

CD36 requires palimitoylation in a selenoprotein K-dependent manner, to localize lipid rafts and function properly.
Keywords: low density lipoprotein, selenium, scavenger receptor, atherosclerosis
Abstract
Selk is an ER transmembrane protein important for calcium flux and macrophage activation, but its role in foam cell formation and atherosclerosis has not been evaluated. BMDMs from Selk−/− mice exhibited decreased uptake of modLDL and foam cell formation compared with WT controls, and the differences were eliminated with anti-CD36 blocking antibody. CD36 expression was decreased in TNF-α-stimulated Selk−/− BMDMs compared with WT controls. Fluorescence microscopy revealed TNF-α-induced clustering of CD36 in WT BMDMs indicative of lipid raft localization, which was absent in Selk−/− BMDMs. Fractionation revealed lower levels of CD36 reaching lipid rafts in TNF-α-stimulated Selk−/− BMDMs. Immunoprecipitation showed that Selk−/− BMDMs have decreased CD36 palmitoylation, which occurs at the ER membrane and is crucial for stabilizing CD36 expression and directing its localization to lipid rafts. To assess if this phenomenon had a role in atherogenesis, a HFD was fed to irradiated Ldlr−/− mice reconstituted with BM from Selk−/− or WT mice. Selk was detected in aortic plaques of controls, particularly in macrophages. Selk−/− in immune cells led to reduction in atherosclerotic lesion formation without affecting leukocyte migration into the arterial wall. These findings suggest that Selk is important for stable, localized expression of CD36 in macrophages during inflammation, thereby contributing to foam cell formation and atherogenesis.
Introduction
Atherosclerosis is the underlying cause of myriad cardiovascular diseases, including coronary artery disease, stroke, and many cases of heart failure. Although morbidity and mortality from atherosclerosis have decreased in recent years, the health impact and financial burden of this disease still rank among the highest of all diseases worldwide [1, 2]. Complex chronic inflammatory processes drive atherosclerosis in the arterial wall, and its pathogenesis is modulated by immune responses. Monocytes/macrophages play key roles in the initiation and progression of atherosclerosis, with T cells and DCs also contributing to the development of atherosclerotic lesions [3, 4].
The early stages of atherogenesis involve conditions of hyperlipidemia, followed by a rapid influx of circulating monocytes into the arterial intima [5, 6]. Recruited inflammatory monocytes differentiate into macrophages that express scavenger receptors, such as CD36 and SR-A that promote ingestion of the atherogenic-modified cholesterol ester-rich lipoproteins in the intima of the vessel wall [7]. Experiments using these receptors to promote foam cell formation have revealed that CD36 is the principal receptor for ingesting oxLDL [8], whereas SR-A is mainly involved in uptake of AcLDL [9]. The accumulation of cholesterol-loaded macrophages in the arterial wall, termed foam cell formation, is a hallmark of early atherosclerotic lesions. Rupture of foam cells can lead to an increase in the necrotic core and progression of atherosclerotic plaques. LDL particles are internalized by macrophages in the arterial wall via the LDLR on the cell surface, and LDL is hydrolyzed to free cholesterol within lysosomes [10]. Lipid uptake by macrophages is enhanced by inflammatory cytokines, such as TNF-α, which plays a key role in the formation of atherosclerotic plaques [11].
Whereas progress has been made in determining the role of cholesterol and lipid metabolism in atherogenesis, pervasive gaps remain regarding cellular and molecular factors that regulate the immune cells and drive the pathology of atherosclerosis. Selenium is an essential micronutrient that influences cardiovascular health. We have recently identified Selk as a protein expressed at particularly high levels in immune cells, which is important for their activation [12]. Selk is one of 25 selenoproteins that incorporate dietary selenium into their protein structure, as the amino acid, selenocysteine, and levels of Selk in immune tissues are sensitive to selenium intake [13]. Selk is localized to the ER membrane in T cells, neutrophils, and macrophages, and its expression is required for optimal calcium flux triggered during engagement of the TCR, FcγRs, chemokines, and TLRs. Several functions are particularly impaired in Selk−/− macrophages, including chemokine-induced migration, proinflammatory cytokine secretion in response to TLR ligands LPS and polyinosinic:polycytidylic acid, and the phagocytosis of IgG-coated particles [12, 14].
As Selk is important for optimal activation and functionality of immune cells, particularly macrophages, we evaluated its role in the uptake of modLDL and the formation of foam cells that contribute to atherogenesis. With the use of macrophages generated from BM of Selk−/− mice and WT controls, we demonstrated that Selk−/− caused reduced uptake of modLDL and foam cell formation. With the use of a HFD fed to lethally irradiated Ldlr−/− mice reconstituted with BM from Selk−/− mice and from WT controls, we found that Selk−/− also reduced the formation of atherosclerotic lesions. Selk−/− macrophages exhibited reduced expression of CD36 as a result of reduced palmitoylation, which was required for its clustered expression in the plasma membrane. These findings provide important information regarding the role of Selk in atherogenesis, offering novel insight into a potential therapeutic target, as well as mechanisms by which dietary selenium may affect cardiovascular disease.
MATERIALS AND METHODS
Mice and reagents
C57BL/6J and Ldlr−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA), and generation of Selk−/− mice was described previously [12]. All animal protocols were approved by the University of Hawaii Institutional Animal Care and Use Committee. Chemicals were obtained from Sigma (St. Louis, MO, USA), unless otherwise noted. Human lipoproteins (AcLDL, oxLDL, with or without DiI labeling) were obtained from Biomedical Technologies (Stoughton, MA, USA).
BMDM culture
BMDMs were cultured from BM of Selk−/− and WT control mice as described previously [14]. In brief, femurs, tibias, and pelvic bones of mice were removed and cleaned aseptically and BM flushed using a 25-gauge syringe. BM cells were washed with HBSS and seeded at 4 × 105 cells/mL in BMDM media consisting of DMEM/F12 (Gibco/Invitrogen, Carlsbad, CA, USA) containing 10% FBS, 1% penicillin-streptomycin, 10 mM l-glutamine, and 20% L929 conditioned medium for 6 days. Differentiated macrophages were counted and replated in DMEM/F12 with 10% FBS and 1% penicillin-streptomycin for various experiments.
modLDL uptake and foam cell formation assays
For cholesterol uptake assays, BMDMs were seeded at a density of 5 × 104 cells/well (sterile black 96-well plate, Corning, Corning, NY, USA), 1 day prior to treatment. Cells were treated using 10 ng/mL murine TNF-α for 1 h and treated with 20 μg/mL DiI-labeled AcLDL or oxLDL (collectively known as modLDL). In some cases, after TNF-α treatment for 1 h, 2 μg/mL anti-CD36 IgA-blocking antibody (JC63.1; Cayman Chemical, Ann Arbor, MI, USA) or IgA control (eBioscience, San Diego, CA, USA) was added to cells for 30 min and then modLDL added. After an incubation time of 4 h, cells were washed thoroughly and covered with equal amounts of HBSS. Fluorescence intensity was measured immediately using a SpectraMax 340 (Molecular Devices, Sunnyvale, CA, USA), set with excitation at 541 nm and emission at 550 nm. Fluorescence data were analyzed using GraphPad Prism. Additionally, cells were fixed using 4% PFA, and images of cells were captured using an Axiovert microscope (Carl Zeiss, Germany). For foam cell formation assays, BMDMs were seeded and treated as described earlier. AcLDL (50 μg/mL) was added, and cells were incubated for 24 h and 48 h. Foam cell formation was evaluated by measuring lipid content of macropages using Oil Red O stain and quantified using ImageJ software. Importantly, equivalent cell viability between WT and Selk−/− BMDM after incubation with modLDL was confirmed by microscopic evaluation of PI-stained cells on coverslips.
Western blot analysis
BMDMs or frozen tissues were lysed in ice-cold buffer containing 150 mM NaCl, 50 mM Tris, 1% Triton X-100, 1% sodium deoxycholate, and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA) at 4°C for 1 h. Insoluble material was removed by centrifugation at maximum speed for 5 min, and total protein in the supernatant was determined using a Bradford assay reagent (Bio-Rad, Hercules, CA, USA). After adjusting to equal protein concentration, cell lysates were boiled in SDS sample buffer and then separated by SDS-PAGE, followed by transfer of the proteins onto nitrocellulose membranes. Blots were incubated with primary anti-Selk (1:1000; Epitomics, Burlingame, CA, USA), anti-CD36 (1:500; Novus Biologicals, Littleton, CO, USA), anti-FLAG (1:1000; OriGene, Rockville, MD, USA), or anti-β-actin (1:1000; Sigma) for 1.5 h, washed, incubated with appropriate HRP-conjugated secondary antibody (1:20,000; Li-Cor, Lincoln, NE, USA), and visualized using the Odyssey Scanner (Li-Cor).
Analyses of cell fractions
For fractionation, 5 × 106 BMDMs were stimulated for 18 h with TNF-α (20 ng/mL) or PBS as a negative control. The cells were removed from the culture plates using Cellstripper solution (Cellgro Mediatech, Manassas, VA, USA) and resuspended at 0.5 × 106 cells/200 μl FACS buffer (PBS containing 2% FBS). Lipid fractions were generated as using a sucrose gradient ultracentrifugation [15]. In brief, cells were lysed on ice for 30 min in 250 μl MES-buffered saline (MBS: 25 mm MES, pH 6.5, 0.15 m NaCl, 5 mm EDTA, and 1× proteases inhibitor cocktail) containing 0.5% Triton X-100. Cell lysates and 80% sucrose were mixed 1:1 for a final concentration of 40% sucrose and then placed at the bottom of an ultracentrifugation tube. A 5–30% discontinuous sucrose gradient (in MBS) was overlaid and centrifuged at 48,000 rpm at 4°C for 18 h in an Optima ultracentrifuge (Beckman Instruments, Palo Alto, CA, USA). Fractions of 0.5 mL were collected from the top of the tube to yield a total of 10 fractions. Each fraction was loaded into separate wells of a SDS-polyacrylamide gel and Western blot performed as described above.
Detection of CD36 palmitoylation
BMDMs (107 cells) were transfected with 3.75 μg FLAG-CD36 pDNA (OriGene) using a Neon nucleofector (Invitrogen). After 2 h recovery, cells were metabolically labeled with Click-iT azide-conjugated palmitic acid, per the manufacturer's instructions (Invitrogen) for 5 h. Cells were lysed as described above, and lysates were precleared using streptavidin Sepharose beads (Invitrogen) to remove biotin. The precleared lysates were then incubated with anti-FLAG-coated magnetic beads (OriGene) overnight, followed by Click-iT conjugation of immunoprecipitated, azide-labeled protein with an alkyne biotin compound (Invitrogen). The resulting biotinylated proteins were disassociated from the beads by incubating in reduced Laemmli buffer at 95°C for 10 min, separated by PAGE, and transferred to PVDF membranes, as described above. Streptavidin-conjugated IRDye 680 (Li-Cor) was used to detect biotinylated proteins with the Odyssey Scanner (Li-Cor).
Flow cytometry and confocal microscopy
BMDMs were harvested after different timepoints, after TNF-α stimulation and 1 μl Fc Block (BD Biosciences, San Jose, CA, USA) were added for 15 min, followed by 1 μl PE anti-CD36 (BioLegend, San Diego, CA, USA) or AlexaFluor488 anti-CD204 (AbD Serotec, Raleigh, NC, USA), and cells were incubated for 30 min on ice. Cells were then washed three times with FACS buffer and analyzed by flow cytometry using a FACSCaliber (BD Biosciences). Data were analyzed using FlowJo software. For microscopic examination of CD36, BMDMs were plated on glass coverslips and stimulated with TNF-α or IL-10, as described above. The cells were then incubated with Fc Block, followed by PE anti-CD36 on ice for 30 min. The cells were washed and fixed with 4% PFA, permeabilized with 0.02% Triton X-100, and stained for intracellular CD36 with PE anti-CD36 for 18 h. Cells were stained for F-actin using Alexa488-phalloidin (Invitrogen) and coverslip-mounted onto glass slides for confocal imaging using a Zeiss Axiovert 200M attached to a Zeiss LSM 5 Pascal imaging system.
Atherosclerosis model and BMT
Ldlr−/− male mice of 8 weeks of age were placed on a standard chow diet or an atherogenic HFD containing 15.8% (wt/wt) fat and 1.25% cholesterol (diet 94,059; Teklad, Harlan Laboratories, Indianapolis, IN, USA). BMTs were performed as described previously [16]. Briefly, BM cells (2×106 in PBS) from donor mice were administered to recipient mice by i.v. tail-vein injection, 24 h after an ablative dose of whole-body irradiation (10.5 Gy) using an X-ray irradiator (CP160; Faxitron, Tucson, AZ, USA). After transfer, the immune systems of recipient mice were allowed to reconstitute for 4 weeks, after which, the mice were fed the HFD for 12 weeks. Some Ldlr−/− male and/or female mice were fed a normal chow or the HFD for 0, 2, 12, or 36 weeks to generate different stages of atherosclerosis. Upon sacrifice of these mice, the hearts and aortas were collected and used for further analysis.
Quantification and immunohistochemical analysis of atherosclerosis
The extent of atherosclerosis was assessed in aortas by staining for lipid depositions with Oil Red O and quantified using ImageJ software as described [16]. Briefly, the aortas were opened longitudinally, and the percentage of aortic surface covered by lipid deposition was calculated by dividing the stained area over the total aortic surface. The aortic sinus was sectioned to use for various immunohistochemical staining as described below.
The presence of macrophages, CD3+ T cells, SMCs, and Selk-positive cells in the aortic sinus plaque was determined by mAb staining for monocyte/macrophage marker antibody-2 (ab33451; AbCam, Cambridge, MA, USA), CD3 (PC3/188A, Santa Cruz Biotechnology, Santa Cruz, CA, USA), SM22α (10,493-1-AP; Proteintech, Chicago, IL, USA), and Selk (custom rabbit mAb; Epigenomics, Seattle, WA, USA), respectively, and detection with Cy2- and Cy3-conjugated secondary antibodies (AbCam). A TUNEL assay kit (Roche Applied Science) was used, according to the manufacturer's recommendation. For all assays, nuclei were labeled using DAPI, and images were recorded using an Axiovert microscope (Carl Zeiss).
Lipid analyses and lipoprotein profile measurement
Mice were fasted for 12–14 h prior to blood collection into tubes coated with EDTA. Plasma was separated by centrifugation and stored at −80°C. When all of the samples were collected, triglyceride and cholesterol levels were measured with their respective assay kits (Cayman Chemical), according to the manufacturer's protocol. The lipid distribution in plasma lipoprotein fractions was assessed by FPLC gel filtration with a Superose 6 10/300 GL column (GE Healthcare Biosciences, Piscataway, NJ, USA), as described [17].
Statistical analysis
PRISM software (GraphPad Software, San Diego, CA, USA) was used to compare means between two groups with the Student's t-test and among three or more groups with a one-way ANOVA, followed by Tukey's mulitple comparison test. Data are presented as mean ± sem, and statistical significance was accepted at the level of P < 0.05.
Online supplemental material
Supplemental Fig. 1 shows that Selk−/− does not affect growth or differentation of BMDMs. Supplemental Fig. 2 shows that Ldlr−/− mice, which were X-ray-irradiated with Wt or Selk knockout BM, were equivalent-reconstituted. Supplemental Fig. 3 includes data showing that Selk−/− does not affect lipid profiles in mice, including triglycerides, total cholesterol, VLDL/LDL ratios, and HDL levels. Supplemental Fig. 4 demonstrates that Selk−/−does not influence cellular composition in aortic plaques with fluorescence microscopic evaluation of macrophages, SMCs, and T cells.
RESULTS
Selk−/− inhibits the uptake of modLDL and decreases macrophage foam cell formation
Our previous studies have shown that Selk−/− macrophages exhibit functional impairments that depend on efficient receptor-mediated calcium flux. However, it is unknown whether Selk is involved in modLDL uptake and the formation of foam cells that drive atherogenesis. Thus, we assayed the uptake of fluorescent Dil-AcLDL and Dil-oxLDL by WT and Selk−/− BMDMs. Plate-based fluorimetry and fluorescent microscopy showed that the internalization of Dil-AcLDL or Dil-oxLDL in Selk−/− BMDMs was decreased compared with WT controls, suggesting that Selk−/− led to impaired uptake of both types of modLDL (Fig. 1A). Stimulation of the BMDMs with TNF-α enhanced the differences between WT and Selk−/− BMDMs (Fig. 1B). Interestingly, the reduction in uptake resulting from Selk−/− was more pronounced with oxLDL compared with AcLDL. Importantly, WT and Selk−/− BMDMs showed equivalent morphology, surface marker expression, and capacity to differentiate in response to TNF-α as well as IL-4 (Supplemental Fig. 1). Thus, the lower modLDL uptake by Selk−/− BMDMs was not a result of differences in growth or differentiation of these macrophages compared with WT BMDMs.
Figure 1. Selk−/− in macrophages decreases uptake of modLDL.
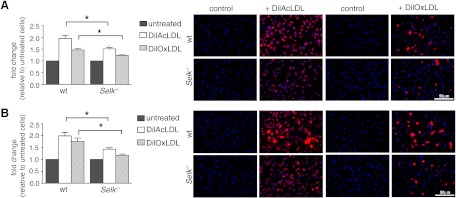
(A) WT or Selk−/− BMDMs were incubated with Dil-AcLDL or Dil-OxLDL, and levels of ingested, fluorescent modLDL determined by plate-based fluorimetry (bar graphs) and fluorescent microscopy (representative images). (B) BMDMs from WT or Selk−/− mice were treated with TNF-α for 4 h, followed by the addition of Dil-AcLDL or Dil-OxLDL, and uptake measured similar untreated BMDMs. Data represent mean ± se (n=3). *P < 0.05.
As modLDL uptake by macrophages is known to affect macrophage lipid homeostasis, we next investigated the role of Selk in this important process. TNF-α-treated Selk−/− and WT BMDMs were subjected to foam cell formation, followed by staining with Oil Red O and assessment by microscopy. Foam cell formation was decreased significantly in Selk−/− BMDMs compared with controls at both time-points (Fig. 2). Taken together, these results suggest that Selk−/− inhibits the uptake of modLDL, which may lead to foam cell formation in primary macrophages.
Figure 2. Selk−/− leads to decreased foam cell formation.

BMDMs from WT or Selk−/− mice were treated with TNF-α for 4 h, followed by incubation with AcLDL for 24 or 48 h. Levels of ingested AcLDL were determined by fluorescent microscopic evaluation of Oil Red O. Representative images are shown for lipid content at 48 h. Data represent mean ± se (n=3). *P < 0.05.
Selk−/− inhibits TNF-α-induced CD36 surface expression
Subsequently, we analyzed the expression of the two scavenger receptors—CD36 and SR-A—involved in modLDL uptake using flow cytometry. A lower level of cell-surface CD36 expression was detected on Selk−/− BMDMs compared with WT controls treated for 24 h with TNF-α (Fig. 3A). In contrast, there were no differences in CD36 when Selk−/− and WT BMDMs were stimulated with IL-10. When the TNF-α-stimulated BMDMs were evaluated for levels of SR-A, no differences were found between WT and Selk−/− BMDMs (Fig. 3B), suggesting that Selk is more important for surface expression of CD36 compared with SR-A. Moreover, the effect of Selk−/− on surface expression of CD36 was surprisingly rapid, with increased CD36, detectable within 3 h of TNF-α stimulation (Fig. 3C). This was consistent with Western blot analyses of total CD36 showing lower CD36 protein in Selk−/− BMDMs within 3 h of TNF-α treatment compared with WT BMDMs (Fig. 3D). This rapid increase of CD36 protein levels suggested that this rise was not a result of up-regulation of CD36 transcription induced by TNF-α stimulation. Indeed, real-time PCR analysis of CD36 mRNA confirmed that TNF-α stimulation did not enhance transcription of the CD36 gene (Fig. 3E). In fact, levels of CD36 were decreased slightly over time in a similar manner for WT and Selk−/− BMDMs.
Figure 3. Selk−/− reduces TNF-α-induced surface expression of CD36.

(A) Flow cytometry was used to detect surface CD36 levels on BMDMs from WT (wt; blue) or Selk−/− (green) BMDMs with no treatment, TNF-α treatment, or IL-10 treatment. Several different time-points after TNF-α or IL-10 treatment were evaluated for effects on CD36 surface expression, and representative histograms are shown for 18 h poststimulation. (B) Flow cytometry was used to detect surface SR-A levels on BMDMs from WT (blue) or Selk−/− (green) BMDMs with no treatment or with TNF-α treatment. (C) Mean fluorescence for CD36 was measured by flow cytometry for WT or Selk−/− BMDMs, with or without TNF-α treatment for 3 h. (D) Western blot was performed on whole cell lysates from WT and Selk−/− BMDMs treated with TNF-α for increasing time to determine levels of CD36 protein. (E) Real-time PCR was used to evaluate CD36 mRNA levels normalized to β-actin mRNA in WT or Selk−/− BMDMs stimulated with TNF-α for increasing time. Data represent mean ± se (n=3). *P < 0.05.
Selk−/− macrophages exhibit impaired CD36 surface aggregation as a result of defective palmitoylation
The results above suggested rapid, post-translational regulation of CD36 protein by Selk during TNF-α treatment. Thus, we next examined untreated and TNF-α-treated WT and Selk−/− BMDMs by confocal microscopy for qualitative differences in surface expression of CD36 (Fig. 4A). Unstimulated WT BMDMs exhibited a diffuse staining of CD36, and upon TNF-α stimulation, there was an increase in surface CD36 organized in clusters. In contrast, levels of CD36 fluorescence on TNF-α-treated Selk−/− BMDMs were lower than WT BMDMs, and the CD36 that was detectable in Selk−/− BMDMs was found in a similar clustered pattern of lower intensity. Interestingly, organization of filamentous actin induced with TNF-α stimulation did not differ between WT and Selk−/− BMDMs. This suggests an effect of Selk−/− on CD36 trafficking and/or stability that is unlikely to be a result of differences in reorganization of the actin cytoskeleton.
Figure 4. Selk is required for palmitoylation and membrane clustering of CD36 during TNF-α stimulation.

(A) Unstimulated or TNF-α-stimulated WT and Selk−/− BMDMs were stained with PE anti-CD36 (red) and F-actin stained using AlexaFluor488-phalloidin (green). Staining was evaluated using confocal microscopy. Original scale bar: 10 μm. (B) Cell lysates from unstimulated or TNF-α-stimulated WT and Selk−/− BMDMs were separated into fractions by sucrose gradient centrifugation and proteins separated and analyzed by Western blot. CD36 was detected in several fractions and was particularly enriched in those fractions exhibiting the marker for lipid rafts, Cav-1. wcl, whole cell lysate. (C) Western blot was used to analyze expression levels of FLAG-CD36 in WT or Selk−/− [knockout (KO)] BMDMs with β-actin used as a loading control. (D) Metabolic labeling of palmitoylated FLAG-CD36 and coupling to biotin showed higher levels of palmitoylation in WT BMDMs compared with Selk−/− BMDMs (upper panel). As a control, immunoprecipitated (I.P.) FLAG-CD36 was detected by Western blot using anti-FLAG, and similar levels of protein were pulled down for WT and Selk−/− BMDMs (lower panel). Note that bands below the 75-kD marker band correspond to the palmitoylated FLAG-CD36 protein, whereas the bands above the 75-kD marker band likely represent glycosylated FLAG-CD36 that has not been palmitoylated.
The clustered pattern of CD36 on the surface of macrophages observed above and previous studies showing increased lipid raft association of CD36 promoting fatty acid uptake [18, 19] led us to investigate the membrane localization of CD36 in WT and Selk−/− BMDMs. Sucrose gradient separation of cell lysates from WT BMDMs demonstrated that CD36 was dispersed in several fractions, with enrichment of CD36 in fractions containing the lipid raft marker Cav-1 (Fig. 4B). TNF-α stimulation of the WT BMDMs did not overtly change the distribution of CD36 or Cav-1 but increased the overall levels of both proteins. Selk−/− BMDMs exhibited much lower levels of CD36 compared with WT BMDMs. Interestingly, the CD36 that was detected in TNF-α-stimulated Selk−/− BMDMs was only found in the lipid raft fractions. This is consistent with the punctate staining displayed in Fig. 4A for surface expression of CD36. A recent study demonstrated that palmitoylation of CD36 at the ER membrane is a crucial step in stabilizing this molecule, and nonpalmitoylated CD36 outside of lipid rafts is vulnerable to degradation [20]. Given that Selk is an ER membrane protein and that stable CD36 in Selk−/− BMDMs was found only within lipid rafts, we next investigated whether Selk−/− affected palmitoylation of CD36. When a plasmid construct encoding FLAG-CD36 was transfected into WT and Selk−/− BMDMs, Selk−/− led to lower levels of FLAG-CD36 expression, consistent with the data above (Fig. 4C). A protein-labeling system was next used, in which BMDMs were metabolically labeled with azide-tagged palmitic acid, followed by FLAG-CD36 immunoprecipitation. The FLAG-CD36, pulled-down on the beads, was then labeled with alkyne-biotin, followed by several washes and elution, proteins separated by electrophoresis and transferred to PVDF, and levels of palmitoylated CD36 detected with fluorescently labeled streptavidin. As lower levels of FLAG-CD36 were expressed in Selk−/− BMDMs, twice the amount of lysate protein from Selk−/− BMDMs (200 μg) compared with WT BMDM lysate protein (100 μg) was incubated with the anti-FLAG-coated beads prior to biotin labeling. Results show that despite slightly more immunoprecipitated FLAG-CD36 from the Selk−/− BMDM lysate, there was much less palmitate detected on this protein compared with WT BMDM FLAG-CD36 (Fig. 4D). Overall, these results suggest that Selk−/− leads to decreased stability of CD36 as a result of reduced palmitoylation at the ER.
Differences in modLDL uptake by Selk−/− macrophages are attributable to reduced CD36 expression
To ensure that the differences in modLDL uptake were a result of the differences in CD36 surface expression, as outlined in detail above, we used a blocking IgA against CD36 in modLDL uptake experiments. Pretreatment with anti-CD36 IgA lowered Dil-AcLDL uptake in WT BMDMs to levels equivalent to those exhibited by Selk−/− BMDMs (Fig. 5). In addition, pretreatment of Selk−/− BMDMs with the anti-CD36-blocking IgA had no effect on Dil-AcLDL uptake compared with untreated or control IgA-treated Selk−/− BMDMs. Overall, these results strongly indicate that the differences in modLDL uptake between WT and Selk−/− BMDMs are indeed a result of the presence of functional CD36 on the cell surface.
Figure 5. Decreased uptake of modLDL by Selk−/− macrophages is a result of TNF-α-induced CD36 expression.

Untreated or TNF-α-treated WT and Selk−/− BMDMs were preincubated with anti-CD36 IgA to block this scavenger receptor or with a control IgA. Dil-AcLDL was then added for 4 h and levels of ingested Dil-AcLDL determined by plate-based fluorimetry. *P < 0.05.
Macrophage-associated Selk is increased in atherosclerotic lesions
The effects of Selk−/− on inflammatory stimulation of macrophages and foam cell formation led us to investigate whether Selk is involved in the chronic inflammatory processes underlying atherosclerotic lesion formation. We first investigated the levels of Selk in aortas containing atherosclerotic plaques compared with healthy aortas. Atherosclerotic tissues from Ldlr−/− mice fed an atherogenic HFD from 0 to 36 weeks were analyzed for the presence of Selk using immunofluorescence microscopy. Selk-positive cells were detected at higher frequency in aortic roots of Ldlr−/− mice fed HFD compared with those fed normal chow, with Selk-positive cells particularly evident in the intima and media of Ldlr−/− mice with HFD-induced atherosclerosis (Fig. 6A). Similarly, Western blot data indicated that full-length Selk (∼16 kDa) was expressed in large arteries of mice fed normal chow or HFD (Fig. 6B). However, there was an increase in the expression of the truncated form of Selk (∼12 kDa) in mice fed the HFD compared with controls. This is important given our previous work showing that truncated Selk is mainly present in macrophages but not in lymphocytes, and truncated Selk does not function to promote macrophage migration-like, full-length Selk [21]. Taken together, these data suggest a correlation between levels of macrophage-associated Selk and atherosclerotic lesion formation and/or progression, which led to subsequent studies to determine the role of Selk in these processes.
Figure 6. Macrophage-associated Selk is detected in atherosclerotic plaques.

(A) Immunofluorescence microscopy was performed on aortic roots from Ldlr−/− mice fed a HFD for 2, 12, and 36 weeks. Representative images of Selk-positive cells (green) within the aortic intima and in the plaque. Nuclei (blue) were stained with DAPI (original scale bar: 100 μm). (B) Western blot analyses of aorta tissue from Ldlr−/− mice fed a normal chow or a HFD indicated higher levels of the lower band (12 kDa) that corresponds to truncated Selk, found predominantly in macrophages. *P < 0.05.
Selk−/− reduces atherosclerosis in Ldlr−/− mice
We next investigated the role of Selk in immune cells during atherosclerotic lesion formation using a BMT model combined with HFD. Equivalent reconstitution of Ldlr−/− mice with WT or Selk−/− BM was confirmed (Supplemental Fig. 2), and the formation of atherosclerotic lesions was then evaluated. Consistent with the results described above, Selk-positive cells were detected in advanced plaques in the aortic roots of chimeric Ldlr−/− mice transplanted with WT BM fed a HFD (Fig. 7A). As expected, Selk-positive cells were not found in Ldlr−/− mice reconstituted with Selk−/− BM fed a HFD. Oil Red O staining was used to analyze the extent of atherosclerotic lesion formation after 12 weeks of HFD feeding. Plaque formation in the aorta of Ldlr−/− mice reconstituted with Selk−/− BM was reduced significantly compared with Ldlr−/− mice transplanted with WT BM (Fig. 7B). The differences in plaque formation were not a result of changes in systemic lipid metabolism, as indicated by similar levels of total plasma cholesterol, triglycerides, VLDL/LDL, and HDL between Ldlr−/− mice reconstituted with WT versus Selk−/− BM (Supplemental Fig. 3). Immune cell migration is one function for which Selk is important, so we next performed quantitative immunofluorescence analysis of the cellular composition in aortic root plaques of Ldlr−/− mice reconstituted with WT BM or Selk−/− BM. Surprisingly, numbers of macrophages, SMCs, and T cells did not differ between WT and Selk−/− BM-reconstituted mice (Supplemental Fig. 4). Furthermore, the relative numbers of TUNEL-positive apoptotic cells, as well as the necrotic core size in atherosclerotic lesions of these mice, showed no significant differences (Supplemental Fig. 4). Taken together, these data suggest that Selk−/− did not influence migration of monocytes/macrophages and T cells into the arterial wall and did not affect SMC proliferation. Thus, the attenuation of atherosclerosis that we observed in the Selk−/− BM-transplanted mice is likely a result of reduced foam cell formation in Selk−/− macrophages caused by impaired palmitoylation of CD36.
Figure 7. Decreased atherosclerotic plaque in Selk BMT mice.
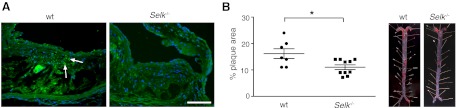
(A) Selk-positive cells (green) detected in atherosclerotic plaques from mice reconstituted with WT BM versus mice reconstituted with Selk−/− BM. (B) Quantification of Oil Red O+ lipid depositions in the aorta (left) of Ldlr−/− mice reconstituted with WT BM or with Selk−/− BM (right; representative images). *P < 0.05.
DISCUSSION
Our previously published work revealed a role for Selk in receptor-mediated calcium flux in immune cells that affects calcium-dependent signaling and effector functions. In particular, macrophages stimulated through FcγRs with IgG-coated particles, through the TLR4 with LPS, or through chemokine receptors with specific chemokines, such as MCP-1, trigger a rapid increase in cytosolic calcium that is dependent on Selk [12, 13, 21]. Surprisingly, in the present study, Selk−/− did not decrease the presence of immune cells, including monocytes/macrophages, in the atherosclerotic plaques. In contrast to other proinflammatory mediators, immune cell influx in atherogenesis likely occurs in a Selk-independent manner. The data presented in the current study, however, demonstrate a new role of Selk, namely in macrophage uptake of modLDL and foam cell formation. More specifically, Selk promotes modLDL uptake via increased surface expression of aggregated CD36 with a requirement for Selk in the palmitoylation of CD36. These findings provide a new link among the ER selenoprotein, Selk, and regulation of CD36 expression that contributes to foam cell formation and atherosclerosis.
S-palmitoylation of cysteine residues is a common, post-translational modification occurring on integral and peripheral membrane proteins [22] that contribute to membrane association. Unlike prenylation or myristoylation, however, palmitoylation can be reversed, thus providing additional mechanisms to regulate the functional activities of integral and peripheral membrane proteins [23]. The palmitoylation of CD36 occurs on N- and C-termini and is a requirement for its stabilization within the cytoplasm [20, 24]. Whereas our data implicate Selk in the palmitoylation and stable expression of CD36, it is not clear whether Selk is involved directly or indirectly in the transfer of palmitate to CD36. Selk may act to recruit palmitoyl acyl transferases, which are required for palmitoylation, to the surface of the ER. It is interesting to note that Selk contains a SH3-binding domain, and one palmitoyl acyl transferase, ZDHHC6, contains a SH3 domain. Thus, Selk may act to tether this enzyme to the ER membrane and facilitate palmitoylation of newly synthesized CD36 at this site. In this manner, Selk may be involved in the palmitoylation of those proteins that serve as substrates for ZDHHC6 and not proteins targeted by other palmitoyl acyl transferases. This may explain why Cav-1 levels were not affected as strongly by Selk−/− as CD36 in the membrane fractionation studies shown in Fig. 6. It has been shown that Cav-1 is palmitoylated, and this event is required for its localization and interactions with certain proteins [25]. Further studies are underway to determine the precise role of Selk in palmitoylation and which proteins rely on Selk for this important modification to occur.
TNF-α plays multiple roles in the development of plaque formation, including induced expression of vascular cell adhesion molecules and activation of macrophages [10, 26, 27]. A recent study has linked TNF-α stimulation of macrophages to calcium-dependent signaling [28]. An important aspect of macrophage activation contributing to foam cell formation and pathology is the high expression of the CD36 scavenger receptor on lipid-laden macrophages [29]. TNF-α has been implicated in the regulation of CD36 expression on macrophages within the atherosclerotic plaque [30], as have other cytokines, such as IL-10 [31]. Our data suggest that Selk functions in response to TNF-α stimulation of macrophages to enhance the expression and distribution of CD36 on the plasma membrane. As a result, Selk−/− lowers LDL uptake and foam cell formation. This effect was not observed for macrophages stimulated with IL-10, which is consistent with our previous work showing an important role for Selk only upon proinflammatory stimulation of immune cells, particularly macrophages [12]. Whereas our data implicate the glycoprotein CD36 as a key scavenger receptor that is regulated by Selk, uptake of modLDL is not the only physiological role for this surface receptor. CD36 is also known as glycoprotein IIIb/IV or fatty acid translocase, and in addition to monocytes/macrophages, it is expressed on the surface of platelets, microvascular endothelial cell, SMCs, cardiomyocytes, and other cells of the cardiovascular system [32]. Our data demonstrate a novel process of TNF-α-induced clustering of CD36 on the surface of cells, which promotes uptake of oxLDL. Similar types of dynamic reorganization of CD36 have been shown in other cell types, with CD36 shown to translocate from intracellular stores, presumably endosomes, to the plasma membrane upon various physiological stimuli [33, 34]. The Rab and Raf protein families are involved in vesicle transport, and Rab11, in particular, has been linked to CD36 transport [35]. In addition to the effects of Selk on palmitoylation and stabilization of this protein within the cell, Selk-dependent calcium flux may be required for directing Rab or Raf proteins for CD36 translocation to the cell surface upon stimulation.
Aside from vesicular trafficking, surface CD36 levels are regulated by protein degradation [36], and Selk may be required to retain proper levels of CD36 protein in the cell, as suggested by our Western blot analysis of whole cell lysate. Expression of CD36 on macrophages can also be regulated by a variety of other molecules, such as peroxisome proliferator-activated receptor γ-mediated oxLDLR1 [37] and adenosine A(2A) receptor, a GPCR known to regulate inflammatory responses by raising cAMP synthesis [38]. Interestingly, activation of the integrin αMβ2 (CD11b/CD18) on alternatively activated macrophages was recently shown to inhibit CD36 expression by blocking tyrosine kinase 2- and Jak2-mediated pathways [39]. Overall, the findings described herein provide new insight into the role of Selk and potentially dietary selenium in foam cell formation and atherosclerosis. Studies are ongoing to further define molecular mechanisms by which this may occur.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by U.S. National Institutes of Health grants R21AT004844 and R01AI089999 to P.R.H. and R01HL075677 and R01HL081663, as well as Hawaii Community Foundation grant 10ADVC-47037 to W.A.B. Core facilities were supported by P20GM103516, P20RR016453, G12RR003061, and G12MD007601. This research was also supported by the NSFC (20975045) and the NSFC-U.S. National Institutes of Health Biomedical Collaborative Research grant (81261120408).
The authors thank Monica Montgomery and Eric Collier for excellent technical assistance.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- −/−
- deficient
- AcLDL
- acetylated LDL
- BM
- bone marrow
- BMDM
- bone marrow-derived macrophage
- BMT
- bone marrow transplantation
- Cav-1
- caveolin 1
- DiI
- 1,1'-dioctadecyl – 3,3,3',3'-tetramethyl-indocarbocyanine perchlorat
- FLAG-CD36
- FLAG-tagged CD36
- HFD
- high-fat diet
- MBS
- MES buffered saline
- modLDL
- modified LDL
- NSFC
- National Natural Science Foundation of China
- oxLDL
- oxidized LDL
- Selk
- Selenoprotein K
- SH3
- Src homology 3
- SMC
- smooth muscle cell
- SR-A
- scavenger receptor A
AUTHORSHIP
S.M. performed the cholesterol homeostasis studies and mouse BMT studies, as well as contributed to the preparation of the manuscript. Y.B. helped with the in vivo studies and performed the microscopy. Experiments were performed by Z.H., F.W.H., G.J.F., A.H.R., and R.L.N., and Z.H. helped prepare the manuscript. W.A.B. and P.R.H. conceived of the studies and prepared the manuscript.
DISCLOSURES
The authors have no conflict of interest to disclose.
REFERENCES
- 1. Roger V. L., Go A. S., Lloyd-Jones D. M., Adams R. J., Berry J. D., Brown T. M., Carnethon M. R., Dai S., de Simone G., Ford E. S.., et al. (2011) Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 123, e18–e209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lloyd-Jones D., Adams R. J., Brown T. M., Carnethon M., Dai S., De Simone G., Ferguson T. B., Ford E., Furie K., Gillespie C.., et al. (2010) Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation 121, e46–e215 [DOI] [PubMed] [Google Scholar]
- 3. Hansson G. K., Robertson A. K., Soderberg-Naucler C. (2006) Inflammation and atherosclerosis. Ann. Rev. Pathol. 1, 297–329 [DOI] [PubMed] [Google Scholar]
- 4. Weber C., Zernecke A., Libby P. (2008) The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat. Rev. Immunol. 8, 802–815 [DOI] [PubMed] [Google Scholar]
- 5. Hansson G. K., Libby P. (2006) The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 6, 508–519 [DOI] [PubMed] [Google Scholar]
- 6. Ross R. (1999) Atherosclerosis is an inflammatory disease. Am. Heart J. 138, S419–S420 [DOI] [PubMed] [Google Scholar]
- 7. Yoshida H., Quehenberger O., Kondratenko N., Green S., Steinberg D. (1998) Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 18, 794–802 [DOI] [PubMed] [Google Scholar]
- 8. Febbraio M., Podrez E. A., Smith J. D., Hajjar D. P., Hazen S. L., Hoff H. F., Sharma K., Silverstein R. L. (2000) Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 105, 1049–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki H., Kurihara Y., Takeya M., Kamada N., Kataoka M., Jishage K., Ueda O., Sakaguchi H., Higashi T., Suzuki T.., et al. (1997) A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 386, 292–296 [DOI] [PubMed] [Google Scholar]
- 10. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohta H., Wada H., Niwa T., Kirii H., Iwamoto N., Fujii H., Saito K., Sekikawa K., Seishima M. (2005) Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180, 11–17 [DOI] [PubMed] [Google Scholar]
- 12. Verma S., Hoffmann F. W., Kumar M., Huang Z., Roe K., Nguyen-Wu E., Hashimoto A. S., Hoffmann P. R. (2011) Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J. Immunol. 186, 2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Z., Rose A. H., Hoffmann P. R. (2012) The role of selenium in inflammation and immunity: from molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 16, 705–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Z., Hoffmann F. W., Fay J. D., Hashimoto A. C., Chapagain M. L., Kaufusi P. H., Hoffmann P. R. (2012) Stimulation of unprimed macrophages with immune complexes triggers a low output of nitric oxide by calcium-dependent neuronal nitric-oxide synthase. J. Biol. Chem. 287, 4492–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matsui T., Nagoshi T., Hong E. G., Luptak I., Hartil K., Li L., Gorovits N., Charron M. J., Kim J. K., Tian R., Rosenzweig A. (2006) Effects of chronic Akt activation on glucose uptake in the heart. Am. J. Physiol. Endocrinol. Metab. 290, E789–E797 [DOI] [PubMed] [Google Scholar]
- 16. Han X., Kitamoto S., Wang H., Boisvert W. A. (2010) Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 24, 2869–2880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boisvert W. A., Black A. S., Curtiss L. K. (1999) ApoA1 reduces free cholesterol accumulation in atherosclerotic lesions of ApoE-deficient mice transplanted with ApoE-expressing macrophages. Arterioscler. Thromb. Vasc. Biol. 19, 525–530 [DOI] [PubMed] [Google Scholar]
- 18. Ehehalt R., Sparla R., Kulaksiz H., Herrmann T., Fullekrug J., Stremmel W. (2008) Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains (lipid rafts). BMC Cell Biol. 9, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eyre N. S., Cleland L. G., Tandon N. N., Mayrhofer G. (2007) Importance of the carboxyl terminus of FAT/CD36 for plasma membrane localization and function in long-chain fatty acid uptake. J. Lipid Res. 48, 528–542 [DOI] [PubMed] [Google Scholar]
- 20. Thorne R. F., Ralston K. J., de Bock C. E., Mhaidat N. M., Zhang X. D., Boyd A. W., Burns G. F. (2010) Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta 1803, 1298–1307 [DOI] [PubMed] [Google Scholar]
- 21. Huang Z., Hoffmann F. W., Norton R. L., Hashimoto A. C., Hoffmann P. R. (2011) Selenoprotein K is a novel target of m-calpain, and cleavage is regulated by Toll-like receptor-induced calpastatin in macrophages. J. Biol. Chem. 286, 34830–34838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rocks O., Gerauer M., Vartak N., Koch S., Huang Z. P., Pechlivanis M., Kuhlmann J., Brunsveld L., Chandra A., Ellinger B., Waldmann H., Bastiaens P. I. (2010) The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–471 [DOI] [PubMed] [Google Scholar]
- 23. Bijlmakers M. J., Marsh M. (2003) The on-off story of protein palmitoylation. Trends Cell Biol. 13, 32–42 [DOI] [PubMed] [Google Scholar]
- 24. Tao N., Wagner S. J., Lublin D. M. (1996) CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 271, 22315–22320 [DOI] [PubMed] [Google Scholar]
- 25. Di Vizio D., Adam R. M., Kim J., Kim R., Sotgia F., Williams T., Demichelis F., Solomon K. R., Loda M., Rubin M. A., Lisanti M. P., Freeman M. R. (2008) Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle 7, 2257–2267 [DOI] [PubMed] [Google Scholar]
- 26. Modur V., Zimmerman G. A., Prescott S. M., McIntyre T. M. (1996) Endothelial cell inflammatory responses to tumor necrosis factor α. Ceramide-dependent and -independent mitogen-activated protein kinase cascades. J. Biol. Chem. 271, 13094–13102 [DOI] [PubMed] [Google Scholar]
- 27. Springer T. A. (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314 [DOI] [PubMed] [Google Scholar]
- 28. Yarilina A., Xu K., Chen J., Ivashkiv L. B. (2011) TNF activates calcium-nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc. Natl. Acad. Sci. USA 108, 1573–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakata A., Nakagawa Y., Nishida M., Nozaki S., Miyagawa J., Nakagawa T., Tamura R., Matsumoto K., Kameda-Takemura K., Yamashita S., Matsuzawa Y. (1999) CD36, a novel receptor for oxidized low-density lipoproteins, is highly expressed on lipid-laden macrophages in human atherosclerotic aorta. Arterioscler. Thromb. Vasc. Biol. 19, 1333–1339 [DOI] [PubMed] [Google Scholar]
- 30. Boyer J. F., Balard P., Authier H., Faucon B., Bernad J., Mazieres B., Davignon J. L., Cantagrel A., Pipy B., Constantin A. (2007) Tumor necrosis factor α and adalimumab differentially regulate CD36 expression in human monocytes. Arthritis Res. Ther. 9, R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han X., Kitamoto S., Lian Q., Boisvert W. A. (2009) Interleukin-10 facilitates both cholesterol uptake and efflux in macrophages. J. Biol. Chem. 284, 32950–32958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silverstein R. L., Febbraio M. (2009) CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal 2, re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Steinbusch L. K., Schwenk R. W., Ouwens D. M., Diamant M., Glatz J. F., Luiken J. J. (2011) Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell. Mol. Life Sci. 68, 2525–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Coort S. L., Bonen A., van der Vusse G. J., Glatz J. F., Luiken J. J. (2007) Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: role of sarcolemmal substrate transporters. Mol. Cell. Biochem. 299, 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwenk R. W., Luiken J. J., Eckel J. (2007) FIP2 and Rip11 specify Rab11a-mediated cellular distribution of GLUT4 and FAT/CD36 in H9c2-hIR cells. Biochem. Biophys. Res. Commun. 363, 119–125 [DOI] [PubMed] [Google Scholar]
- 36. Tran T. T., Poirier H., Clement L., Nassir F., Pelsers M. M., Petit V., Degrace P., Monnot M. C., Glatz J. F., Abumrad N. A., Besnard P., Niot I. (2011) Luminal lipid regulates CD36 levels and downstream signaling to stimulate chylomicron synthesis. J. Biol. Chem. 286, 25201–25210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dai Y., Su W., Ding Z., Wang X., Mercanti F., Chen M., Raina S., Mehta J. L. (2013) Regulation of MSR-1 and CD36 in macrophages by LOX-1 mediated through PPAR-γ. Biochem. Biophys. Res. Commun. 431, 496–500 [DOI] [PubMed] [Google Scholar]
- 38. Voloshyna I., Carsons S., Littlefield M. J., Rieger J. M., Figler R., Reiss A. B. (2013) Adenosine A(2A) receptor activation supports an atheroprotective cholesterol balance in human macrophages and endothelial cells. Biochim. Biophys. Acta 1831, 407–416 [DOI] [PubMed] [Google Scholar]
- 39. Yakubenko V. P., Hsi L. C., Cathcart M. K., Bhattacharjee A. (2013) From macrophage interleukin-13 receptor to foam cell formation: mechanisms for αMβ2 integrin interference. J. Biol. Chem. 288, 2778–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.