the SEM; values in parentheses represent the number of recordings; ns=no significant difference from XA alone. Blockade of GABA receptors did not change the effects of XA. (g) Average percentage reductions in fEPSPs upon application of XA (3 mM) or LY354740 (30 nM) or co-application of the agonist LY354740 (30 nM) together with XA, taken from the same experiments. Note that XA did not potentiate the submaximal effect of LY354740.
In the presence of LY354740, a significant change in the paired-pulse ratio was observed with a shift from paired-pulse depression in all recordings toward paired-pulse facilitation. LY354740 (30 nM) resulted in a 17±3% (n=4; P<0.05; Figure 3) change compared with control, and in the presence of 1 μM LY354740 the change in the paired pulse ratio was 36±5% (n=14; P<0.001). In the presence of XA, no significant change in the paired-pulse ratio was observed at either 3 mM (0±2% n=11; P>0.05) or 10 mM (−3±1% n=4; P>0.05; Figure 3).
Figure 3.

Paired-pulse ratios in the dentate gyrus. (a) Representative averaged fEPSP traces from a single hippocampal slice showing responses to pairs of stimuli and revealing the depression of the second response, characteristic of the perforant path to dentate gyrus pathway. Both LY354740 (1 μM) and XA (10 mM) reduced the fEPSP amplitudes, but had different effects on the relative amplitudes of the first and second responses, as shown by the scaled traces in the presence of the compounds: LY354740 reduced the paired-pulse depression, whereas XA did not have any effect. For clarity, the stimulus artifacts have been truncated. (b) Average changes in paired-pulse ratios from control values in the presence of XA, Congo Red (Red), Rose Bengal (Rose), or LY354740, Error bars represent the SEM; values in parentheses represent the number of recordings; ns=no significant difference from control; *P<0.05; ***P<0.001. LY354740 caused a dose-related change in paired-pulse ratio, whereas the other compounds had little or no effect.
Modulation of Synaptic Transmission in the DG by VGLUT Inhibitors
The VGLUT inhibitors Rose Bengal (1–30 μM) and Congo Red (1 mM) decreased the amplitude of the fEPSP recorded in the DG (Figure 1). In the presence of 1 mM of the prototypical VGLUT inhibitor (Ahmed et al, 2011) Congo Red, the fEPSP amplitude was reduced to 61±2% of control (n=3; P<0.01), with no significant change in the paired-pulse ratio (6±2% of control; n=3; P>0.05). In the presence of Rose Bengal, the fEPSP amplitude was reduced in a concentration-dependent manner, with 30 μM, the highest concentration tested, reducing the amplitude by 80±12% compared with the control amplitude (n=4; P<0.01). At a concentration of 10 μM, Rose Bengal depressed the fEPSP amplitude by 42±% and produced only a small increase in the paired-pulse ratio of 6±1% (n=7; P<0.001) (Figure 3).
XA Modulation of Synaptic Transmission in the CA1 Region
Application of XA (3 mM) produced a statistically significant decrease in CA1 fEPSP amplitude to 18±5% (P<0.05; Figure 4) from control in five out of six recordings. In four of the six recordings, XA application resulted in changes to the fEPSP waveform, indicative of increases in overall excitability (Figure 4a). In one of these recordings, spiking on the peak of the fEPSP meant it was omitted from the peak amplitude measurements. In contrast, application of LY354740 (1 μM) resulted in no significant change to the fEPSP (Figure 4).
Figure 4.

Actions of XA and LY354704 in the CA1 region. (a) Representative averaged fEPSP traces recorded from the CA1 region in a single hippocampal slice experiment taken under control conditions and during application of either LY354740 (1 μM) or XA (3 mM). LY354740 had no effect on fEPSP amplitude or waveform, whereas XA reduced the amplitude of the fEPSP and produced changes in the later components of the fEPSP (see Results). For clarity, the stimulus artifacts have been truncated. (b) Time course of the effects of LY354740 (1 μM) and XA (3 mM) on the amplitude of the CA1 fEPSP. The agents were applied as indicated by the bars above the trace. (c) Average percentage reductions in fEPSPs upon application of LY354740 (1 μM) or XA (3 mM). Error bars represent the SEM; values in parentheses represent the number of recordings; ns=no significant difference from control; *P<0.05.
XA Binding to the mGlu2 Receptor
In membranes stably expressing the human mGlu2 receptor, XA (1 μM–1 mM) had no effect on specific binding of 1 nM [3H]LY341495. In contrast, 100 μM LY354740 was able to significantly displace [3H]LY341495 binding (2.6±1.0% of control-specific binding; n=8; Figure 5).
Figure 5.
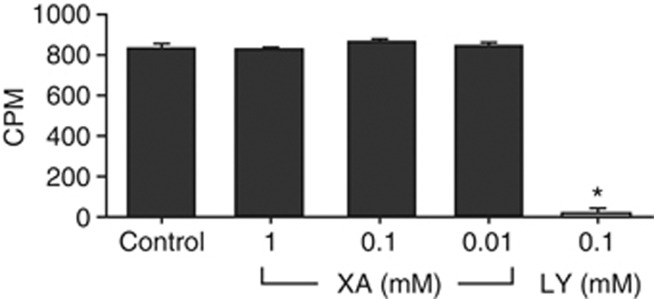
Effects of XA and LY354740 in mGlu2 binding assay. Results of binding experiments using [3H]LY341495: XA (0.01–1 mM) was not able to displace LY341495 binding, whereas LY354740 (0.1 mM) displaced binding completely. Error bars represent the SEM; *P<0.05; CPM, counts per minute.
DISCUSSION
In the present study, we have characterized the actions of XA on synaptic transmission in the DG and CA1 regions of the mouse hippocampus and find evidence of XA modulating neurotransmission in both regions. We also explored the possibility of XA modulation of Group II mGlu receptors and, in contrast to previous reports (Fazio et al, 2012; Mauro et al, 2010), find little evidence of interaction with these receptors under our assay conditions. We do, however, observe modulation of synaptic transmission in the DG with similar characteristics by other VGLUT inhibitors. This suggests that XA may exert its effects via this molecular target, rather than via the Group II mGlu receptors.
Modulation of Hippocampal Synaptic Transmission by XA
In the DG and CA1 regions of the mouse hippocampus, XA induced a depression of the fEPSP. In the DG, this effect was unchanged in the presence of GABA receptor antagonists, suggesting an action of XA independent of any effects on inhibition. In the CA1 region, the actions of XA were more complex and, in addition to the depression of synaptic transmission, we observed alterations in the fEPSP waveform consistent with increases in excitability. This observation concurs with previous reports that XA may be proconvulsant (Lapin, 1978a, 1978b) and that XA reduces sensory inhibition in the thalamus (Copeland et al, 2013).
XA is reportedly an agonist of Group II mGlu receptors (Fazio et al, 2012; Mauro et al, 2010). We, therefore, sought to determine whether the depression of synaptic transmission produced by XA was mediated via an interaction with these receptors. Consistent with previous reports (Macek et al, 1996; Kew et al, 2002), activation of Group II mGlu receptors with the Group II selective agonist LY354740 depressed transmission in the DG and this effect was reversed by the Group II selective antagonist LY341495; however, LY341495 did not affect the depression of synaptic transmission produced by XA, suggesting that XA is unlikely to be acting as a conventional orthosteric agonist of the Group II mGlu receptors. The depression of synaptic transmission by LY354740 at the perforant path-DG synapse is mediated predominately by activation of mGlu2, rather than mGlu3 receptors (Kew et al, 2002). A number of mGlu2 receptor-selective PAMs have been developed (Fell et al, 2011) and are reported to enhance the action of orthosteric agonists on synaptic transmission in the DG (Galici et al, 2006). We tested the possibility that XA was a PAM, but saw no evidence of such an action. Similarly, we previously reported that XA modulates sensory processing in the rat thalamus in vivo, but the PAM LY487379 was found to not potentiate this effect (Copeland et al, 2013), whereas the effects of a Group II agonist were enhanced by the PAM (Copeland et al, 2012). In the CA1 region, consistent with previous reports (Gereau and Conn, 1995), application of LY354740 had no effect on synaptic transmission at the Schaffer collateral-CA1 synapse, whereas XA produced clear effects. In total, these data suggest the actions of XA observed in the hippocampus are unlikely to be a result of Group II mGlu receptor activation, or PAM activity. Consistent with this conclusion, in the radioligand binding assay XA was found to not bind to the mGlu2 receptor orthosteric binding site.
In addition to pharmacological differences, further evidence that the effect of XA was not via Group II mGlu receptors was provided by the observed mechanistic difference between the mode of action of XA and the effect of Group II mGlu receptor activation. At the medial perforant path-DG synapse, responses to pairs of stimuli typically exhibit paired-pulse depression (Macek et al, 1996). This represents a form of short-lived synaptic plasticity believed to arise from changes in the probability of presynaptic neurotransmitter release (Kamiya and Zucker, 1994). In our experiments, LY354740 produced a clear change in the paired-pulse ratio, an observation consistent with presynaptic Group II mGlu receptors reducing the probability of transmitter release (Macek et al, 1996); however, in the presence of XA we observed no such change. This might suggest that XA modulates glutamatergic neurotransmission postsynaptically (Bartlett et al, 1998); however, XA is known to inhibit VGLUTs (Carrigan et al, 2002; Bartlett et al, 1998), the transport system thought to be responsible for ℒ-glutamate accumulation into synaptic vesicles in presynaptic terminals. There are three known VGLUT subtypes (VGLUT1–3; Shigeri et al, 2004; Takamori, 2006), which have been localized in the hippocampus, with VGLUT1 and VGLUT2 particularly associated with excitatory synapses (Herzog et al, 2006; Shigeri et al, 2004; Takamori, 2006). Electrophysiological studies in neuronal tissue where VGLUT expression has been silenced genetically demonstrate a decrease in amplitude of the postsynaptic response at glutamatergic synapses (Wojcik et al, 2004; Fremeau et al, 2004). Mice with genetic deletion of VGLUT also show minimal change in release probability (Fremeau et al, 2004; Wojcik et al, 2004), with synapses lacking VGLUT having a tendency toward depression in response to trains of stimuli (Fremeau et al, 2004; Smear et al, 2007); thus, despite a presynaptic location of VGLUT, VGLUT inhibition might not be expected to affect the paired-pulse ratio, consistent with XA exerting minimal effects on the paired-pulse ratio. In the current study, we have further investigated the effect of VGLUT modulation on synaptic transmission with two VGLUT inhibitors, Congo Red (Ahmed et al, 2011) and Rose Bengal (Pietrancosta et al, 2010). These compounds both depressed synaptic transmission in the DG and, as with XA, showed little effect on the paired-pulse ratio compared with the Group II mGlu receptor agonist, despite attenuating the fEPSP amplitude to a similar degree. In total, therefore, an effect of XA at VGLUT would be a likely explanation for the reduction in synaptic transmission seen with XA.
XA and the Kynurenine Pathway
XA occurs naturally in the mammalian CNS (Gobaille et al, 2008) and the data presented here, and in our previous publication (Copeland et al, 2013), demonstrate modulation of neuronal function by XA in multiple brain regions. Thus, XA can join the list of naturally occurring kynurenines, such as the NMDA receptor agonist quinolinic acid and the ionotropic glutamate receptor antagonist kynurenic acid (Stone, 1993), which modulate synaptic transmission. It is noticeable that XA is reported to be present in the brain in low micromolar concentrations (Gobaille et al, 2008), yet we required millimolar concentrations to depress synaptic transmission; however, mechanisms exist to allow XA accumulation in vesicles, and in vivo XA is released in the cortex in response to electrical stimulation in a manner requiring sodium channel activity (Gobaille et al, 2008). It is therefore likely that surveying gross tissue concentrations does not necessarily provide an accurate prediction of the concentration at the release site, which may be orders of magnitude higher than the concentration sampled in the extracellular fluid. This is the case for neurotransmitters such as glutamate, which may be present at the synapse in millimolar concentrations (Clements et al, 1992). Similarly, it has recently been reported that antipsychotics may accumulate in synaptic vesicles leading to higher than expected local concentrations in the synaptic cleft than might be predicted (Tischbirek et al, 2012). In addition, there are reportedly active transport systems for XA in neurones (Gobaille et al, 2008) and in intact neuronal tissue these systems may reduce the concentration of exogenously applied XA reaching the synapse, again, as is seen with established transmitters such as glycine (Berger et al, 1998) and glutamate (Herman and Jahr, 2007). It is therefore likely that XA levels occurring at the synapse may be in the range at which we see effects on synaptic transmission. This reinforces the notion that XA changes can affect synaptic function.
Possible Involvement in Disease Mechanisms
In this study, we have provided evidence that XA can modulate glutamatergic neurotransmission in the mouse hippocampus and, previously, we demonstrated modulation of sensory processing by XA in the rat thalamus, an effect likely involving modulation of GABAergic signaling (Copeland et al, 2012). Both Glutamate and GABA receptor signaling are believed to be altered in schizophrenia (Hahn et al, 2006; Maher and LoTurco, 2012; Uhlhaas and Singer, 2010; Coyle et al, 2003; Sodhi et al, 2011) and expression of VGLUT, a likely molecular target of XA, is also affected in this disease (Eastwood and Harrison, 2005; Varea et al, 2012; Uezato et al, 2009; Oni-Orisan et al, 2008). Furthermore, there is considerable evidence suggesting that levels of kynurenines, such as kynurenic acid, are altered in psychiatric disorders, including schizophrenia, bipolar disorder, and depression (Schwarcz et al, 2012; Myint, 2012) and that XA metabolism may be altered in patients (Price et al, 1959; Benassi et al, 1961). In schizophrenia, a number of studies demonstrate impairments in both the gene expression and activity of kynurenine pathway enzymes, and increases in kynurenine metabolite levels in cerebrospinal fluid (Schwarcz et al, 2012; Myint, 2012). Changes in kynurenine levels have been observed in drug-naïve first-episode schizophrenic patients, suggesting that changes might be linked to pathology, rather than drug-treatment (Erhardt et al, 2001). Together, these observations point to disruption of the kynurenine pathway as a feature of schizophrenia, and further studies are required to establish whether these changes also result in altered XA levels. In total, our data demonstrating effects of XA on synaptic transmission and sensory processing, the ability of XA to alter neuronal activity via VGLUT, and the possibility of XA levels being altered in disease raise the intriguing possibility that XA may have a role in the abnormal neuronal activity underlying schizophrenia (Pinault, 2011; Uhlhaas and Singer, 2010).
In conclusion, our data show that XA can be added to the list of kynurenines, which are able to modulate synaptic transmission and that this action is independent of Group II mGlu receptor modulation. In addition, we have demonstrated depression of glutamatergic synaptic transmission by three structurally unrelated VGLUT inhibitors. Given the role of glutamatergic signaling in a wide range of CNS disorders, this raises the possibility of VGLUT being worthy of further investigation as a target for development of novel therapeutics.
Acknowledgments
This work was supported by grants from the BBSRC (UK) and Merck. We would like to thank J Bruno and R Robinson for their help in performing the in vitro biochemical assays.
Dr Neale is a director of Neurexpert Ltd., Ms Copeland is supported by the BBSRC and Merck, Dr Uebele and Dr Thomson are employees of Merck & Co., Inc., and may own stock and/or stock options in the company. Dr Salt is in receipt of funding from Merck and the BBSRC, and is a director of Neurexpert Ltd.
References
- Ahmed SK, Etoga JL, Patel SA, Bridges RJ, Thompson CM. Use of the hydantoin isostere to produce inhibitors showing selectivity toward the vesicular glutamate transporter versus the obligate exchange transporter system x(c)(-) Bioorg Med Chem Lett. 2011;21:4358–4362. doi: 10.1016/j.bmcl.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ. Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles. Neuropharmacology. 1998;37:839–846. doi: 10.1016/s0028-3908(98)00080-x. [DOI] [PubMed] [Google Scholar]
- Benassi CA, Benassi P, Allegri G, Ballarin P. Tryptophan metabolism in schizophrenic patients. J Neurochem. 1961;7:264–270. [Google Scholar]
- Berger AJ, Dieudonne S, Ascher P. Glycine uptake governs glycine site occupancy at NMDA receptors of excitatory synapses. J Neurophysiol. 1998;80:3336–3340. doi: 10.1152/jn.1998.80.6.3336. [DOI] [PubMed] [Google Scholar]
- Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, et al. Synthesis and in vitro pharmacology of substituted quinoline-2,4-dicarboxylic acids as inhibitors of vesicular glutamate transport. J Med Chem. 2002;45:2260–2276. doi: 10.1021/jm010261z. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Copeland CS, Neale SA, Salt TE. Actions of xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology. 2013;66:133–142. doi: 10.1016/j.neuropharm.2012.03.009. [DOI] [PubMed] [Google Scholar]
- Copeland CS, Neale SA, Salt TE. Positive allosteric modulation reveals a specific role for mGlu2 receptors in sensory processing in the thalamus. J Physiol. 2012;590:937–951. doi: 10.1113/jphysiol.2011.218065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Tsai G, Goff D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann NY Acad Sci. 2003;1003:318–327. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr Res. 2005;73:159–172. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Blennow K, Nordin C, Skogh E, Lindstrom LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001;313:96–98. doi: 10.1016/s0304-3940(01)02242-x. [DOI] [PubMed] [Google Scholar]
- Fazio F, Molinaro G, Iacovelli L, Bernabucci M, Mauro G, Di Menna L, et al. Xanthurenic acid as a novel putative agonist of Group II metabotropic glutamate receptors. Curr Neuropharmacol. 2012;9 (Suppl 1:19. [Google Scholar]
- Fell MJ, McKinzie DL, Monn JA, Svensson KA. Group II metabotropic glutamate receptor agonists and positive allosteric modulators as novel treatments for schizophrenia. Neuropharmacology. 2011;62:1473–1483. doi: 10.1016/j.neuropharm.2011.06.007. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, et al. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Galici R, Jones CK, Hemstapat K, Nong Y, Echemendia NG, Williams LC, et al. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- Gereau RW, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobaille S, Kemmel V, Brumaru D, Dugave C, Aunis D, Maitre M. Xanthurenic acid distribution, transport, accumulation and release in the rat brain. J Neurochem. 2008;105:982–993. doi: 10.1111/j.1471-4159.2008.05219.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF, et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog E, Takamori S, Jahn R, Brose N, Wojcik SM. Synaptic and vesicular co-localization of the glutamate transporters VGLUT1 and VGLUT2 in the mouse hippocampus. J Neurochem. 2006;99:1011–1018. doi: 10.1111/j.1471-4159.2006.04144.x. [DOI] [PubMed] [Google Scholar]
- Heyliger SO, Goodman CB, Ngong JM, Soliman KF. The analgesic effects of tryptophan and its metabolites in the rat. Pharmacol Res. 1998;38:243–250. doi: 10.1006/phrs.1998.0362. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- Kew JN, Pflimlin MC, Kemp JA, Mutel V. Differential regulation of synaptic transmission by mGlu2 and mGlu3 at the perforant path inputs to the dentate gyrus and CA1 revealed in mGlu2-/- mice. Neuropharmacology. 2002;43:215–221. doi: 10.1016/s0028-3908(02)00084-9. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, et al. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lapin IP. Convulsions and tremor in immature rats after intraperitoneal injection of kynurenine and its metabolites. Pharmacol Res Commun. 1978a;10:81–84. doi: 10.1016/s0031-6989(78)80065-4. [DOI] [PubMed] [Google Scholar]
- Lapin IP. Stimulant and convulsive effects of kynurenines injected into brain ventricles in mice. J Neural Transm. 1978b;42:37–43. doi: 10.1007/BF01262727. [DOI] [PubMed] [Google Scholar]
- Lapin IP, Oxenkrug GF. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet. 1969;1:132–136. doi: 10.1016/s0140-6736(69)91140-4. [DOI] [PubMed] [Google Scholar]
- Macek TA, Winder DG, Gereau RW, Ladd CO, Conn PJ. Differential involvement of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapses. J Neurophysiol. 1996;76:3798–3806. doi: 10.1152/jn.1996.76.6.3798. [DOI] [PubMed] [Google Scholar]
- Maher BJ, LoTurco JJ. Disrupted-in-schizophrenia (DISC1) functions presynaptically at glutamatergic synapses. PLoS One. 2012;7:e34053. doi: 10.1371/journal.pone.0034053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauro G, Fazio F, Molinaro G, Iacovelli L, Ngomba R, Di Menna L, et al. 2010Xanthurenic acid, as a novel endogenous ligand for metabotropic glutamate receptorsAnnual Meeting of the Society for Neuroscience, San Diego, CA.
- Millan MJ. N-methyl-d-aspartate receptors as a target for improved antipsychotic agents: novel insights and clinical perspectives. Psychopharmacology (Berl) 2005;179:30–53. doi: 10.1007/s00213-005-2199-1. [DOI] [PubMed] [Google Scholar]
- Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Johnson BG, et al. Design, synthesis, and pharmacological characterization of (+)-2- aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J Med Chem. 1997;40:528–537. doi: 10.1021/jm9606756. [DOI] [PubMed] [Google Scholar]
- Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J. 2012;279:1375–1385. doi: 10.1111/j.1742-4658.2012.08551.x. [DOI] [PubMed] [Google Scholar]
- Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia. Biol Psychiatry. 2008;63:766–775. doi: 10.1016/j.biopsych.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrancosta N, Kessler A, Favre-Besse FC, Triballeau N, Quentin T, Giros B, et al. Rose Bengal analogs and vesicular glutamate transporters (VGLUTs) Bioorg Med Chem. 2010;18:6922–6933. doi: 10.1016/j.bmc.2010.06.069. [DOI] [PubMed] [Google Scholar]
- Pinault D. Dysfunctional thalamus-related networks in schizophrenia. Schizophr Bull. 2011;37:238–243. doi: 10.1093/schbul/sbq165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J, Brown R, Peters H. Tryptophan metabolism in porphyria, schizophrenia, and a variety of neurologic and psychiatric diseases. Neurology. 1959;9:456–468. doi: 10.1212/wnl.9.7.456. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci. 2012;13:465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004;45:250–265. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Smear MC, Tao HW, Staub W, Orger MB, Gosse NJ, Liu Y, et al. Vesicular glutamate transport at a central synapse limits the acuity of visual perception in zebrafish. Neuron. 2007;53:65–77. doi: 10.1016/j.neuron.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2001;158:1393–1399. doi: 10.1176/appi.ajp.158.9.1393. [DOI] [PubMed] [Google Scholar]
- Sodhi MS, Simmons M, McCullumsmith R, Haroutunian V, Meador-Woodruff JH. Glutamatergic gene expression is specifically reduced in thalamocortical projecting relay neurons in schizophrenia. Biol Psychiatry. 2011;70:646–654. doi: 10.1016/j.biopsych.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- Takamori S. VGLUTs: ‘exciting' times for glutamatergic research. Neurosci Res. 2006;55:343–351. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Tischbirek CH, Wenzel EM, Zheng F, Huth T, Amato D, Trapp S, et al. Use-dependent inhibition of synaptic transmission by the secretion of intravesicularly accumulated antipsychotic drugs. Neuron. 2012;74:830–844. doi: 10.1016/j.neuron.2012.04.019. [DOI] [PubMed] [Google Scholar]
- Uezato A, Meador-Woodruff JH, McCullumsmith RE. Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar Disord. 2009;11:711–725. doi: 10.1111/j.1399-5618.2009.00752.x. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–113. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- Varea E, Guirado R, Gilabert-Juan J, Marti U, Castillo-Gomez E, Blasco-Ibanez JM, et al. Expression of PSA-NCAM and synaptic proteins in the amygdala of psychiatric disorder patients. J Psychiatr Res. 2012;46:189–197. doi: 10.1016/j.jpsychires.2011.10.011. [DOI] [PubMed] [Google Scholar]
- Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, et al. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci USA. 2004;101:7158–7163. doi: 10.1073/pnas.0401764101. [DOI] [PMC free article] [PubMed] [Google Scholar]