

Abstract
Frontotemporal dementia (FTD) is a clinical syndrome with heterogeneous molecular basis. Until recently, our knowledge was limited to a minority of cases associated with abnormalities of the tau protein or gene (MAPT). However, in 2006, mutations in progranulin (GRN) were discovered as another important cause of familial FTD. That same year, TAR DNA binding protein 43 (TDP-43) was identified as the pathological protein in the most common subtypes of FTD and ALS. Since then, significant efforts have been made to understand the normal functions and regulation of GRN and TDP-43 and their roles in neurodegeneration. More recently, other DNA/RNA binding proteins (FUS, EWS and TAF15) were identified as pathological proteins in most of the remaining cases of FTD. And just six months ago, abnormal expansion of a hexanucleotide repeat in C9ORF72 was found to be the most common genetic cause of both FTD and ALS. With this remarkable progress, it appears that all the common FTD-causing genes have now been discovered and the major pathological proteins identified. This review highlights recent advances in the molecular aspects of FTD, which will provide the basis for improved patient care through the future development of more targeted diagnostic tests and therapies.
Introduction
Frontotemporal dementia (FTD) accounts for 5–15% of all dementia and is the second commonest cause in the presenile age group.1,2 FTD is a clinical syndrome, characterized by progressive deterioration in behavior, personality and/or language, with relative preservation of memory.3–5 Clinical subtypes include the behavioral variant (bvFTD) and two forms of primary progressive aphasia; progressive non-fluent aphasia (PNFA) and semantic dementia (SD). In addition, FTD is often associated with an extrapyramidal movement disorder (parkinsonism or corticobasal syndrome) and/or motor neuron disease (MND).6,7 Given the variability in phenotype, it is not surprising that the molecular basis of FTD is also heterogeneous (Table 1).
Table 1.
Molecular classification of FTLD with genetic and clinical correlations
| major molecular class | pathological subtype* | associated genes† | associated clinical phenotypes | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| bvFTD | PNFA | SD | park | MND | |||
|
| |||||||
| FTLD-tau |
|
+ | (+) | (+) | + | ALS, PLS | |
|
+ | + | (+) | ||||
|
+ | + | + | PLS | |||
|
+ | + | + | PLS | |||
|
+ | ||||||
|
+ | ||||||
|
+ | + | PLS | ||||
|
+ | + | PLS | ||||
| FTLD-TDP |
|
(+) | + | ALS | |||
|
|
+ | + | + | |||
|
|
+ | + | (+) | + | ALS | |
|
+ | + | |||||
|
|
+ | (+) | ALS | |||
| FTLD-FUS |
|
(+) | ALS | ||||
|
+ | ||||||
|
+ | + | PLS | ||||
|
+ | + | ALS | ||||
| FTLD-UPS | |||||||
|
|
+ | (+) | (ALS) | |||
| FTLD-ni | |||||||
aFTLD-U, atypical frontotemporal lobar degeneration with ubiquitinated inclusions; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; BIBD, basophilic inclusion body disease; bvFTD, behavioral variant FTD; C9ORF72, chromosome 9 open reading frame 72 gene; CBD, corticobasal degeneration; CHMP2B, charged multivescicular body protein 2B gene; FTD, frontotemporal dementia; FTD-3, FTD linked to chromosome 3; FTLD, frontotemporal lobar degeneration; FUS, fused in sarcoma; GRN, progranulin gene; MAPT, microtubule associated protein tau gene; MND, motor neuron disease; MSTD, multiple system tauopathy with dementia; NFT-dementia, neurofibrillary tangle predominant dementia; ni, no inclusions; NIFID, neuronal intermediate filament inclusion disease; park, parkinsonism; PiD, Pick’s disease; PLS, primary lateral sclerosis; PNFA, progressive non-fluent aphasia; PSP, progressive supranuclear palsy; SD, semantic dementia; TARDBP, transactive response DNA binding protein gene; TDP, TDP-43; UPS, ubiquitin proteasome system; VCP, valosin containing protein gene; WMT-GGI, white matter tauopathy with globular glial inclusions.
indicates the characteristic pattern of pathology, not the clinical syndrome.
genes in which variation may cause or increase the risk of FTD with the corresponding FTLD pathological subtype.
(+) rare cause or unusual phenotype.
A family history of FTD is present in 25–50% of cases, often with an autosomal dominant pattern of inheritance, indicating a strong genetic component.8,9 In 1998, mutations in the microtubule associated protein tau gene (MAPT) on chromosome 17 were identified in a number of families with FTD and parkinsonism.10–12 Since then, 44 different MAPT mutations have been reported, accounting for 5–20% of familial FTD (www.molgen.ua.ac.be/ftdmutations).13 However, there remained a number of chromosome 17 linked FTD families that were not explained by MAPT mutations. A major breakthrough occurred in 2006 when progranulin (GRN) was identified as the second FTD-related gene on chromosome 17, with GRN mutations responsible for an even larger proportion of FTD families.14,15 Much less common are mutations in the valosin containing protein gene (VCP) which cause the rare familial syndrome of inclusion body myopathy with Paget disease of bone and FTD16 and a mutation in the gene for charged multivesicular body protein 2B (CHMP2B) found in a large Danish FTD pedigree.17 In addition, several families with a combination of FTD and amyotrophic lateral sclerosis (ALS) have been reported with genetic linkage to a locus on chromosome 9p.18–26 Despite years of intense effort by many research groups worldwide, the identity of the FTD/ALS gene on 9p remained elusive until just last year, when two independent studies identified the defect as being an expanded hexanucleotide repeat in a non-coding region of the chromosome 9 open reading frame 72 gene (C9ORF72).27,28 Discovery of the C9ORF72 mutation has generated tremendous excitement in the FTD and ALS research communities, as it appears to be the most common genetic cause of both conditions (see below).
The neuropathology associated with clinical FTD is also heterogeneous.29 A common feature is the relatively selective degeneration of the frontal and temporal lobes, and the term “frontotemporal lobar degeneration” (FTLD) is often used for those pathological conditions that predominantly or commonly present with FTD. In addition, most cases of FTLD are found to have abnormal intracellular accumulation of some disease-specific protein and it has become popular to classify FTLD into broad categories, based on the molecular defect thought to be most characteristic.30,31 Until quite recently, the only FTLD subgroup we knew much about were those conditions characterized by the aggregation of hyperphosphorylated tau protein in neurons and glia (FTLD-tau) (Table 1). However, most FTD is not associated with tau pathology, but is characterized by neuronal inclusions that were originally identified with ubiquitin immunohistochemistry (FTLD-U).32,33 Just months after publication of the GRN mutation discovery, another landmark paper reported identification of the transactive response DNA binding protein with molecular weight 43 kD (TDP-43) as the ubiquitinated pathological protein in most cases of FTLD-U (subsequently renamed FTLD-TDP), as well as the vast majority of ALS.34,35 This provided strong evidence that FTD and ALS are closely related conditions with overlapping molecular pathogenesis. This concept was further strengthened in 2009 when, following the discovery that mutations of the fused in sarcoma gene (FUS) cause autosomal dominant ALS,36,37 it was shown that most of the ~10% of FTLD that do not have either tau or TDP-43 based pathology are characterized by inclusions that are immunoreactive for FUS (FTLD-FUS).38–40 More recently, it has been shown that the FUS-positive inclusions in FTLD-FUS also label for the other members of the FET protein family (including Ewing’s sarcoma protein and TATA-binding protein-associated factor 15).41
The recent pace with which knowledge of the molecular genetics and neuropathology of FTD has advanced has been truly remarkable (Box 1). In just over five years, we have gone from knowing virtually nothing of the molecular basis of most cases of FTD, to now being able to determine the genetic cause in the majority of autosomal dominant families and being able to assign virtually all cases of FTLD to one of three major pathological subtypes (FTLD-tau, FTLD-TDP, FTLD-FUS).31 This insight is a crucial step towards improved FTD patient care, as it provides the basis for more informed counseling and the potential for more specific diagnostic tests and targeted therapies. In this review, we will highlight several recent advances in our understanding of molecular aspects of FTD, focusing on the recent discovery of the C9ORF72 mutation and the roles of GRN, TDP-43, FUS and the other FET proteins in disease pathogenesis.
Box 1. Important events in the molecular pathogenesis of FTD.
1892: Arnold Pick describes lobar atrophy in a patient with presenile dementia and aphasia.145
1911: Alois Alzheimer characterizes Pick bodies using silver stains.146
1960’s: descriptions of PSP and CBD clinicopathological syndromes.147,148
1974: different pathological subtypes of PiD disease described.149
mid 1980’s - early 1990’s: identification of tau as major component of pathological lesions in AD, PiD, PSP and CBD (reviewed in Lee et al.).150
1990: description of FTD cases without specific histopathology (DLDH).151
mid 1990’s: identification of subset of FTD with FTLD-U pathology.152
1998: MAPT mutations identified in some families with FTD and parkinsonism genetically linked to chromosome 17.10–12
2004–06: recognition that most cases of DLDH are really FTLD-U and that FTLD-U is the most common FTD-associated pathology.33
2006: description of different patterns of FTLD-U that correlate with clinical phenotypes, genetic abnormalities and biochemical properties of inclusions.115,117
2006: discovery that GRN mutations cause autosomal dominant FTD and explain all remaining chromosome 17 linked families.14,15
2006: TDP-43 identified as pathological protein in most cases of FTLD-U and ALS.34,35
2008: identification of a subset of FTLD-U cases that lack TDP-43-immunoreactive pathology (aFTLD-U).153,154
2009: discovery that most cases of tau/TDP-43-negative FTLD have FUS-immunoreactive pathology (FTLD-FUS).38–40
2011: discovery that FTLD-FUS shows accumulation of other FET protein members TAF15 and EWS.41
2011: FTD/ALS associated gene defect on chromosome 9p identified as repeat expansion in C9ORF72.27,28
Abbreviations: AD, Alzheimer’s disease; aFTLD-U, atypical FTLD-U; C9ORF72, chromosome 9 open reading frame 72; CBD, corticobasal degeneration; DLDH, dementia lacking distinctive histopathology; EWS, Ewing’s sarcoma; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; FTLD-U, FTLD with ubiquitin immunoreactive inclusions; FUS, fused in sarcoma protein; PSP, progressive supranuclear palsy; PiD, Pick’s disease; TAF15, TATA-binding protein-associated factor 15; TDP-43, transactive response DNA binding protein with molecular weight 43 kD.
Advances in the molecular genetics of FTD
GRN variants and its biological regulators in FTD
In less than six years, 69 different pathogenic GRN mutations have been reported in more than 230 families worldwide, accounting for 5–20% of familial FTD and 1–5% of sporadic cases (www.molgen.ua.ac.be/ftdmutations).42 Progranulin (GRN) is a multifunctional secreted growth factor, expressed by many cell types including neurons.43 Pathogenic mutations are of various types and occur throughout the gene, but all cause disease via haploinsufficiency.14,15 As a result, significantly reduced levels of GRN are consistently observed in plasma, serum and CSF samples of symptomatic and asymptomatic GRN mutation carriers.44–46 Based on these findings, GRN enzyme-linked immunosorbent assays (ELISA) are now being developed as an inexpensive alternative to classical sequencing analyses for diagnostic testing of FTD patients.
Genetic modifiers and regulators of GRN expression
The clinical phenotype associated with GRN mutations is quite variable47–52 and penetrance is incomplete.53 Understanding the factors that modify the expression of GRN mutations or regulate the normal GRN gene is of potential therapeutic importance. One such genetic factor is the uncharacterized transmembrane protein 106B (TMEM106B), which was recently identified in a genome-wide association study (GWAS) of cases with known FTLD-TDP pathology.54 Genetic variants in and near TMEM106B appear to protect or delay the onset of FTD in individuals with pathogenic GRN mutations, possibly by increasing GRN levels.54–57 A number of microRNAs, including miR-29b and miR-107 have also been implicated in GRN regulation.58,59 In addition, the minor T-allele of genetic variant rs5848, located in the 3′ untranslated region of GRN, increases the binding of miR-659 to GRN, thereby reducing GRN levels.60 Genetic association studies have shown that carriers homozygous for the T-allele of rs5848 have a three-fold increased risk to develop FTLD-TDP compared with homozygous C-allele carriers60, supporting a role for GRN in sporadic FTD and possibly other neurodegenerative diseases such as Alzheimer’s disease.61–63 GRN expression may also be modified by certain exogenous factors. It was recently shown that GRN transcription can be enhanced by small molecules including suberoylanilide hydroxamic acid (SAHA),64 while inhibitors of the vacuolar ATPase and some alkalizing drugs increase GRN production and secretion through a translational mechanism.65
GRN cellular biology
Significant progress has also been made in recent years towards our understanding of GRN biology and its neuroprotective function. The addition of GRN to stressed or GRN-depleted neuronal cells promotes neurite outgrowth.66–69 The neuroprotective effects of GRN might be due, at least in part, to the activation of cell signaling pathways involved in cell survival.66,70–73 A role for GRN in excitotoxicity and synaptic transmission has also been suggested.68,74 Importantly, sortilin-1 (SORT1), a known receptor for other neurotrophic factors in the brain, was identified in two independent studies as the first known receptor for GRN.75,76 SORT1 has been shown to mediate GRN endocytosis and regulate the levels of GRN in vivo in mouse brain and in human plasma. More recently, tumor necrosis factor receptors were reported to directly interact with GRN.77 The identification of GRN receptors is exciting as it opens new avenues in GRN cell biology research and another potential route to FTD therapy.
C9ORF72 mutation
Since 2006, increasing evidence has suggested the presence of a major locus for the combined phenotype of FTD and ALS on chromosome 9p21, but the disease mutation has remained elusive (Box 2).18–26 The key to the identification of the disease-causing mutation came from the apparent non-Mendelian inheritance of a GGGGCC hexanucleotide repeat located in a non-coding region of C9ORF72 (Figure 1a) in a large FTD-ALS family designated VSM-20 (Vancouver, San Francisco and Mayo family 20).27 Using primers flanking the repeat region, all affected individuals appeared homozygous by fluorescent PCR, while affected children seemed not to inherit an allele from their affected parent. This finding suggested the presence of a repeat expansion that was too large to be amplified by the PCR method, which was confirmed using a repeat-primed PCR assay (Figure 1b) and southern blot analysis. The polymorphic nature of this GC-rich hexanucleotide repeat was independently recognized in a Welsh FTD-ALS family using next-generation sequencing, also implicating this genomic region in disease pathogenesis.28
Box 2. History of the chromosome 9p FTD-ALS locus.
Since 2006, at least 10 autosomal dominant families in which patients were affected with FTD, ALS, or both were published with conclusive or suggestive linkage to chromosome 9p.18–26 The minimal candidate region shared by all families was a 3.7Mb region containing only 10 known or predicted genes. In 2010, three genome-wide association studies (GWAS) in sporadic ALS populations identified a novel susceptibility locus on chromosome 9p which completely overlapped with the candidate region for familial FTD-ALS.155–157 Strongest association was identified in a ~80kb haplotype block containing only three genes; MOBKL2B, IFNK and C9ORF72. An independent GWAS study in patients with pathologically confirmed FTLD-TDP nominated the same chromosomal region, implicating the chromosome 9p gene defect in sporadic forms of both FTD and ALS.54 However, despite concentrated efforts by the FTD and ALS research communities, in-depth candidate-gene sequencing and targeted next-generation sequencing of the minimal candidate region failed to identify the causative mutation, suggesting that a complex mutational mechanism may be involved. In 2011, an expanded hexanucleotide repeat in the non-coding region of C9ORF72 was found to be the long sought-after cause of FTD and ALS on chromosome 9p.27,28
Figure 1.
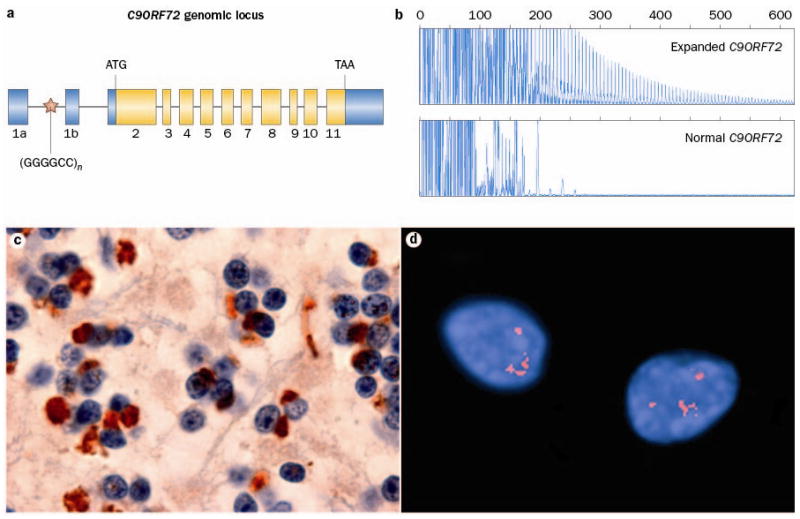
Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes FTD and ALS linked to chromosome 9p. a Overview of the genomic structure of C9ORF72. Numbered boxes represent coding (white) and non-coding (gray) exons and the position of the start codon (ATG) and stop codon (TAA) are indicated. The position of the (GGGGCC)n repeat in the intronic region between exons 1a and 1b is indicated with a red star. b PCR products of repeat-primed PCR reactions separated on an ABI3730 DNA Analyzer and visualized by GENEMAPPER software. Electropherograms are zoomed to 2,000 relative fluorescence units to show stutter amplification. One FTD patient with a pathogenic expanded C9ORF72 repeat (top) and one FTD patient with a C9ORF72 normal repeat length (bottom) are shown. c In addition to FTLD-TDP and ALS pathology, all patients with the C9ORF72 mutation show a unique pattern of ubiquitin-positive (brown), TDP-43-negative neuronal inclusions in the cerebellar granule layer and other specific neuroanatomical regions. This disease-specific finding implies the mis-metabolism and accumulation of some yet unidentified protein(s). d RNA foci, visualized using a Cy3-labeled (GGCCCC)4 oligonucleotide probe (red), in the nuclei of two lower motor neurons from an FTD-ALS patient carrying the expanded GGGGCC repeat in C9ORF72.
Mutation frequency
In the six months since the original discovery, numerous FTD and ALS patients have been screened for the presence of GGGGCC repeat expansions in C9ORF72 using the repeat-primed PCR assay (Table 2).27,28,78–94 The mutation frequency has varied significantly among populations, with the highest being in genetically isolated populations from Finland and Sardinia, and in cohorts where all patients had a pathological diagnosis of FTLD-TDP (with or without ALS).27,28,81,85,88 The average mutation frequencies in North American and European populations reported are 37% for familial ALS, 6% for sporadic ALS, 21% for familial FTD and 6% for sporadic FTD patients. In all series, the C9ORF72 mutation has been the most common genetic cause of familial ALS (more frequent than SOD1 mutations) and at least similar in frequency to GRN mutations in FTD families. To date, most of the patients included in the mutation screenings have been Caucasian; however, C9ORF72 repeat expansions have also been identified in patients of African American, Middle Eastern and Asian race.86,88,94 Interestingly, independent of the clinical presentation or ethnic origin, all C9ORF72 mutation carriers inherit the expansion on the same genetic background, suggesting the presence of a common ancestor or, alternatively, the occurrence of multiple independent expansions on a fragile predisposing disease haplotype.88,95,96
Table 2.
Frequency of the C9ORF72 repeat expansion in FTD and ALS patient populations
| Familial FTD | Sporadic FTD | Familial ALS | Sporadic ALS | Cases | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Study regiona | Ref. | N | C9ORF72 mutation | N | C9ORF72 mutation | N | C9ORF72 mutation | N | C9ORF72 mutation | excluded h |
| Europe | ||||||||||
| Finland | 28,88 | 27 | 13 (48.1%) | 48 | 9 (18.8%) | 112 | 52 (46.4%) | 289 | 61 (21.1%) | Yes |
| Ireland | 80 | - | - | - | - | 47 | 18 (38.3%) | 386 | 19 (4.9%) | NS |
| UKb | 82 | - | - | - | - | 63 | 27 (42.9%) | 500 | 35 (7.0%) | No |
| UKb | 87 | 93 | 12 (12.9%) | 163 | 6 (3.7%) | - | - | - | - | No |
| UKb | 93 | 161 | 20 (12.4%) | 209 | 16 (7.7%) | - | - | - | - | No |
| Netherlands | 88,92 | 129 | 37 (28.7%) | 224 | 5(2.2%) | - | - | - | - | Yes |
| Belgiumc | 84 | 75 | 12 (16.0%) | 230 | 9 (3.9%) | 15 | 7 (46.7%) | 122 | 6 (4.9%) | Yes |
| France | 88 | 50 | 22 (44.0%) | 150 | 14 (9.3%) | - | - | - | - | |
| Germany | 28,81,88 | 29 | 4(13.8%) | - | - | 69 | 15 (21.7%) | 421 | 22 (5.2%) | Yes |
| Italy (mainland) | 28,81,88,91 | - | - | - | - | 120 | 45 (37.5%) | 1523 | 55 (3.6%) | Yes |
| Italy (Sardinia) | 81,88,91 | - | - | - | - | 21 | 12 (57.1%) | 133 | 9 (6.8%) | Yes |
| Italy (Sicily) | 91 | - | - | - | - | - | - | 101 | 5 (4.9%) | Yes |
| Greece | 89 | - | - | - | - | 10 | 5 (50.0%) | 136 | 11 (8.1%) | NS |
| North America | ||||||||||
| US | 27,78 | 171 | 20 (11.7%) | 203 | 6 (3.0%) | 34 | 8 (23.5%) | 195 | 8 (4.1%) | No |
| USd | 28,88 | - | - | - | - | 163 | 59 (36.2%) | 1014 | 56 (5.5%) | NS |
| Canada | 94 | - | - | - | - | 62 | 17 (27.4%) | 169 | 6 (3.6%) | No |
| Canadae | 27,85 | 26 | 16 (61.5%) | 3 | 0 (0.0%) | - | - | - | - | No |
| USe | 27 | 40 | 9 (22.5%) | 53f | 8 (15.1%) | - | - | - | - | No |
| USe | 79 | 18 | 6 (33.3%) | 6 | 0 (0.0%) | 14 | 6 (42.9%) | 43f | 5 (11.6%) | NS |
| Other regions | ||||||||||
| Israel | 88 | - | - | - | - | 14 | 3 (21.4%) | - | - | NS |
| India | 88 | - | - | 31 | 0 (0.0%) | - | - | 31 | 0 (0.0%) | NS |
| Asiag | 88 | 3 | 2 (66.7%) | 10 | 0 (0.0%) | 20 | 1 (5.0%) | 238 | 0 (0.0%) | NS |
| Guam | 88 | - | - | - | - | - | - | 90 | 0 (0.0%) | NS |
| Australia | 88 | - | - | - | - | - | - | 263 | 14 (5.3%) | NS |
ALS, amyotrophic lateral sclerosis; FTD, frontotemporal dementia; C9ORF72, chromosome 9 open reading frame 72; UK, United Kingdom; US, Unites States.
Only geographical regions with at least 10 FTD or ALS patients are listed.
Cohorts are part of a larger series of UK patients which are grouped in Majounie et al. with highly comparable mutation frequencies.56
Numbers do not include 23 patients with both FTD and ALS of which 85.7% of familial and 6.3% of sporadic patients were C9ORF72 mutation carriers.
In the sporadic ALS cohort, 5.4% (48 of 890) of white and 4.1% (2 of 49) of African American patients carried a C9ORF72 repeat expansion.
Only included patients with a pathological diagnosis of FTLD-TDP.
Includes individuals for which no information on family history was available.
The geographical origin of the Asian patients was not reported in detail; however, the familial ALS patient carrying the C9ORF72 repeat expansion was Japanese.
In studies where cases known to carry mutations in other genes were excluded, the frequency of C9ORF72 repeat expansions are overestimated (NS, not stated).
Clinical phenotypes
Several groups from North America and Europe have now published descriptions of the demographic, clinical and neuropathological features of their cohorts of patients with the C9ORF72 mutation.27,28,78–94 The clinical presentation is heterogeneous and highly variable between and within families. Patients may present with FTD, ALS or features of both. The FTD subtype is most often bvFTD with PNFA less frequent. ALS usually has early involvement of both upper and lower motor neurons and bulbar presentation is particularly common.79,81,82,94 Several studies have found that ALS patients with the mutation have a slightly earlier onset and shorter disease duration than those without the mutation.79,81,82,84,91,94 In addition to FTD and ALS, other features may include memory disorder,78,85,87,90,92,93 psychosis,78,83,85,92,93 extrapyramidal movement disorder (usually an akinetic-rigid syndrome)78,82,83,85,90,92 and cerebellar signs.85 Symptoms tend to accumulate and phenotypes converge with disease progression, so that most patients eventually develop at least some abnormalities of behavior, language and motor function.78,80,82,85,94 There is wide variation in the age at onset (27 – 83 years, mean = 50s) and disease duration (1 – 22 years) and several studies have noted earlier disease onset in subsequent generations, consistent with genetic anticipation.78,81,82,84,85,94 Structural neuroimaging tends to show symmetric bilateral atrophy, primarily affecting frontotemporal regions, but also involving other cerebral lobes and the cerebellum.78,85,87,92,97
Neuropathology
The neuropathology associated with the C9ORF72 mutation is a combination of FTLD-TDP and classical ALS.27,78,79,82,84,85,87,90,92–94,98,99 Regardless of the clinical phenotype, postmortem examination usually shows TDP-43 positive inclusions in a wide range of neuroanatomical regions including the extramotor cerebral cortex, hippocampus, basal ganglia, substantia nigra and lower motor neurons of the brainstem and spinal cord. In addition, a unique and highly characteristic feature of cases with the mutation is the presence of neuronal inclusions in the cerebellar granule cell layer, hippocampal pyramidal neurons and other neuroanatomical sites, that label for proteins of the ubiquitin proteasome system (ubiquitin, ubiquilins and p62) but that are negative for TDP-43 (Figure 1c).27,78,79,82,84,85,87,90,92–94,98,99 This consistent finding supports the abnormal metabolism and accumulation of some, as yet unidentified molecule(s) that could include the mutant RNA, RNA-binding proteins or protein products of aberrant splicing. To date, immunohistochemical studies using commercial antibodies against C9ORF72 have failed to demonstrate any abnormal distribution or accumulation of the protein.27,79,82,85,87,93,94
Repeat size
All C9ORF72 mutation screenings performed to date have used the repeat-primed PCR method to detect the presence of a pathogenic GGGGCC repeat expansion. However, it is important to note that this method is only semi-quantitative and that the characteristic stutter pattern observed (Figure 1b) cannot be used to determine the exact number of repeats. In one family, southern blot analyses performed using DNA extracted from lymphoblast cell lines showed pathogenic repeat expansions of 700–1600 repeat units;27 however, the minimal repeat size associated with disease may be considerably smaller. Similar to other non-coding repeat expansion disorders, there is evidence for somatic instability of the C9ORF72 repeat.27 This means that repeat lengths may vary among different tissues within the same individual, making it difficult to accurately size the repeat and determine genetic/clinical/pathological correlations.100–102 Our current lack of knowledge of the minimal pathogenic repeat-size combined with the technical challenges mentioned, raise important questions regarding genetic testing for this common mutation; particularly in the context of predictive genetic testing. Accurate sizing of the expanded repeat in larger FTD and ALS patient series will be crucial to establish reliable cut-off sizes needed to counsel individuals undergoing genetic testing. Future studies also need to determine whether the repeat-length contributes to the variability in onset age and clinical presentation or whether other genetic and/or environmental modifiers are involved.
Disease mechanism
C9ORF72 is a completely uncharacterized protein whose function is presently unknown. Two different C9ORF72 isoforms are predicted to be generated from a total of three different C9ORF72 transcripts;27 however, the relative expression of each of these transcripts in relevant brain regions has not yet been studied. Several groups have shown ~50% loss of at least one C9ORF72 transcript in expanded repeat carriers, presumably resulting from the interference of the expanded GC-rich repeat with C9ORF72 transcription regulation.27,28,84 Although these findings support a possible loss-of-function disease mechanism, the accumulation of GGGGCC repeat containing transcripts as nuclear RNA foci in frontal cortex and spinal cord of C9ORF72 mutation carriers has also been demonstrated (Figure 1d), suggesting a possible toxic RNA gain-of function disease mechanism.27 Based on commonalities with other non-coding repeat expansion disorders, these RNA foci may alter the function of one or more RNA-binding proteins resulting in downstream changes in gene expression and/or alternative splicing of a range of transcripts.103 A number of cellular and animal models, either eliminating C9ORF72 expression or overexpressing human C9ORF72 containing expanded GGGGCC repeats, are currently being generated to determine the relative contribution of each disease mechanism to neurodegeneration and TDP-43 aggregation.
Other FTD genes and genetic risk factors
With the identification of the repeat expansion in C9ORF72, all previously published FTD families with genome-wide linkage have now been accounted for. While it is unlikely that any other common FTD-causing genes exist, rare mutations in other genes may each explain a small number of the remaining families and combinations of genetic variants and environmental factors are likely to be responsible for disease in the majority of sporadic FTD patients. The use of exome and whole-genome sequencing will greatly facilitate the discovery of rare genetic defects in the future. This was recently demonstrated with the identification of the colony stimulating factor 1 receptor gene (CSF1R) as the cause of hereditary diffuse leukoencephalopathy with spheroids (HDLS), 104 a disorder with variable clinical presentation that includes features of FTD. Additional GWAS, such as the large collaborative study currently underway, which includes more than 2500 FTD patient samples, may identify additional genetic risk factors.
ALS-related genes in FTD
Other rare genetic causes of FTD that have been identified in recent years include TARDBP and FUS, although mutations in each of these genes usually cause a pure ALS phenotype.36,37,105,106 This past year, UBQLN2, which encodes a member of the ubiquilin family that is involved in the degradation of ubiquitinated proteins, was also added to the list of ALS-FTD genes.107 In about 20% of UBQLN2 mutation carriers, progressive dementia with abnormalities in both behavior and executive functions were reported; however, none of these patients presented with FTD alone.
Advances in the molecular pathology of FTD
TDP-43
TDP-43 is a highly conserved, predominantly nuclear protein, able to shuttle between the nucleus and cytoplasm. It has a number of well-described functions in RNA regulation such as control of splicing, mRNA transport and stability; however, the complexity of TDP-43 functions is just emerging.108–110
FTLD-TDP
Abnormal accumulation of TDP-43 in neuronal and glial inclusions is the characteristic neuropathological feature in approximately 50% of FTD patients (FTLD-TDP) and the vast majority of ALS cases.30,34,35 Pathological modifications of TDP-43 in the disease state include a redistribution from the nucleus to the cytoplasm in cells with inclusions, hyperphosphorylation, ubiquitination and N-terminal truncation.35 FTLD-TDP includes sporadic and genetic forms with mutations in GRN, VCP, TARDBP and the recently recognized C9ORF72 repeat expansion (see above).27,28,111–114 Based on the morphology and anatomic distribution of TDP-43 pathology, four distinct FTLD-TDP subtypes are recognized.115–117 The relevance of this heterogeneity is supported by clinical and genetic correlations (Table 1), as well as emerging evidence for distinct biochemical properties of TDP-43 in the different subtypes.35,117,118
Pathogenesis of TDP-43 proteinopathies
The neuropathological findings in FTLD-TDP implicate that both loss and gain-of-function mechanisms might be involved in TDP-43 associated cell death. Addressing these fundamental questions is the focus of numerous research activities worldwide and detailed discussions of TDP-43 pathogenesis are published elsewhere.119–121 Briefly, current in vivo models provide evidence for both scenarios by demonstrating that either reduced or increased expression levels of the physiologically tightly autoregulated TDP-43 are not well tolerated.119–121 However, no model has fully recapitulated the neuropathological and biochemical features of human TDP-43 related diseases. While the identification of TARDBP mutations is a clear indicator that dysfunction of TDP-43 is directly linked to neurodegeneration, the functional consequences of TARDBP mutations are still unresolved. There is no solid evidence that TARDBP mutations act through a toxic-gain-of function mechanism and no functional consequences of TARDBP mutations on processing of the few RNA targets studied to date have been reported. However, studies using crosslinking immunoprecipitation and high-throughput sequencing have recently identified more than 6000 RNA targets of TDP-43, and a major challenge now is to dissect specific pathways regulated by TDP-43 and to identify possible disease-relevant RNA targets.109,110
Another important but unresolved issue is the role of TDP-43 in the other genetic forms of FTLD-TDP. The fact that mutations in GRN, VCP and C9ORF72 are all consistently characterized by TDP-43 pathology suggests that dysregulation of TDP-43 might be a crucial common downstream mechanism leading to cell death in all of them. However, the significance of TDP-43 accumulation in C9ORF72 mutation carriers has recently been challenged by the identification of additional TDP-43 negative, ubiquitin-positive pathology that is more abundant than TDP-43 pathology in distinct brain regions (Figure 1c), raising the possibility that another unidentified protein(s) might be more important in the pathogenesis in these cases (see above).27,78,79,82,84,85,87,90,92–94,98,99
FUS and other FET proteins
FUS belongs to the FET protein family that also includes Ewing’s sarcoma (EWS), TATA-binding protein-associated factor 15 (TAF15) and the drosophila orthologue cabeza. They are highly conserved, ubiquitously expressed, predominantly nuclear (Figure 2a), multifunctional DNA/RNA binding proteins,122 that can bind to a large number of partially overlapping RNA targets.123
Figure 2.
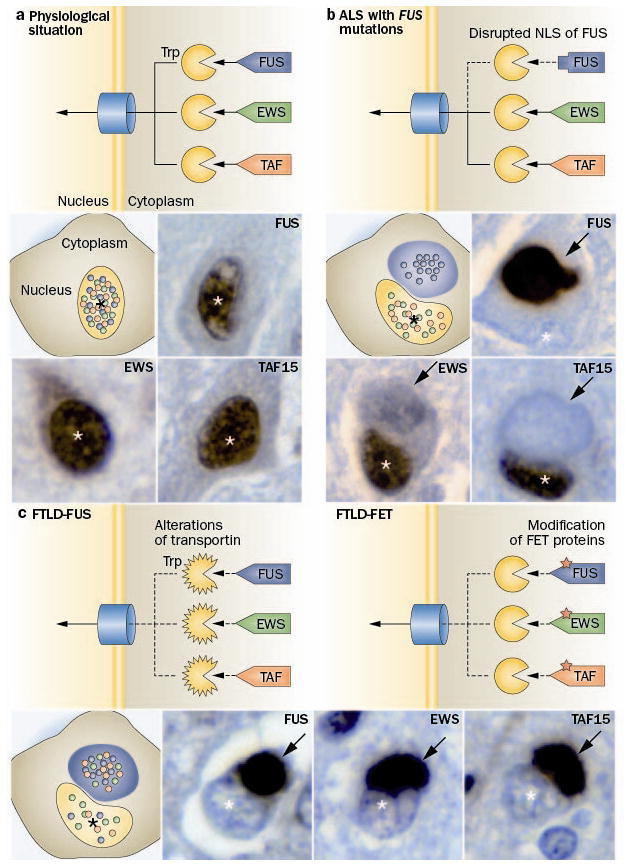
Distinct pathomechanisms of ALS-FUS and FTLD-FUS. a The FET protein members, FUS, EWS and TAF15 contain a proline-tyrosine nuclear localization signal (PY-NLS, triangle) which is bound by the receptor protein Transportin (Trp). This mediates the transport of these proteins across the nuclear pore complex into the nucleus, resulting in a predominant nuclear localization of all three proteins under physiological conditions. b In ALS with FUS mutations, the PY-NLS of FUS is disrupted due to mutations (rectangle) leading to an impaired interaction with Trp and nuclear import of FUS, while TAF15 and EWS are normally transported to the nucleus. This results in a selective accumulation of FUS into cytoplasmic inclusion in ALS-FUS patients, with retained nuclear localization of TAF15 and EWS. c In contrast, FTLD-FUS patients show co-accumulation of all FET proteins into cytoplasmic inclusions accompanied with their nuclear depletion. This complex dysregulation of all FET proteins in FTLD-FUS might be explained by either of two broad scenarios: c-1 alterations of Trp itself, e. g. by genetic variations, reduced expression levels or posttranslational modifications; or c-2 posttranslational modifications of FET proteins, which interfere with proper Trp binding. Each photomicrograph shows a single neuron with a cytoplasmic inclusion (arrow) and the nucleus indicated by the asterisk (*), immunostained for the FET protein indicated (brown stain).
FTLD-FUS / FTLD-FET
In early 2009, FUS mutations were reported to be the cause of ~3 % of familial ALS cases, in which the associated pathology is characterized by inclusions that are FUS positive but TDP-43 negative (ALS-FUS).36,37 Subsequently, FUS was found to be the most characteristic marker for the pathology in most of the remaining tau/TDP-negative FTLD cases, which include three closely related but distinct clinicopathological entities; atypical FTLD-U (aFTLD-U), neuronal intermediate filament inclusion disease (NIFID) and basophilic inclusion body disease (BIBD).38–40,124 The identification of FTLD-FUS as a new molecular subgroup31 provided further evidence that FTD and ALS are closely related conditions and emphasized the pathogenic role of RNA binding proteins. However, despite there being some overlap in the phenotype and pathological features of FTLD-FUS and ALS-FUS, significant differences were also observed.125,126 Moreover, the publication of additional cases made it evident that ALS with FUS pathology is almost always caused by a FUS mutation; whereas, cases of FTLD-FUS tend to be sporadic and none has yet been associated with any genetic abnormality of FUS.38–40,124 Further evidence for different pathomechanisms has been provided by a recent study that investigated the other FET protein members in a series of ALS-FUS and FTLD-FUS cases.41 In cases of ALS-FUS with a range of different mutations, there was no co-accumulation of other FET proteins into FUS-positive inclusions and cells retained the physiological nuclear staining of TAF15 and EWS (Figure 2b). In striking contrast, in all FTLD-FUS subtypes TAF15 and EWS were also found to co-accumulate in FUS-positive inclusions and inclusion-bearing cells showed a reduction in the normal nuclear staining of all three FET proteins, particularly TAF15 (Figure 2c). The addition of TAF15 and EWS to the growing list of RNA binding proteins involved in neurodegeneration is further supported by studies in which TAF15 was predicted as a potential candidate through an independent approach using a yeast functional screen aimed to identify RNA binding proteins with similar function to TDP-43 and FUS127 and descriptions of genetic variants (of undetermined pathogenic significance) in TAF15 and EWSR1 in a small number of ALS cases.128,129 Although the respective roles of FUS, TAF15 and EWS in FTLD-FUS remains to be elucidated, the term FTLD-FET now seems more appropriate for this molecular FTLD subgroup.
Pathogenesis of FUS-proteinopathies
The above described differences in the molecular pathology of ALS-FUS and FTLD-FUS imply different pathological processes underlying inclusion formation and cell death, with ALS-FUS being restricted to dysfunction of FUS, while FTLD-FUS might involve dysfunction of all FET proteins (Figure 2).
In ALS-FUS, mutations in the C-terminus of the protein disrupt a region characterized as non-classical nuclear localization sequence. This results in an impaired transportin-mediated nuclear import with redistribution of FUS to the cytoplasm (Figure 2b).130,131 Importantly, no alteration of other FET proteins is seen under these conditions.41 The degree of FUS nuclear transport impairment varies between different FUS mutations, but correlates with the observed variability in disease course associated with different mutations and with distinct pathological patterns of ALS-FUS pathology,125 thereby providing strong evidence that impaired nuclear import of FUS is a key event in disease pathogenesis of ALS-FUS.
In FTLD-FUS, a more general defect of transportin-mediated nuclear import is postulated that affects the distribution of all FET proteins, with two broad scenarios plausible (Figure 2c). First, a primary defect of transportin itself, either resulting from genetic variations in TNPO1, posttranslational modifications, or altered expression levels of transportin, could result in reduced efficiency of nuclear import of all FET proteins. However, in this scenario one might also expect alterations in the subcellular distribution of other transportin cargos, such as hnRNPA1, which is not supported by preliminary data.41 Second, the proper nuclear import of FET proteins might be affected by abnormal posttranslational modifications of FET proteins, such as arginine methylation or phosphorylation, that have been shown to modulate nucleocytoplasmic transport, protein-protein interaction and protein stability.122,132–138 So far, biochemical analysis of protein extracted from FTLD-FUS brains has revealed only increased insolubility of all FET proteins, without other obvious disease-associated changes, such as truncation or abnormal phosphorylation;39,41,139 however, more detailed analysis is required.
The downstream effects of redistributed FUS or all FET proteins in the pathogenesis of ALS-FUS and FTLD-FUS, respectively, have not yet been determined. Similar to TDP-43, both a gain of toxic properties and a loss of functions via their sequestration in aggregates are plausible. Results from initial in vivo models of ALS-FUS have been inconsistent and the mechanisms remain unresolved.121,140
Molecular correlates of FTD phenotypes
Table 1 lists the molecular subtypes of FTLD pathology with the associated genetic defects and common clinical features. Each genetic cause is associated with a specific neuropathology. However, predicting the underlying molecular pathology or genetics, based on the pattern of inheritance and clinical features, is often imprecise.141,142 SD is usually sporadic and associated with FTLD-TDP type C with fewer cases having the pathology of classical PiD. Cases of sporadic PNFA are somewhat more likely to have FTLD-tau than FTLD-TDP, but bvFTD may be associated with any of the major pathologies. Early-onset bvFTD with severe psychobehavioral abnormality but minimal motor features or aphasia is characteristic of the aFTLD-U subtype of FTLD-FUS. When FTD is combined with ALS, the pathology is usually FTLD-TDP; whereas, FTD with prominent parkinsonism is more often FTLD-tau (PSP or CBD). In families with autosomal dominant inheritance of bvFTD or PNFA without significant motor dysfunction, the underlying gene defect may be a mutation in C9ORF72, GRN or MAPT. When parkinsonism or primary lateral sclerosis (PLS) are also prominent features, a MAPT mutation is more likely; whereas, coexistence of classical ALS in a family strongly suggests a C9ORF72 mutation.
Conclusions and future directions
The past six years have seen remarkable progress in our understanding of the molecular basis of FTD. It appears that all the common FTD-causing genes have now been discovered and the major pathological proteins identified. Although many aspects of the specific pathogenic mechanisms still need to be resolved, we are already in position to begin translating this newly acquired knowledge into improved FTD patient care. The recent discoveries of GRN and C9ORF72 mutations allow for more informed genetic counseling. Knowledge of the signature pathological proteins is prompting attempts to develop more disease-specific, molecular-based diagnostic tests, such as the quantification of total or pathological protein species in biofluids.143,144 Recognition of GRN insufficiency as an important mechanism in familial and some sporadic forms of FTD, combined with improved understanding of GRN regulation and cell biology, has already led to initial plans for GRN-based clinical trials (http://www.alzforum.org/new/pdf/FTLDSeries.pdf). The identification of TDP-43, FET proteins and C9ORF72 has open up new avenues of research related to RNA regulation. Finally, a greater appreciation of the overlap between FTD and ALS is now bringing these two areas of research and patient care closer together. Hopefully, patients with FTD will soon experience real benefits from these and future advances.
Key points.
All common FTD-causing genes and signature proteins have now been discovered
Regulation of GRN is one potential therapeutic strategy for FTD
Expansion of a GGGGCC hexanucleotide repeat in a non-coding region of C9ORF72 is the most common genetic cause of FTD and ALS
The pathomechanism of C9ORF72 mutation may include haploinsufficiency and/or toxic RNA foci
Most cases of tau/TDP-negative FTLD are characterized by inclusions that are immunoreactive for FUS (FTLD-FUS) and the other FET proteins (EWS and TAF15)
Differential involvement of the FET proteins implies different pathomechanisms in ALS with FUS mutations versus FTLD-FUS
Acknowledgments
Rosa Rademakers is funded by NIH grants P50 AG016574, R01 NS065782 and R01 AG026251, the ALS Therapy Alliance and the Consortium for Frontotemporal dementia (CFR). Manuela Neumann is funded by the Swiss National Science Foundation grants 31003A-132864 and CRSII3 136222, the German Federal Ministry of Education and Research grant 01GI1005B, the Stavros-Niarchos Foundation, the Synapsis Foundation, and the Hans and Ilse Breuer Foundation. Ian Mackenzie is funded by the Canadian Institutes of Health Research grants 179009 and 74580 and the Pacific Alzheimer’s Research Foundation center grant C06-01.
Contributor Information
Rosa Rademakers, Department of Neuroscience, Mayo Clinic Florida, Jacksonville, FL, USA.
Manuela Neumann, Institute of Neuropathology, University Hospital of Zurich, Zurich, Switzerland.
Ian R. A. Mackenzie, Department of Pathology and Laboratory Medicine, University of British Columbia, Vancouver, Canada
References
- 1.Bird T, et al. Epidemiology and genetics of frontotemporal dementia/Pick’s disease. Ann Neurol. 2003;54 (Suppl 5):S29–31. doi: 10.1002/ana.10572. [DOI] [PubMed] [Google Scholar]
- 2.Feldman H, et al. A Canadian cohort study of cognitive impairment and related dementias (ACCORD): study methods and baseline results. Neuroepidemiology. 2003;22:265–274. doi: 10.1159/000071189. [DOI] [PubMed] [Google Scholar]
- 3.The Lund and Manchester Groups. Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatr. 1994;57:416–418. doi: 10.1136/jnnp.57.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKhann GM, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 5.Neary D, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 6.Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134:2582–2594. doi: 10.1093/brain/awr195. [DOI] [PubMed] [Google Scholar]
- 7.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 8.Rohrer JD, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seelaar H, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71:1220–1226. doi: 10.1212/01.wnl.0000319702.37497.72. [DOI] [PubMed] [Google Scholar]
- 10.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 11.Poorkaj P, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 12.Spillantini MG, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mut. 2004;24:277–295. doi: 10.1002/humu.20086. [DOI] [PubMed] [Google Scholar]
- 14.Baker M, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 15.Cruts M, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 16.Watts GD, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 17.Skibinski G, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 18.Boxer AL, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatr. 2011;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gijselinck I, et al. Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. doi: 10.1001/archneurol.2010.82. [DOI] [PubMed] [Google Scholar]
- 20.Le Ber I, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- 21.Luty AA, et al. Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Momeni P, et al. Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 2006;6:44. doi: 10.1186/1471-2377-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morita M, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 24.Pearson JP, et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258:647–655. doi: 10.1007/s00415-010-5815-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valdmanis PN, et al. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–245. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- 26.Vance C, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 27.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns NJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackenzie IR, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackenzie IR, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipton AM, White CL, 3rd, Bigio EH. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379–385. doi: 10.1007/s00401-004-0900-9. [DOI] [PubMed] [Google Scholar]
- 33.Mackenzie IR, et al. Dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol. 2006;112:551–559. doi: 10.1007/s00401-006-0123-3. [DOI] [PubMed] [Google Scholar]
- 34.Arai T, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 35.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 36.Kwiatkowski TJ, Jr, et al. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 37.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz DG, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–627. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 39.Neumann M, et al. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neumann M, et al. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118:605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neumann M, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134:2595–2609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gijselinck I, Van Broeckhoven C, Cruts M. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: an update. Hum Mut. 2008;29:1373–1386. doi: 10.1002/humu.20785. [DOI] [PubMed] [Google Scholar]
- 43.Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- 44.Finch N, et al. Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain. 2009;132:583–591. doi: 10.1093/brain/awn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. doi: 10.1212/01.wnl.0000325058.10218.fc. [DOI] [PubMed] [Google Scholar]
- 46.Sleegers K, et al. Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol. 2009;65:603–609. doi: 10.1002/ana.21621. [DOI] [PubMed] [Google Scholar]
- 47.Beck J, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain. 2008;131:706–720. doi: 10.1093/brain/awm320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gass J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 49.Le Ber I, et al. Progranulin null mutations in both sporadic and familial frontotemporal dementia. Human Mut. 2007;28:846–855. doi: 10.1002/humu.20520. [DOI] [PubMed] [Google Scholar]
- 50.Moreno F, et al. “Frontotemporoparietal” dementia: clinical phenotype associated with the c.709-1G>A PGRN mutation. Neurology. 2009;73:1367–1374. doi: 10.1212/WNL.0b013e3181bd82a7. [DOI] [PubMed] [Google Scholar]
- 51.Snowden JS, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006;129:3091–3102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- 52.Masellis M, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129:3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- 53.Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24:186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 54.Van Deerlin VM, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cruchaga C, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68:581–586. doi: 10.1001/archneurol.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finch N, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Zee J, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–815. doi: 10.1093/brain/awr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiao J, Herl LD, Farese RV, Gao FB. MicroRNA-29b regulates the expression level of human progranulin, a secreted glycoprotein implicated in frontotemporal dementia. PLoS One. 2010;5:e10551. doi: 10.1371/journal.pone.0010551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang WX, et al. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am J Pathol. 2010;177:334–345. doi: 10.2353/ajpath.2010.091202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rademakers R, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Gene t. 2008;17:3631–3642. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brouwers N, et al. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. 2008;71:656–664. doi: 10.1212/01.wnl.0000319688.89790.7a. [DOI] [PubMed] [Google Scholar]
- 62.Lee MJ, Chen TF, Cheng TW, Chiu MJ. rs5848 variant of progranulin gene is a risk of Alzheimer’s disease in the Taiwanese population. Neurodegener Dis. 2011;8:216–220. doi: 10.1159/000322538. [DOI] [PubMed] [Google Scholar]
- 63.Viswanathan J, et al. An association study between granulin gene polymorphisms and Alzheimer’s disease in Finnish population. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:747–750. doi: 10.1002/ajmg.b.30889. [DOI] [PubMed] [Google Scholar]
- 64.Cenik B, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. 2011;286:16101–16108. doi: 10.1074/jbc.M110.193433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Capell A, et al. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao X, et al. Progranulin promotes neurite outgrowth and neuronal differentiation by regulating GSK-3beta. Protein Cell. 2010;1:552–562. doi: 10.1007/s13238-010-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ryan CL, et al. Progranulin is expressed within motor neurons and promotes neuronal cell survival. BMC Neurosci. 2009;10:130. doi: 10.1186/1471-2202-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tapia L, et al. Progranulin deficiency decreases gross neural connectivity but enhances transmission at individual synapses. J Neurosci. 2011;31:11126–11132. doi: 10.1523/JNEUROSCI.6244-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van Damme P, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol. 2008;181:37–41. doi: 10.1083/jcb.200712039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kleinberger G, et al. Increased caspase activation and decreased TDP-43 solubility in progranulin knockout cortical cultures. J Neurochem. 2010;115:735–747. doi: 10.1111/j.1471-4159.2010.06961.x. [DOI] [PubMed] [Google Scholar]
- 71.Nedachi T, Kawai T, Matsuwaki T, Yamanouchi K, Nishihara M. Progranulin enhances neural progenitor cell proliferation through glycogen synthase kinase 3beta phosphorylation. Neuroscience. 2011;185:106–115. doi: 10.1016/j.neuroscience.2011.04.037. [DOI] [PubMed] [Google Scholar]
- 72.Rosen EY, et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron. 2011;71:1030–1042. doi: 10.1016/j.neuron.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xu J, et al. Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol Aging. 2011;32:2326 e2325–2316. doi: 10.1016/j.neurobiolaging.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo A, Tapia L, Bamji SX, Cynader MS, Jia W. Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 2010;1366:1–8. doi: 10.1016/j.brainres.2010.09.099. [DOI] [PubMed] [Google Scholar]
- 75.Carrasquillo MM, et al. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am J Hum Genet. 2010;87:890–897. doi: 10.1016/j.ajhg.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu F, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang W, et al. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 2011;332:478–484. doi: 10.1126/science.1199214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boeve BF, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012;135:765–783. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brettschneider J, et al. Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012 doi: 10.1007/s00401-012-0970-z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Byrne S, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chio A, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. 2012;135:784–793. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cooper-Knock J, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain. 2012;135:751–764. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Floris G, et al. Frontotemporal dementia with psychosis, parkinsonism, visuo-spatial dysfunction, upper motor neuron involvement associated to expansion of C9ORF72: a peculiar phenotype? J Neurol. 2012 doi: 10.1007/s00415-012-6444-3. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gijselinck I, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 85.Hsiung GY, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135:709–722. doi: 10.1093/brain/awr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kandiah N, et al. Case Report of an Asian Patient with FTD-ALS due to C9ORF72 mutation. Can J Neurol Sci. 2012 in press. [Google Scholar]
- 87.Mahoney CJ, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012;135:736–750. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Majounie E, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mok KY, et al. High frequency of the expanded C9ORF72 hexanucleotide repeat in familial and sporadic Greek ALS patients. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.02.021. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Murray ME, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sabatelli M, et al. C9ORF72 hexanucleotide repeat expansions in the Italian sporadic ALS population. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Simon-Sanchez J, et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain. 2012;135:723–735. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- 93.Snowden JS, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stewart H, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012;123:409–417. doi: 10.1007/s00401-011-0937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mok K, et al. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging. 2012;33:209 e203–208. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rademakers R. C9orf72 repeat expansions in patients with ALS and FTD. Lancet Neurol. 2012;11:297–298. doi: 10.1016/S1474-4422(12)70046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whitwell JL, et al. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135:794–806. doi: 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Al-Sarraj S, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 99.Troakes C, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2011 doi: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- 100.Lavedan C, et al. Myotonic dystrophy: size- and sex-dependent dynamics of CTG meiotic instability, and somatic mosaicism. Am J Hum Genet. 1993;52:875–883. [PMC free article] [PubMed] [Google Scholar]
- 101.Matsuura T, et al. Somatic and germline instability of the ATTCT repeat in spinocerebellar ataxia type 10. Am J Hum Genet. 2004;74:1216–1224. doi: 10.1086/421526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moseley ML, et al. SCA8 CTG repeat: en masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum Mol Genet. 2000;9:2125–2130. doi: 10.1093/hmg/9.14.2125. [DOI] [PubMed] [Google Scholar]
- 103.Renoux AJ, Todd PK. Neurodegeneration the RNA way. Prog Neurobiol. 2011 doi: 10.1016/j.pneurobio.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rademakers R, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2012;44:200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kabashi E, et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum Mol Genet. 2010;19:671–683. doi: 10.1093/hmg/ddp534. [DOI] [PubMed] [Google Scholar]
- 106.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng HX, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buratti E, Baralle FE. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010;7:420–429. doi: 10.4161/rna.7.4.12205. [DOI] [PubMed] [Google Scholar]
- 109.Polymenidou M, et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tollervey JR, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Van Deerlin VM, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kovacs GG, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- 113.Neumann M, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007;66:152–157. doi: 10.1097/nen.0b013e31803020b9. [DOI] [PubMed] [Google Scholar]
- 114.Cairns NJ, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171:227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mackenzie IR, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–549. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mackenzie IR, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sampathu DM, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–1352. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hasegawa M, et al. Molecular Dissection of TDP-43 Proteinopathies. J Mol Neurosci. 2011;45:480–485. doi: 10.1007/s12031-011-9571-x. [DOI] [PubMed] [Google Scholar]
- 119.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 121.Da Cruz S, Cleveland DW. Understanding the role of TDP-43 and FUS/TLS in ALS and beyond. Curr Opin Neurobiol. 2011;21:904–919. doi: 10.1016/j.conb.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol. 2009;1:82–92. doi: 10.1093/jmcb/mjp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hoell JI, et al. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol. 2011;18:1428–1431. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Urwin H, et al. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mackenzie IR, et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011;122:87–98. doi: 10.1007/s00401-011-0838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mackenzie IR, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol. 2011;121:207–218. doi: 10.1007/s00401-010-0764-0. [DOI] [PubMed] [Google Scholar]
- 127.Couthouis J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Couthouis J, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ticozzi N, et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet. 2011;156:285–290. doi: 10.1002/ajmg.b.31158. [DOI] [PubMed] [Google Scholar]
- 130.Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2010 doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 132.Belyanskaya LL, Delattre O, Gehring H. Expression and subcellular localization of Ewing sarcoma (EWS) protein is affected by the methylation process. Exp Cell Res. 2003;288:374–381. doi: 10.1016/s0014-4827(03)00221-0. [DOI] [PubMed] [Google Scholar]
- 133.Jobert L, Argentini M, Tora L. PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res. 2009;315:1273–1286. doi: 10.1016/j.yexcr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 134.Rappsilber J, Friesen WJ, Paushkin S, Dreyfuss G, Mann M. Detection of arginine dimethylated peptides by parallel precursor ion scanning mass spectrometry in positive ion mode. Anal Chem. 2003;75:3107–3114. doi: 10.1021/ac026283q. [DOI] [PubMed] [Google Scholar]
- 135.Fronz K, et al. Arginine methylation of the nuclear poly(a) binding protein weakens the interaction with its nuclear import receptor, transportin. J Biol Chem. 2011;286:32986–32994. doi: 10.1074/jbc.M111.273912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deloulme JC, Prichard L, Delattre O, Storm DR. The prooncoprotein EWS binds calmodulin and is phosphorylated by protein kinase C through an IQ domain. J Biol Chem. 1997;272:27369–27377. doi: 10.1074/jbc.272.43.27369. [DOI] [PubMed] [Google Scholar]
- 137.Perrotti D, et al. TLS/FUS, a pro-oncogene involved in multiple chromosomal translocations, is a novel regulator of BCR/ABL-mediated leukemogenesis. EMBO J. 1998;17:4442–4455. doi: 10.1093/emboj/17.15.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leemann-Zakaryan RP, Pahlich S, Grossenbacher D, Gehring H. Tyrosine Phosphorylation in the C-Terminal Nuclear Localization and Retention Signal (C-NLS) of the EWS Protein. Sarcoma. 2011;2011:218483. doi: 10.1155/2011/218483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Page T, et al. FUS immunogold labeling TEM analysis of the neuronal cytoplasmic inclusions of neuronal intermediate filament inclusion disease: a frontotemporal lobar degeneration with FUS proteinopathy. J Mol Neurosci. 2011;45:409–421. doi: 10.1007/s12031-011-9549-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lanson NA, Jr, Pandey UB. FUS-related proteinopathies: Lessons from animal models. Brain Res. 2012 doi: 10.1016/j.brainres.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 141.Josephs KA, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122:137–153. doi: 10.1007/s00401-011-0839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol. 2011;24:542–549. doi: 10.1097/WCO.0b013e32834cd442. [DOI] [PubMed] [Google Scholar]
- 143.Hu WT, Trojanowski JQ, Shaw LM. Biomarkers in frontotemporal lobar degenerations--progress and challenges. Prog Neurobiol. 2011;95:636–648. doi: 10.1016/j.pneurobio.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Noto Y, et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: specificity, sensitivity, and a possible prognostic value. Amyotroph Lateral Scler. 2011;12:140–143. doi: 10.3109/17482968.2010.541263. [DOI] [PubMed] [Google Scholar]
- 145.Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager medicinische Wochenschrift. 1892;17:165–167. [Google Scholar]
- 146.Alzheimer A. Uber eigenartige Krankheitsfaelle des spaeteren Alters. Z Gesamte Neurol Psychiatrie. 1911;4:356–385. [Google Scholar]
- 147.Rebeiz JJ, Kolodny EH, Richardson EP., Jr Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol. 1968;18:20–33. doi: 10.1001/archneur.1968.00470310034003. [DOI] [PubMed] [Google Scholar]
- 148.Steele JC, Richardson JC, Olszewski J. Progressive Supranuclear Palsy. A Heterogeneous Degeneration Involving the Brain Stem, Basal Ganglia and Cerebellum with Vertical Gaze and Pseudobulbar Palsy, Nuchal Dystonia and Dementia. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 149.Constantinidis J, Richard J, Tissot R. Pick’s disease. Histological and clinical correlations. Eur Neurol. 1974;11:208–217. doi: 10.1159/000114320. [DOI] [PubMed] [Google Scholar]
- 150.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 151.Knopman DS, Mastri AR, Frey WH, 2nd, Sung JH, Rustan T. Dementia lacking distinctive histologic features: a common non-Alzheimer degenerative dementia. Neurology. 1990;40:251–256. doi: 10.1212/wnl.40.2.251. [DOI] [PubMed] [Google Scholar]
- 152.Jackson M, Lennox G, Lowe J. Motor neurone disease-inclusion dementia. Neurodegeneration. 1996;5:339–350. doi: 10.1006/neur.1996.0046. [DOI] [PubMed] [Google Scholar]
- 153.Mackenzie IR, Foti D, Woulfe J, Hurwitz TA. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain. 2008;131:1282–1293. doi: 10.1093/brain/awn061. [DOI] [PubMed] [Google Scholar]
- 154.Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 2008;116:147–157. doi: 10.1007/s00401-008-0395-x. [DOI] [PubMed] [Google Scholar]
- 155.Laaksovirta H, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shatunov A, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.van Es MA, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]