

TLR2 signaling modulates K/BxN serum transfer arthritis by enhancing the expression of immune complex-induced IL-10.
Keywords: macrophage, inflammation, cytokine, IgG Fc receptor, osteoclastogenesis
Abstract
RA is a chronic inflammatory disease characterized by the persistent expression of inflammatory cytokines from macrophages, which may be mediated, in part, through TLR2 signaling. Earlier studies demonstrate a role for TLR2 signaling in dampening the arthritis in IL-1Ra−/− mice, which was mediated through T cells. This study was performed to determine whether TLR2 signaling plays a role in the pathogenesis of T cell-independent arthritis triggered by transferring serum from K/BxN mice. We documented more severe arthritis in Tlr2−/− mice compared with WT controls. The Tlr2−/− mice also demonstrated increased inflammation, erosion, pannus formation, and osteoclastogenesis, as well as increased IL-1β and decreased IL-10 within the joints. In vitro bone marrow-differentiated macrophages expressed comparable levels of activating and inhibitory FcγRs, however when stimulated with immune complexes, the Tlr2−/− macrophages expressed decreased IL-10 and reduced activation of Akt and ERK. Our findings indicate that Tlr2−/− promotes the effector phase of arthritis through decreased IL-10 by macrophages, which is important, not only as an anti-inflammatory cytokine but also in restraining the differentiation and activation of osteoclasts.
Introduction
RA is a chronic inflammatory disease in which the release of inflammatory cytokines, such as TNF-α and IL-6, by synovial tissue macrophages is critically important [1]. In patients who are shared epitope-positive, ACPA form immune complexes that are capable of promoting disease pathogenesis [2–4]. Specifically, ACPA-containing immune complexes are capable of complement activation and the induction of proinflammatory cytokines from macrophages, including those isolated from the RA joint [2, 3, 5]. Clinical studies document that once structural joint damage occurs, patients become more resistant to therapeutic intervention [6, 7]. Although the mechanisms for this resistance and the persistent activation of joint macrophages in RA are not fully understood, the engagement of TLRs by endogenously expressed Toll ligands may contribute to disease progression [8, 9].
The expression of TLR2 is increased on macrophages from the synovial fluid of patients with RA [10]. These macrophages demonstrate increased activation by microbial TLR ligands compared with in vitro-differentiated control macrophages, as well as macrophages from the joints of patients with other forms of inflammatory arthritis, such as psoriatic arthritis [10]. Further, endogenous TLR2 ligands, such as gp96 and Snapin, are highly expressed in the RA joint [11, 12]. These endogenous ligands are capable of activating macrophages, including those obtained from RA synovial fluid, to induce the expression of proinflammatory cytokines, such as TNF-α and IL-8 [12, 13]. These observations suggest that the TLR2 signaling pathway is a potential therapeutic target in RA.
However, the potential value of TLR2 as a therapeutic target is uncertain. Studies with Tlr2−/− mice suggest that TLR2 may suppress arthritis. IL-1Ra−/− mice demonstrate a spontaneous arthritis that is mediated through TLR4 and IL-17 [14]. However, when IL-1Ra−/− mice are crossed with Tlr2−/− mice, the arthritis is more severe [14], although arthritis induced by the overexpression of IL-1β is independent of TLR2 [15]. The increased severity of the arthritis is associated with increased IFN-γ and reduced TGF-β and T regulatory cell function, suggesting a role for T cells [14]. Further, TLR2 signaling, in addition to inducing proinflammatory cytokines, has been implicated in the expression of anti-inflammatory IL-10 [16].
To determine if the effector phase of arthritis is affected by deletion of TLR2, we performed studies with the K/BxN serum transfer model, which is mediated by antibodies to GPI, resulting in an immune complex-mediated arthritis [17]. The transfer of anti-GPI serum resulted in significantly worse arthritis in Tlr2−/− mice compared with WT controls. Histologic examination demonstrated more inflammation and joint destruction. Examination of the joint extracts revealed increased IL-1β and decreased IL-10 in the Tlr2−/− mice. There was no difference in the expression of inhibitory or activating FcγR in the Tlr2−/− mice. However, activation of Tlr2−/− macrophages with immune complexes resulted in significantly reduced IL-10, whereas there was no difference in TNF-α compared with the controls. Macrophages from the Tlr2−/− mice demonstrated decreased activation of Akt, GSK3, and ERK but not p38 following activation with immune complexes. These observations demonstrate that deletion of TLR2 promotes the effector phase of arthritis, mediated by a decreased IL-10, resulting from reduced immune complex-mediated activation of ERK and Akt.
MATERIALS AND METHODS
Mouse lines
WT and homozygote Tlr2tm1Kir mutation (Tlr2−/−) mice on the C57Bl/6 background are from The Jackson Laboratory (Bar Harbor, ME, USA). The Tlr2−/− mice are viable, normal-sized, and display neither gross physical nor behavioral abnormalities. WT and Tlr2−/− lines were maintained by inbreeding in the Northwestern University Barrier Animal Facility for more than 6 months. All animal procedures were approved by the Office of Research Safety and the Institutional Animal Care and Use Committee of Northwestern University.
K/BxN serum transfer arthritis model
K/BxN mice were generated and anti-GPI serum collected at 8–9 weeks of age [17]. Arthritis was induced by injecting 100 μl anti-GPI serum i.p. The development of arthritis was assessed by measuring the hind-ankle thickness by a caliper and by grading the clinical index of the scale of 0–3 of each for all four ankles/paws (maximum=12), as described [18, 19]. At the time of harvesting, ankles were dissected quickly and skin removed and were stored in 10% neutral-buffered formalin for histological and osteoclast analysis or at −80°C for ELISA.
Histologic analysis
Paraffin-embedded ankle sections were prepared as described [18, 19] and subjected to H&E staining for histology and TRAP staining for osteoclasts [20]. The numbers of osteoclasts were countered around the region of talus bone and presented as number/mm2 bone surface. Ankle histopathology was evaluated by blinded observers, scoring 0–5 for inflammation, pannus formation, and bone erosion, as described previously [18, 19].
Quantification of cytokines/chemokines/ligands in ankle homogenates
Day 7 postarthritis ankles were homogenized and supernatants collected for analysis as described [18, 19, 21]. The total protein concentration of the ankle homogenates was determined by BCA protein assay reagents (Thermo Scientific, Rockford, IL, USA). Levels of IL-1β, IL-10, RANKL, and OPG were determined by ELISA (DuoSets; R&D Systems, Minneapolis, MN, USA), following the manufacturer's instructions. The concentrations of each indicated protein tested were adjusted to milligrams of total ankle protein.
Macrophages differentiation
GM-CSF-mediated bone marrow-derived macrophages were generated from whole bone marrow cells isolated from the femur and tibia of WT and Tlr2−/− mice, followed by in vitro differentiation in RPMI media, supplemented with 10% (v/v) FBS, 20 ng/ml mouse GM-CSF (R&D Systems), and 0.1% (v/v) of 2-ME (Gibco/Invitrogen, Carlsbad, CA, USA), as described [22]. At Days 7–9, cells were collected for experiments.
Detection of FcγR-mediated macrophage activation
Mouse IgG (Equitech-Bio, Kerrville, TX, USA) was diluted in PBS, supplemented with 10 μg/ml Polymyxin B, and used to coat 96-well microtiter plates at 10 or 30 μg/ml or 12-well plates at 50 μg/ml overnight at room temperature [23, 24]. Plates were washed with PBS three times before cells were added. The IgG-coated wells treated in this manner demonstrated no detectable TLR2 or TLR4 ligand contamination, as determined by activation of HEK cells expressing TLR2 or TLR4, whereas the TLR2 ligand Pam3CSK4 and the TLR4 ligand LPS served as the positive controls. Bone marrow-derived macrophages were collected by gentle pipetting, washing with PBS, and resting for 1 h prior to addition to the wells at 1 × 105/well for 96-well plate or 2 × 106/well for 12-well plates in RPMI medium supplemented with 10% FBS and 1 μg/ml Polymyxin B. The supernatants or cell lysates were harvested at the times indicated in the figures.
Macrophage-secreted cytokines
The FcγR-mediated macrophage activation in 96-well plates was assessed by measuring the TNF-α and IL-10 in 4-h culture supernatants by ELISA (DuoSets; R&D Systems). At the time of terminating activation, cell numbers and viability were determined by MTT cleavage assay, as instructed by the manufacturer (Sigma-Aldrich, St. Louis, MO, USA). The concentrations of TNF-α and IL-10 were adjusted by the unit of MTT (OD 490 nm).
Immunoblotting
The FcγR-mediated macrophage signaling was assessed by immunoblot analysis. Whole cell extracts were harvested at indicated time-points in cell lysis buffer, supplemented with 1× protease inhibitor and 1× phosphatase inhibitor cocktails (Sigma-Aldrich). The protein extracts (10 μg) were resolved by 10% SDS-PAGE and transfered to a PVDF membrane (Immobilon-P; Millipore, Bedford, MA, USA), as described [11, 25]. The blots were first probed with phospho-antibodies to Akt, p38, ERK, and GSK3 (Cell Signaling Technology, Danvers, MA, USA). After stripping in stripping buffer (Thermo Scientific), the same blot was reprobed with antibodies to GAPDH (Cell Signaling Technology). The immunoblots were developed using an ECL Prime Western blotting detection reagent (GE Healthcare, Buckinghamshire, UK) and image acquisition system (UltraLum). Densitometry analysis of immunoblots was performed by ImageJ software.
FcγR expression
FcγR expression on the cell membrane was determined by immunophenotyping using antibody mixture to CD16/CD32, which recognizes FcγII and FcγIII, followed by flow cytometry [11, 25]. FcγR isoform expression at the mRNA level was determined by quantitative real-time RT-PCR using primers of FcγRI, FcγRIIb, FcγRIII, FcγRIV, and FcϵRI (TaqMan gene expression essay, Applied Biosystems, Life Technologies, Carlsbad, CA, USA).
Statistical analysis
The two-sided t-test was used to analyze differences between two groups. For samples that failed the normality test, Mann-Whitney rank test was performed. Correlations were determined by Pearson linear correlation or Spearman's nonparametric correlation if the data analyzed had a non-Gaussian distribution. Significance levels were set at 0.05.
RESULTS
Arthritis is increased in Tlr2−/− mice
Studies were performed to determine the role of TLR2 in the effector phase of serum transfer arthritis induced by anti-GPI antibody obtained from K/BxN mice [17–19]. WT mice demonstrated arthritis that peaked at Day 7 and then plateaued (Fig. 1A and B). The Tlr2−/− mice exhibited a similar clinical course, however the arthritis was more severe, beginning on the first clinical examination on Days 2–3 (Fig. 1A and B). Histological examination on Day 9 revealed more severe disease in the joints of the Tlr2−/− mice (Fig. 2A). Specifically, there was a significant increase of inflammation, pannus formation, and erosion in the Tlr2−/− mice compared with the WT controls (Fig. 2B).
Figure 1. Increased anti-GPI serum transfer-induced arthritis in Tlr2−/− mice.
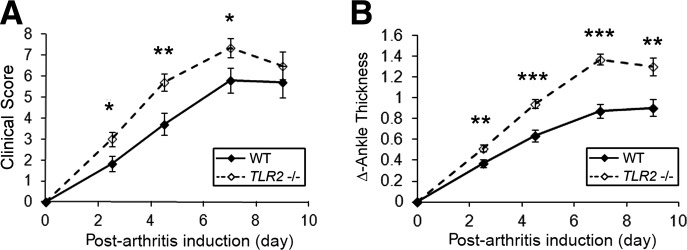
(A) Tlr2−/− and WT mice were subjected to serum transfer-induced arthritis by i.p. administration of 100 μl K/BxN anti-GPI serum. The course of the arthritis was determined by the clinical activity score (maximum 12) and (B) the change of ankle swelling (Δ) in millimeters of the two hind ankles (ankle thickness). Data represent five independent experiments, presented as the mean ± se of 31 WT and 37 Tlr2−/− mice on Days 0–7 and 24 mice in each group on Day 9. *P < 0.05; **P < 0.01; ***P < 0.001 between the Tlr2−/− and WT mice on the indicated days.
Figure 2. Increased inflammation, bone erosion, and pannus formation in Tlr2−/− mice.

(A) Representative H&E image from the ankles of WT and Tlr2−/− mice harvested on Day 9 postarthritis induction. The areas in the boxes are enlarged in the panel below. B, Bone and arrows indicate pannus and bone erosion. (B) The histologic analysis of ankles (n=12/group) harvested on Day 9. The mean ± se is indicated for each group. **P < 0.01; ***P < 0.001 between the Tlr2−/− and WT mice. The arrows identify bone erosion and pannus formation.
To identify potential mechanisms for the enhanced arthritis in Tlr2−/− mice, we examined cytokine expression in joint extracts. Consistent with the known role of IL-1β in anti-GPI-mediated arthritis, IL-1β levels were increased significantly in the joints of Tlr2−/− mice compared with the controls (Fig. 3A). Furthermore, IL-10, which dampens inflammation, was reduced significantly in the Tlr2−/− compared with the control mice (Fig. 3B). The concentration of TNF-α, which is not critical to the pathogenesis of serum transfer arthritis, was not different in the ankles between the Tlr2−/− and control mice (Fig. 3C). When the Tlr2−/− and WT mice were examined together, the concentration of IL-1β in the joints positively correlated (Fig. 3D; P<0.002) with change in ankle thickness, whereas IL-10 demonstrated a negative relationship (Fig. 3E; P<0.001), and there was no significant correlation with TNF-α (Fig. 3F). These observations suggest that Tlr2−/− results in unbalanced pro- and anti-inflammatory cytokine production, induced by immune complexes in this model.
Figure 3. Increased IL-1β and decreased IL-10 in Tlr2−/− mice.
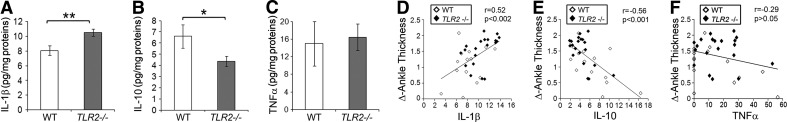
Ankles from Day 7 postarthritis were homogenized and the concentration of IL-1β (A), IL-10 (B), and TNF-α (C) was measured and presented as pg/mg protein for Tlr2−/− (n=20) and WT (n=14) mice. The data represent the mean ± se[scap]; *P < 0.05; **P < 0.01 between the groups. The correlations between the clinical arthritis (Δ of ankle thickness) and IL-1β (D), IL-10 (E), and TNF-α (F) are presented. The r and the P value are indicated in the right corner of D–F.
Increased osteoclast formation in joints of arthritic Tlr2−/− mice
IL-1β stimulates, whereas IL-10 inhibits osteoclastogenesis [26, 27]. Their unbalanced expression in joints of arthritic Tlr2−/− mice led us to examine whether osteoclast formation is altered in the absence of TLR2 during inflammation. TRAP staining of joint sections from Tlr2−/− and WT mice at 9 days post-K/BxN serum injection demonstrated significantly increased osteoclasts in Tlr2−/− mice (Fig. 4A), which was confirmed by histomorphormetric analysis (Fig. 4B). Interestingly, we did not detect differences in RANKL levels in the ankle extracts by ELISA between the Tlr2−/− and control mice (Fig. 4C). However, there was a trend (P=0.056) toward reduction of OPG levels in the Tlr2−/− mice (Fig. 4D), and there was a significant negative correlation (r=−0.72; P<0.001) between joint swelling and OPG when both groups of mice were analyzed together (Fig. 4E). These observations suggest that TLR2 deletion results in increased osteoclastogenesis in the K/BxN serum-induced arthritis model, which may contribute to more severe joint bone erosion in these mice.
Figure 4. Increased osteoclastogenesis in Tlr2−/− mice.

(A) Representatives of TRAP-stained sections from mice at Day 9 postarthritis. Numerous TRAP-positive osteoclasts (arrowheads) and severe bone erosions (arrows) were present in the sections from Tlr2−/− mice. (B) The numbers of osteoclast/mm2 bone surface around the talus bone region were counted (n=14 ankles/group). (C and D) Levels of RANKL and OPG were measured on ankle homogenates from mice on Day 7 postarthritis induction. (E) The correlation between Δ of ankle thickness and OPG levels was analyzed in WT and Tlr2−/− mice. The correlation between RANKL and ankle swelling was not significant. The data presented from 20 Tlr2−/− and 14 WT ankles. *P < 0.05 between the groups.
No difference in FcR expression in macrophages
Anti-GPI serum transfer arthritis is dependent on immune complex-mediated FcγR activation [28–31]. Therefore, the expression of various FcγRs by bone marrow-derived macrophages was examined by quantitative RT-PCR and FACS analysis. The expression of F4/80 and CD11b was comparable on the macrophages (Fig. 5A). No difference in the expression levels of surface FcγRII and FcγRIII, which are recognized by the anti-CD16/CD32 antibody, by FACS was observed between Tlr2−/− and control macrophages (Fig. 5B). There was a slight reduction of FcγRIV mRNA expression levels in the Tlr2−/− macrophages but no other difference in activating FcγR isoforms and no decrease of the inhibitory FcγRIIb isoform between the Tlr2−/− and control mice (Fig. 5C). Furthermore, there were no differences in the expression of the FcγR isoforms by peritoneal macrophages isolated from Tlr2−/− and control mice, 9 days following serum transfer (data not shown). These observations demonstrate that the increased arthritis observed in the Tlr2−/− mice is not a result of an alteration in the expression of inhibitory and activating FcγRs in the Tlr2−/− mice.
Figure 5. Decreased immune complex induced IL-10 by Tlr2−/− macrophages.

Bone marrow-derived macrophages from WT and Tlr2−/− mice were used to examine FcγR signaling. (A) Representative flow cytometry using in vitro-differentiated macrophages from WT and Tlr2−/− mice demonstrate no difference in the expression of CD11b or F4/80. (B) The FcγR expression on the cell membrane of macrophages (upper-right quadrant gating of A) was determined using an anti-CD16/CD32 antibody, which recognizes FcγRII and FcγRIII. (C) The expression of FcγR isoforms by mRNA using in vitro-differentiated macrophages, was determined by quantitative real-time RT-PCR using primers of FcγRI, FcγRIIb, FcγRIII, FcγRIV, and FcϵRI. (D and E) These in vitro-differentiated macrophages were activated by model immune complexes using wells coated with mouse IgG at concentrations of 10 or 30 μg/ml for 4 h. Macrophage activation was determined by measuring TNF-α (D) and IL-10 (E) in the culture supernatants by ELISA, and the values were normalized for cell number by MTT. Data in C–E are present as the mean ± se of eight mice/group. *P < 0.05 between the indicated groups.
Decreased IL-10 induction by Tlr2−/− macrophages
As macrophages are important in serum transfer-induced arthritis [32], and they are sensitive to activation by immune complexes, experiments were performed to determine if there was a difference in their response to immune complexes. Bone marrow-derived macrophages from Tlr2−/− and WT mice were cultured on IgG-coated culture plates for 4 h, and the expression levels of TNF-α and IL-10 in the conditioned medium were determined by ELISA. Immune complex-induced TNF-α production was not different between Tlr2−/− and control cells (Fig. 5D). In contrast, IL-10 was reduced significantly (P<0.05) in the culture supernatants of the Tlr2−/− macrophages compared with the controls (Fig. 5E). These observations demonstrate that intact TLR2 signaling promotes immune complex-induced IL-10 expression.
To define a potential mechanism for the reduction of IL-10 by Tlr2−/− macrophages in response to immune complex stimulation, we examined the activation of Akt, ERK, p38, and GSK3, which have all been reported as downstream kinases, by immune complexes. With the use of WT macrophages, immune complexes activated each of these mediators determined by phosphorylation at 15 min, which was reduced by 240 min (Fig. 6A–C, and data not shown). The activation of Akt and ERK by immune complexes was markedly reduced using Tlr2−/− compared with control macrophages (Fig. 6A and C). Furthermore, consistent with reduced activation of Akt, in the Tlr2−/− macrophages, there was a modest decrease in the activation of GSK3, which is downstream of Akt, by immune complexes (Fig. 6B). In contrast, there was no consistent difference in the activation of p38 between Tlr2−/− and control macrophages (data not shown). These observations demonstrate that immune complexes result in reduced activation of Akt and ERK using Tlr2−/− macrophages.
Figure 6. Reduced immune complex-mediated Akt, GSK3, and ERK activation by Tlr2−/− macrophages.

GM-CSF in vitro-differentiated macrophages were collected prior to addition of cells to immune complexes (0) or following addition to immune complexes for 15–240 min. Macrophage cell lysates were collected at indicated time-points and analyzed by immunoblot analysis using antibodies to (A) phospho (p)-Akt and GAPDH; (B) phospho-GSK3α (Ser21) and -β (Ser9) and GAPDH, or (C) phospho-ERK of p44 and p42 and GAPDH. The numbers below each of the phospho-protein represent the fold of change compared with time 0, after adjusted loading with GAPDH. The relative expression of each protein is calculated by densitometry using ImageJ. The gels presented in A–C are representative of blots from greater than or equal to four mice for each protein and are from a single gel for each antibody blot.
DISCUSSION
In this study, we demonstrate that Tlr2−/− mice develop more severe anti-GPI antibody-mediated arthritis. Histologic examination of joint sections revealed that Tlr2−/− mice had more inflammation, pannus formation, bone erosion, and osteoclasts compared with those from the WT controls. The ankles of Tlr2−/− mice also exhibited increased IL-1β and decreased IL-10 compared with the controls. As the pathogenesis of anti-GPI serum transfer-induced arthritis is mediated by the formation of immune complexes locally [33, 34], a model system of immune complexes was used, which demonstrated a reduction of IL-10 by Tlr2−/− macrophages compared with control macrophages. The decreased expression of IL-10 was associated with diminished activation of Akt and ERK and moderately reduced GSK3. These observations suggest that the enhanced arthritis observed in Tlr2−/− mice is mediated by a reduction of local immune complex-mediated IL-10, resulting from a reduction of activation of Akt and ERK.
Previous studies have shown that Tlr2−/− results in more severe arthritis in IL-1Ra−/− mice [14], and IL-1Ra−/− spontaneously develops arthritis associated with increased proinflammatory cytokines, including IL-1β, TNF-α, and IL-6, in the joints [35], which is mediated by IL-17, downstream of IL-1β [36]. Further studies documented that germ-free IL-1Ra−/− mice do not develop arthritis and that TLR4 deletion is protective, associated with reduced Th17 cells and IL-17 production [14]. In contrast, IL-1Ra−/−, Tlr2−/− mice develop more severe arthritis associated with an increase of Th1 and a decrease of Th2 markers, which provides a more proinflammatory environment [14]. The IL-1Ra−/−, Tlr2−/− mice also demonstrated decreased T regulatory cell function [14]. Thus, in contrast to the K/BxN serum transfer model, which does not require lymphocytes, the increased arthritis observed in the IL-1Ra−/−, Tlr2−/− mice is mediated through effects on T cells.
Our study extends these observations by documenting that Tlr2−/− also modulates the effector phase of arthritis. The K/BxN serum transfer model of arthritis is mediated by immune complexes formed by the ubiquitously expressed GPI and antibodies to GPI, dependent on FcγR signaling and complement activation [33, 34]. In addition to neutrophils, inflammatory monocytes and macrophages each have been shown to contribute to this model of arthritis, and each cell type possesses FcγRs [29, 30, 37, 38]. Therefore, in these studies, we used macrophages differentiated in GM-CSF, as deletion of or neutralization of GM-CSF ameliorates K/BxN serum transfer arthritis and reduces the number of inflammatory monocytes and macrophages in the inflamed joints [39]. The activation of Tlr2−/− macrophages by immune complexes resulted in the comparable expression of proinflammatory TNF-α but significantly reduced anti-inflammatory IL-10. The reduced expression of IL-10 by immune complexes suggests that this mechanism contributed to the reduction of IL-10 observed in the joints of Tlr2−/− mice.
IL-10 is critical in modulating RA and CIA. IL-10 is highly expressed in RA synovial fluid and peripheral blood [40], while IL-10 production by RA peripheral blood T cells correlates inversely with radiographic joint damage [41]. IL-10 is expressed in ex vivo RA synovial tissue cultures, and neutralization of IL-10 increases, whereas addition of exogenous IL-10 decreases proinflammatory cytokine production, including TNF-α and IL-1β [42]. Deletion of IL-10 results in more severe CIA [43, 44], whereas antibody-mediated neutralization accelerates disease onset and severity [45]. Consistent with these observations, the treatment of CIA with IL-10 inhibits the development of and reduces the severity of established disease without a consistent effect on antibody production [46–48]. In contrast, one study demonstrated a reduction in the incidence but not the severity of collagen antibody-mediated arthritis, which includes the addition of LPS, an immune complex- and TLR4-mediated disease, in IL-10−/− mice, [43]. This result appears limited to this specific model of arthritis and the decreased LPS-induced production of TNF-α by macrophages from this specific IL-10−/− murine line [43]. Together, these observations suggest that the reduction of IL-10 observed in the joints of the Tlr2−/− mice in this study, which inversely correlated with disease activity, likely contributed to more severe disease and joint destruction and increased IL-1β.
We observed increased joint destruction and osteoclastogenesis in the Tlr2−/− mice after they were challenged with K/BxN serum. TLR2 expression in osteoclast cells and precursors is not required for osteoclast formation under physiological condition, as Tlr2−/− mice have normal bone volume. Thus, increased osteoclasts in arthritic Tlr2−/− mice are likely because of changes of microenvironments as a result of Tlr2−/−. In RA, increased production of RANKL, TNF-α, IL-1β, and IL-17 by various types of cells in the inflammatory infiltrates contribute to osteoclastogenesis. However, we did not detect significant changes of RANKL, TNF-α, or IL-17 (data not shown) in Tlr2−/− joints, and there was no increase in immune complex-induced TNF-α by Tlr2−/− macrophages, indicating that these cytokines likely do not play a major role. In contrast, we observed significantly decreased levels of anti-inflammatory cytokine IL-10 in Tlr2−/− joint extracts and in Tlr2−/− macrophages. IL-10 suppresses osteoclastogenesis in vivo and in vitro [27, 49, 50]. The effects of IL-10 on osteoclast inhibition are mediated through reduced NF of activated T cells, cytoplasmic 1, expression via triggering receptor expressed on myeloid cells 2 signaling [26], which is important for activation through RANK [51, 52]. These observations suggest that the increased osteoclastogenesis observed in the Tlr2−/− mice may be a result of a combination of increased IL-1β and decreased IL-10 production.
The data suggest that the mechanism responsible for the reduction of IL-10 is the diminished activation of Akt and ERK by immune complexes. TLR2 ligation results in the activation of Akt mediated through PI3K [53]. The p85 subunit of PI3K has been shown to bind constitutively to a docking site on the intracellular domain of TLR2, and this interaction increases following TLR2 ligation [53, 54]. TLR2 ligation results in ERK1/2 and Akt activation mediated through Ras and Rac1 interactions with p85a [55, 56]. ERK is capable of enhancing IL-10 expression induced by TLR2 ligation or immune complex activation by effects on transcription factors, such as CREB [57–59]. The effects of Akt are mediated, at least in part, through phosphorylation and deactivation of GSK3, which then permits the induction of IL-10 by transcription factors, such as AP-1 and CREB [58, 60]. Consistent with this possibility, we identified a modest decrease in the phosphorylation of GSK3 in Tlr2−/− macrophages following activation by immune complexes. These observations support the role of decreased Akt and ERK1/2 signaling in Tlr2−/− mice as a major contributor to the decreased IL-10. We did not examine the effect of complement activation on the interaction between FcγR and TLR2 signaling. It is also possible that increased IFN-γ might contribute to the reduction of IL-10 in the Tlr2−/− mice [61]. Our data suggest that the diminished activation of ERK and Akt in Tlr2−/− cells results in the reduced expression of IL-10, which permits more inflammation and osteoclastogenesis in this serum transfer model of arthritis.
It is possible that low levels of endogenous TLR2 ligands are present constitutively, which permits a more vigorous induction of IL-10 when TLR2 signaling is intact. We demonstrated recently that the endogenous TLR2 ligand gp96 is induced during the course of anti-GPI serum transfer-induced arthritis [13]. These observations suggest that low levels of endogenous TLR2 ligands are present in the serum transfer-induced, inflamed joints, which may promote the expression of IL-10 in this model. Supporting this possibility, the ligation of FcγR has been shown to increase TLR-mediated induction of IL-10, which is regulated by ERK [59, 62]. Furthermore, TLR2 signaling in myeloid cells is critical for the increased IL-10 expression observed in a variety of microbial infections, which is mediated through ERK and Akt [63–65]. Together, these observations identify a critical role for TLR2 signaling for the induction of IL-10 by macrophages [16].
Our observations and those using IL-1Ra−/− mice [14] suggest that deletion of TLR2 promotes more inflammation. Nonetheless, it is well known that TLR2 signaling is capable of promoting inflammation and arthritis. Tlr2−/− mice are protected from arthritis induced by the intra-articular injection of TLR2 ligands peptidoglycan and Pam3CSK4 but not the nucleotide-oligomerization domain 2 ligand muramyl dipeptide [66]. Also, Tlr2−/− mice are protected from zymosan-induced arthritis early (Day 1) and late (Day 24), although not on Day 3 [67, 68], and zymosan-induced arthritis is suppressed by treatment with a neutralizing TLR2 antibody [67]. Additionally, supporting a role for TLR2, we demonstrated recently that neutralizing the endogenous TLR2 ligand gp96 ameliorates anti-GPI serum transfer arthritis [13]. Furthermore, neutralizing antibodies to TLR2 suppress the activation of macrophages by endogenous TLR2 ligands in RA synovial fluid [13] and the constitutive expression of proinflammatory cytokines by RA synovial tissue [69]. Together, these observations identify a complex role of TLR2 signaling in regulating arthritis. In the anti-GPI serum transfer model, an intact TLR2 pathway modulates immune complex-mediated arthritis through the induction of IL-10. The presence of intact TLR2 signaling is important in maintaining immune homeostasis in inflammation. Nonetheless, activation through TLR2 is capable of contributing to disease pathogenesis, supporting a potential role for neutralizing TLR2 as a therapeutic approach in RA.
ACKNOWLEDGMENTS
This study is supported by a Within Our Reach grant from the American College of Rheumatology and U.S. National Institutes of Health grants AR055240 (R.M.P.), AR050250 (H.P.), AR054796 (H.P.), AI092490 (H.P.), HL108795 (H.P.), and AR048697 (L.X.).
Footnotes
- −/−
- deficient
- ACPA
- antibodies against citrullinated proteins
- CIA
- collagen-induced arthritis
- GPI
- glucose-6-phosphate isomerase
- IL-1Ra
- IL-1R antagonist
- OPG
- osteoprotegerin
- Pam3CSK4
- palmitoyl-3-cysteine-serine-sysine-4
- RA
- rheumatoid arthritis
- TRAP
- tartrate-resistant acid phosphatase
AUTHORSHIP
Q-Q.H. and R.M.P. developed this project, including experimental design, data analysis, and manuscript preparation. R.M.P. supervised the project. Q-Q.H., R.E.K., and R.B. performed the experiments and data acquisition. H.P. contributed to the interpretation of the results and manuscript preparation. L.X. contributed to the osteoclastogenesis data acquisition, analysis, and manuscript preparation.
REFERENCES
- 1. McInnes I. B., Schett G. (2011) The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 365, 2205–2219 [DOI] [PubMed] [Google Scholar]
- 2. Laurent L., Clavel C., Lemaire O., Anquetil F., Cornillet M., Zabraniecki L., Nogueira L., Fournie B., Serre G., Sebbag M. (2011) Fcγ receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann. Rheum. Dis. 70, 1052–1059 [DOI] [PubMed] [Google Scholar]
- 3. Sokolove J., Zhao X., Chandra P. E., Robinson W. H. (2011) Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis Rheum. 63, 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao X., Okeke N. L., Sharpe O., Batliwalla F. M., Lee A. T., Ho P. P., Tomooka B. H., Gregersen P. K., Robinson W. H. (2008) Circulating immune complexes contain citrullinated fibrinogen in rheumatoid arthritis. Arthritis Res. Ther. 10, R94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trouw L. A., Haisma E. M., Levarht E. W., van der Woude D., Ioan-Facsinay A., Daha M. R., Huizinga T. W., Toes R. E. (2009) Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 60, 1923–1931 [DOI] [PubMed] [Google Scholar]
- 6. Finckh A., Liang M. H., van Herckenrode C. M., de Pablo P. (2006) Long-term impact of early treatment on radiographic progression in rheumatoid arthritis: a meta-analysis. Arthritis Rheum. 55, 864–872 [DOI] [PubMed] [Google Scholar]
- 7. Emery P., Kvien T. K., Combe B., Freundlich B., Robertson D., Ferdousi T., Bananis E., Pedersen R., Koenig A. S. (2012) Combination etanercept and methotrexate provides better disease control in very early (<=4 months) versus early rheumatoid arthritis (>4 months and <2 years): post hoc analyses from the COMET study. Ann. Rheum. Dis. 71, 989–992 [DOI] [PubMed] [Google Scholar]
- 8. Huang Q., Pope R. M. (2010) Toll-like receptor signaling: a potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. J. Leukoc. Biol. 88, 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang Q. Q., Pope R. M. (2009) The role of Toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 11, 357–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Q., Ma Y., Adebayo A., Pope R. M. (2007) Increased macrophage activation mediated through Toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 56, 2192–2201 [DOI] [PubMed] [Google Scholar]
- 11. Huang Q. Q., Sobkoviak R., Jockheck-Clark A. R., Shi B., Mandelin A. M., 2nd, Tak P. P., Haines G. K., 3rd, Nicchitta C. V., Pope R. M. (2009) Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J. Immunol. 182, 4965–4973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi B., Huang Q., Tak P. P., Vervoordeldonk M. J., Huang C. C., Dorfleutner A., Stehlik C., Pope R. M. (2012) SNAPIN: an endogenous Toll-like receptor ligand in rheumatoid arthritis. Ann. Rheum. Dis. 71, 1411–1417 [DOI] [PubMed] [Google Scholar]
- 13. Huang Q. Q., Koessler R. E., Birkett R., Dorfleutner A., Perlman H., Kenneth Haines G., 3rd, Stehlik C., Nicchitta C. V., Pope R. M. (2012) Gp96 perpetuates the persistent inflammation of rheumatoid arthritis. Arthritis Rheum. 64, 3638–3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdollahi-Roodsaz S., Joosten L. A., Koenders M. I., Devesa I., Roelofs M. F., Radstake T. R., Heuvelmans-Jacobs M., Akira S., Nicklin M. J., Ribeiro-Dias F., van den Berg W. B. (2008) Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J. Clin. Invest. 118, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abdollahi-Roodsaz S., Joosten L. A., Koenders M. I., van den Brand B. T., van de Loo F. A., van den Berg W. B. (2009) Local interleukin-1-driven joint pathology is dependent on Toll-like receptor 4 activation. Am. J. Pathol. 175, 2004–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saraiva M., O'Garra A. (2010) The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 10, 170–181 [DOI] [PubMed] [Google Scholar]
- 17. Korganow A. S., Ji H., Mangialaio S., Duchatelle V., Pelanda R., Martin T., Degott C., Kikutani H., Rajewsky K., Pasquali J. L., Benoist C., Mathis D. (1999) From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity 10, 451–461 [DOI] [PubMed] [Google Scholar]
- 18. Scatizzi J. C., Hutcheson J., Bickel E., Haines G. K., 3rd, Perlman H. (2007) Pro-apoptotic Bid is required for the resolution of the effector phase of inflammatory arthritis. Arthritis Res. Ther. 9, R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scatizzi J. C., Hutcheson J., Pope R. M., Firestein G. S., Koch A. E., Mavers M., Smason A., Agrawal H., Haines G. K., 3rd, Chandel N. S., Hotchkiss R. S., Perlman H. (2010) Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis Rheum. 62, 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q., Badell I. R., Schwarz E. M., Boulukos K. E., Yao Z., Boyce B. F., Xing L. (2005) Tumor necrosis factor prevents alendronate-induced osteoclast apoptosis in vivo by stimulating Bcl-xL expression through Ets-2. Arthritis Rheum. 52, 2708–2718 [DOI] [PubMed] [Google Scholar]
- 21. Pickens S. R., Chamberlain N. D., Volin M. V., Gonzalez M., Pope R. M., Mandelin A. M., 2nd, Kolls J. K., Shahrara S. (2011) Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization. Angiogenesis 14, 443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Satoh T., Takeuchi O., Vandenbon A., Yasuda K., Tanaka Y., Kumagai Y., Miyake T., Matsushita K., Okazaki T., Saitoh T., Honma K., Matsuyama T., Yui K., Tsujimura T., Standley D. M., Nakanishi K., Nakai K., Akira S. (2010) The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 11, 936–944 [DOI] [PubMed] [Google Scholar]
- 23. Luo Y., Pollard J. W., Casadevall A. (2010) Fcγ receptor cross-linking stimulates cell proliferation of macrophages via the ERK pathway. J. Biol. Chem. 285, 4232–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DiMartino S. J., Yuan W., Redecha P., Ivashkiv L. B., Salmon J. E. (2008) Insoluble immune complexes are most effective at triggering IL-10 production in human monocytes and synergize with TLR ligands and C5a. Clin. Immunol. 127, 56–65 [DOI] [PubMed] [Google Scholar]
- 25. Huang Q. Q., Perlman H., Huang Z., Birkett R., Kan L., Agrawal H., Misharin A., Gurbuxani S., Crispino J. D., Pope R. M. (2010) FLIP: a novel regulator of macrophage differentiation and granulocyte homeostasis. Blood 116, 4968–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park-Min K. H., Ji J. D., Antoniv T., Reid A. C., Silver R. B., Humphrey M. B., Nakamura M., Ivashkiv L. B. (2009) IL-10 suppresses calcium-mediated costimulation of receptor activator NF-κ B signaling during human osteoclast differentiation by inhibiting TREM-2 expression. J. Immunol. 183, 2444–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shin H. H., Lee J. E., Lee E. A., Kwon B. S., Choi H. S. (2006) Enhanced osteoclastogenesis in 4-1BB-deficient mice caused by reduced interleukin-10. J. Bone Miner. Res. 21, 1907–1912 [DOI] [PubMed] [Google Scholar]
- 28. Ji H., Korganow A. S., Mangialaio S., Hoglund P., Andre I., Luhder F., Gonzalez A., Poirot L., Benoist C., Mathis D. (1999) Different modes of pathogenesis in T-cell-dependent autoimmunity: clues from two TCR transgenic systems. Immunol. Rev. 169, 139–146 [DOI] [PubMed] [Google Scholar]
- 29. Scatizzi J. C., Hutcheson J., Bickel E., Woods J. M., Klosowska K., Moore T. L., Haines G. K., 3rd, Perlman H. (2006) p21Cip1 is required for the development of monocytes and their response to serum transfer-induced arthritis. Am. J. Pathol. 168, 1531–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Solomon S. (2005) Local inflammation as a mediator of migraine and tension-type headache: a response. Headache 45, 255. [DOI] [PubMed] [Google Scholar]
- 31. Corr M., Crain B. (2002) The role of FcγR signaling in the K/B x N serum transfer model of arthritis. J. Immunol. 169, 6604–6609 [DOI] [PubMed] [Google Scholar]
- 32. Solomon S., Rajasekaran N., Jeisy-Walder E., Snapper S. B., Illges H. (2005) A crucial role for macrophages in the pathology of K/B x N serum-induced arthritis. Eur. J. Immunol. 35, 3064–3073 [DOI] [PubMed] [Google Scholar]
- 33. Ji H., Ohmura K., Mahmood U., Lee D. M., Hofhuis F. M., Boackle S. A., Takahashi K., Holers V. M., Walport M., Gerard C., Ezekowitz A., Carroll M. C., Brenner M., Weissleder R., Verbeek J. S., Duchatelle V., Degott C., Benoist C., Mathis D. (2002) Arthritis critically dependent on innate immune system players. Immunity 16, 157–168 [DOI] [PubMed] [Google Scholar]
- 34. Wipke B. T., Wang Z., Nagengast W., Reichert D. E., Allen P. M. (2004) Staging the initiation of autoantibody-induced arthritis: a critical role for immune complexes. J. Immunol. 172, 7694–7702 [DOI] [PubMed] [Google Scholar]
- 35. Horai R., Saijo S., Tanioka H., Nakae S., Sudo K., Okahara A., Ikuse T., Asano M., Iwakura Y. (2000) Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J. Exp. Med. 191, 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakae S., Saijo S., Horai R., Sudo K., Mori S., Iwakura Y. (2003) IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc. Natl. Acad. Sci. USA 100, 5986–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Monach P. A., Nigrovic P. A., Chen M., Hock H., Lee D. M., Benoist C., Mathis D. (2010) Neutrophils in a mouse model of autoantibody-mediated arthritis: critical producers of Fc receptor γ, the receptor for C5a, and lymphocyte function-associated antigen 1. Arthritis Rheum. 62, 753–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nigrovic P. A., Binstadt B. A., Monach P. A., Johnsen A., Gurish M., Iwakura Y., Benoist C., Mathis D., Lee D. M. (2007) Mast cells contribute to initiation of autoantibody-mediated arthritis via IL-1. Proc. Natl. Acad. Sci. USA 104, 2325–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cook A. D., Turner A. L., Braine E. L., Pobjoy J., Lenzo J. C., Hamilton J. A. (2011) Regulation of systemic and local myeloid cell subpopulations by bone marrow cell-derived granulocyte-macrophage colony-stimulating factor in experimental inflammatory arthritis. Arthritis Rheum. 63, 2340–2351 [DOI] [PubMed] [Google Scholar]
- 40. Cush J. J., Splawski J. B., Thomas R., McFarlin J. E., Schulze-Koops H., Davis L. S., Fujita K., Lipsky P. E. (1995) Elevated interleukin-10 levels in patients with rheumatoid arthritis. Arthritis Rheum. 38, 96–104 [DOI] [PubMed] [Google Scholar]
- 41. Verhoef C. M., van Roon J. A., Vianen M. E., Bijlsma J. W., Lafeber F. P. (2001) Interleukin 10 (IL-10), not IL-4 or interferon-γ production, correlates with progression of joint destruction in rheumatoid arthritis. J. Rheumatol. 28, 1960–1966 [PubMed] [Google Scholar]
- 42. Katsikis P. D., Chu C. Q., Brennan F. M., Maini R. N., Feldmann M. (1994) Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J. Exp. Med. 179, 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johansson A. C., Hansson A. S., Nandakumar K. S., Backlund J., Holmdahl R. (2001) IL-10-deficient B10.Q mice develop more severe collagen-induced arthritis, but are protected from arthritis induced with anti-type II collagen antibodies. J. Immunol. 167, 3505–3512 [DOI] [PubMed] [Google Scholar]
- 44. Finnegan A., Kaplan C. D., Cao Y., Eibel H., Glant T. T., Zhang J. (2003) Collagen-induced arthritis is exacerbated in IL-10-deficient mice. Arthritis Res. Ther. 5, R18–R24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joosten L. A., Lubberts E., Durez P., Helsen M. M., Jacobs M. J., Goldman M., van den Berg W. B. (1997) Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum. 40, 249–260 [DOI] [PubMed] [Google Scholar]
- 46. Lubberts E., Joosten L. A., Van Den Bersselaar L., Helsen M. M., Bakker A. C., Xing Z., Richards C. D., Van Den Berg W. B. (2000) Intra-articular IL-10 gene transfer regulates the expression of collagen-induced arthritis (CIA) in the knee and ipsilateral paw. Clin. Exp. Immunol. 120, 375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Persson S., Mikulowska A., Narula S., O'Garra A., Holmdahl R. (1996) Interleukin-10 suppresses the development of collagen type II-induced arthritis and ameliorates sustained arthritis in rats. Scand J. Immunol. 44, 607–614 [DOI] [PubMed] [Google Scholar]
- 48. Whalen J. D., Lechman E. L., Carlos C. A., Weiss K., Kovesdi I., Glorioso J. C., Robbins P. D., Evans C. H. (1999) Adenoviral transfer of the viral IL-10 gene periarticularly to mouse paws suppresses development of collagen-induced arthritis in both injected and uninjected paws. J. Immunol. 162, 3625–3632 [PubMed] [Google Scholar]
- 49. Owens J. M., Gallagher A. C., Chambers T. J. (1996) IL-10 modulates formation of osteoclasts in murine hemopoietic cultures. J. Immunol. 157, 936–940 [PubMed] [Google Scholar]
- 50. Sasaki H., Hou L., Belani A., Wang C. Y., Uchiyama T., Muller R., Stashenko P. (2000) IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J. Immunol. 165, 3626–3630 [DOI] [PubMed] [Google Scholar]
- 51. Evans K. E., Fox S. W. (2007) Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mohamed S. G., Sugiyama E., Shinoda K., Taki H., Hounoki H., Abdel-Aziz H. O., Maruyama M., Kobayashi M., Ogawa H., Miyahara T. (2007) Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone 41, 592–602 [DOI] [PubMed] [Google Scholar]
- 53. Arbibe L., Mira J. P., Teusch N., Kline L., Guha M., Mackman N., Godowski P. J., Ulevitch R. J., Knaus U. G. (2000) Toll-like receptor 2-mediated NF-κ B activation requires a Rac1-dependent pathway. Nat. Immunol. 1, 533–540 [DOI] [PubMed] [Google Scholar]
- 54. Lu C., Liu L., Chen Y., Ha T., Kelley J., Schweitzer J., Kalbfleisch J. H., Kao R. L., Williams D. L., Li C. (2011) TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J. Immunol. 187, 1458–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen B. C., Kang J. C., Lu Y. T., Hsu M. J., Liao C. C., Chiu W. T., Yeh F. L., Lin C. H. (2009) Rac1 regulates peptidoglycan-induced nuclear factor-κB activation and cyclooxygenase-2 expression in RAW 264.7 macrophages by activating the phosphatidylinositol 3-kinase/Akt pathway. Mol. Immunol. 46, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 56. Chen B. C., Chang Y. S., Kang J. C., Hsu M. J., Sheu J. R., Chen T. L., Teng C. M., Lin C. H. (2004) Peptidoglycan induces nuclear factor-κB activation and cyclooxygenase-2 expression via Ras, Raf-1, and ERK in RAW 264.7 macrophages. J. Biol. Chem. 279, 20889–20897 [DOI] [PubMed] [Google Scholar]
- 57. Gallo P., Goncalves R., Mosser D. M. (2010) The influence of IgG density and macrophage Fc (γ) receptor cross-linking on phagocytosis and IL-10 production. Immunol. Lett. 133, 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hu X., Paik P. K., Chen J., Yarilina A., Kockeritz L., Lu T. T., Woodgett J. R., Ivashkiv L. B. (2006) IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity 24, 563–574 [DOI] [PubMed] [Google Scholar]
- 59. Lucas M., Zhang X., Prasanna V., Mosser D. M. (2005) ERK activation following macrophage FcγR ligation leads to chromatin modifications at the IL-10 locus. J. Immunol. 175, 469–477 [DOI] [PubMed] [Google Scholar]
- 60. Antoniv T. T., Ivashkiv L. B. (2011) Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology 132, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hu X., Chen J., Wang L., Ivashkiv L. B. (2007) Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J. Leukoc. Biol. 82, 237–243 [DOI] [PubMed] [Google Scholar]
- 62. Gerber J. S., Mosser D. M. (2001) Reversing lipopolysaccharide toxicity by ligating the macrophage Fc γ receptors. J. Immunol. 166, 6861–6868 [DOI] [PubMed] [Google Scholar]
- 63. Nandan D., Camargo de Oliveira C., Moeenrezakhanlou A., Lopez M., Silverman J. M., Subek J., Reiner N. E. (2012) Myeloid cell IL-10 production in response to leishmania involves inactivation of glycogen synthase kinase-3β downstream of phosphatidylinositol-3 kinase. J. Immunol. 188, 367–378 [DOI] [PubMed] [Google Scholar]
- 64. Gao Y., Zhang M., Chen L., Hou M., Ji M., Wu G. (2012) Deficiency in TLR2 but not in TLR4 impairs dendritic cells derived IL-10 responses to schistosome antigens. Cell. Immunol. 272, 242–250 [DOI] [PubMed] [Google Scholar]
- 65. Kaji R., Kiyoshima-Shibata J., Nagaoka M., Nanno M., Shida K. (2010) Bacterial teichoic acids reverse predominant IL-12 production induced by certain lactobacillus strains into predominant IL-10 production via TLR2-dependent ERK activation in macrophages. J. Immunol. 184, 3505–3513 [DOI] [PubMed] [Google Scholar]
- 66. Rosenzweig H. L., Jann M. J., Vance E. E., Planck S. R., Rosenbaum J. T., Davey M. P. (2010) Nucleotide-binding oligomerization domain 2 and Toll-like receptor 2 function independently in a murine model of arthritis triggered by intraarticular peptidoglycan. Arthritis Rheum. 62, 1051–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Frasnelli M. E., Tarussio D., Chobaz-Peclat V., Busso N., So A. (2005) TLR2 modulates inflammation in zymosan-induced arthritis in mice. Arthritis Res. Ther. 7, R370–R379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rosenzweig H. L., Clowers J. S., Nunez G., Rosenbaum J. T., Davey M. P. (2011) Dectin-1 and NOD2 mediate cathepsin activation in zymosan-induced arthritis in mice. Inflamm. Res. 60, 705–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ultaigh S. N., Saber T. P., McCormick J., Connolly M., Dellacasagrande J., Keogh B., McCormack W., Reilly M., O'Neill L. A., McGuirk P., Fearon U., Veale D. J. (2011) Blockade of Toll-like receptor 2 prevents spontaneous cytokine release from rheumatoid arthritis ex vivo synovial explant cultures. Arthritis Res. Ther. 13, R33. [DOI] [PMC free article] [PubMed] [Google Scholar]