

Abstract
Rapidly accumulating evidence indicates that clinically used general anesthesia causes massive, widespread neuroapoptotic degeneration in the developing mammalian brain. Susceptibility to anesthesia-induced neurotoxicity has been documented in rats, mice, guinea pigs, primates, and in this study, piglets; in short, anesthesia-induced developmental neuroapoptosis is not species-dependent. Our findings with piglets, like those in other immature mammals, demonstrate that relatively short exposure to anesthesia is just as detrimental to species with long periods of synaptogenesis as it is to those with short periods of synaptogenesis. However, the highly reproducible findings in different species also indicate that the timing of exposure to anesthesia is critically important; that is, brain regions that are at the peak of synaptogenesis are most vulnerable even when the exposure to anesthesia is relatively brief. Because the peak of synaptogenesis is characterized by intense, highly programmed neuronal communication that is vital for the survival and proper function of immature neurons, we conclude that anesthesia causes severe disturbances in the fine equilibrium between excitatory and inhibitory neurotransmission in the developing mammalian brain, ultimately leading to neuronal redundancy and death.
Keywords: piglets, isoflurane, midazolam, nitrous oxide, fentanyl, synaptogenesis
Introduction
Rapidly accumulating evidence indicates that commonly used general anesthetics, administered alone or in clinically relevant cocktails during the early stages of mammalian brain development, cause severe, widespread neuronal degeneration.1–5 Based on several mechanistic studies, it appears that anesthesia-induced neuronal damage is apoptotic and involves several pathways of caspase activation. The intrinsic mitochondria-dependent pathway appears to be activated first,6,7 although others follow shortly. Activation of the extrinsic, death-receptor activated6 and neurotrophic factor-activated (in particular, brain-derived neurotrophic factor)5 pathways results in massive caspase-3 activation and DNA fragmentation.3,6,7 At the ultra-structural level, the initial insult, detected mainly in the nucleus, is marked by clamping of the chromatin, followed by disruption of the nuclear membrane, intermixing of the cytoplasm and nucleoplasm, and the formation of apoptotic bodies.3 Interestingly, despite massive neuronal demise, anesthesia-induced damage does not seem to result in significant gliosis.
A common finding in the animal studies done thus far is that the vulnerability of the immature mammalian brain to anesthesia-induced damage coincides with synaptogenesis.1–4,6,8 Although synapse formation is the hallmark of this period, there are several other key elements. To make meaningful synapses and form functional circuitries, neurons have to migrate to their final destination, mature, and differentiate. This highly programmed series of events must occur in a specific order and in a highly homeostatic environment in which modulation of excitatory and the inhibitory neurotransmission is well balanced.9 It has been proposed that general anesthetics, by creating an imbalance in excitatory and inhibitory neurotransmission, disturb the fine equilibrium and, if administered during the peak of synaptogenesis, can cause significant perturbation of normal synapse formation.3,5,6 Neurons that do not form meaningful interneuronal connections are redundant and therefore die by apoptosis. (The apoptotic machinery in immature neurons is easily activated.) Interestingly, the processes of anesthesia-induced apoptotic activation and neuronal death are virtually the same as those that normally occur during early stages of synaptogenesis, the only difference between them being their magnitude. Normal neuronal pruning is highly controlled, its intensity varying during normal synaptogenesis but leading, ultimately, to the apoptotic death of a fairly low percentage of neurons (on average, approximately 1% with some regional variations).1,4 In contrast, anesthesia-induced neuronal apoptosis results in the death of as many as 70-fold more neurons compared to age-matched controls.3,4,6
Duration of anesthesia versus duration of synaptogenesis
Initial studies of anesthesia-induced neuroapoptosis were done in rodents. Although the findings were both impressive and highly reproducible, they gave rise to the question of whether rodents are especially susceptible.1–5,10 An even more important point was that the initial rodent studies begged the question of whether there is a direct correlation between the duration of anesthesia and the duration of synaptogenesis. For species with a relatively short span of synaptogenesis, anesthesia lasting several hours covers a substantial amount of time when proper communication between neurons is crucial. For example, the initial anesthesia neurotoxicity experiments dealt with mice and rats, in which synaptogenesis is 2–3 weeks long.11 Thus, anesthesia protocols lasting for 6 to 7 h seem like a long exposure. Could exposure to anesthesia for this period be irrelevant to any kind of clinical setting, considering that human brain development spans a couple of years, including the last trimester of in utero and the first 2 or so years of postnatal life? (However, the argument could certainly be that prolonged, deep sedation in neonatal and pediatric intensive care units is often administered for days, thus exposing the human brain in the fragile early stages of its development to intense homeostatic unbalance.)
To address the correlation between the duration of synaptogenesis and anesthesia as potentially important determinants of the susceptibility to anesthesia-induced developmental neuroapoptosis, we and others have done studies using clinically relevant general anesthetics on species having relatively long durations of synaptogenesis: guinea pigs,4 piglets, and primates.8
Guinea pigs
Synaptogenesis in guinea pigs lasts approximately 10 weeks11 and, unlike synaptogenesis in humans, occurs mainly during the prenatal period. It starts early in pregnancy, reaches a peak between 35 and 40 days of gestation, and is virtually completed by the end of prenatal life (68–70 days).12,13 We used three gestational ages and hence, three stages of brain growth: early (20 to 25 days in utero), peak (35 to 40 days in utero) and late (50+ days in utero). Examining anesthesia-induced neuroapoptosis, we found that the commonly used inhalational anesthetic isoflurane, alone or in combination with nitrous oxide and/or midazolam, administered for 4 h, induces severe and widespread apoptotic neurodegeneration in the guinea pig brain.4 Moreover, when we quantified neuronal densities during the first week of postnatal life, when the brain is fully developed we discovered that most vulnerable brain regions, including the IV cortical layer of the occipital, retrosplenial, parietal, cingulated and piriformis cortices, as well as the amygdaloid nuclei, hippocampus, subiculum, and anterior thalamic nuclei showed significant decreases in neuronal densities as compared to those in both “true” controls (no anesthesia) and sham controls (treated with a continuous infusion of an intravenous narcotic, fentanyl, commonly used in pediatric patients). The overall anesthesia-induced decline in neuronal counts varied from 40%–50% as compared with controls.4 Because the most prominent caspase-3 activation occurs between 35–40 days of gestational age, while the immature brain of older fetuses (50+ gestational days) were almost completely insensitive, we concluded that the peak of vulnerability coincided with the peak of synaptogenesis. Interestingly, fentanyl, which is commonly used in intensive care settings and, in some cases is infused for many hours, caused an increase in apoptotic damage as compared to the fetuses of mothers that were not anesthetized (“true” controls) although this increase was not significant compared to “true” controls. Furthermore, continuous infusion of fentanyl did not result in a permanent decrease in neuronal densities during later development.4
Piglets
Synaptogenesis in piglets lasts approximately 20 weeks11 and, much like the process in humans, occurs both pre- and postnatally. Like guinea pigs, piglets are much larger than rat pups, which makes it technically feasible to obtain close control and continuous invasive and noninvasive monitoring of physiological parameters.
We used 5-day-old Yorkshire piglets for all experiments (average weight around 1700 g), because synaptogenesis in piglets reaches a plateau during the last 5 weeks of prenatal life and remains at that level during the first 5 weeks of postnatal life.11 All efforts were made to minimize the number of animals used and their suffering. All experiments were approved by The Animal Use and Care Committee of the University of Virginia Health System and were conducted in accordance with the USA Public Health Service’s Policy on Human Care and Use of Laboratory Animals. The animals were randomly designated as “true” control (no anesthesia, no monitoring); sham control (fentanyl given as an intravenous bolus at 30 μg/kg, followed by continuous infusion at 15 μg/kg/h for 4 h); and experimental (1 mg/kg of midazolam injected i.m., followed by 4 h of nitrous oxide at 75-vol% + isoflurane at 0.55-vol% + 24-vol% oxygen). For isoflurane, the minimum alveolar anesthetic concentration (MAC), which prevents purposeful movement in response to supramaximal noxious stimulation in 50% of subjects, is about 2.0-vol% in piglets.14 Although the MAC for nitrous oxide anesthesia in piglets is not known, those for other mammals are between 104 and 180-vol%.15,16
We delivered nitrous oxide (N2O) and oxygen (GTS, Allentown, PA) to piglets using a calibrated flowmeter. Isoflurane (Abbott Laboratories, Chicago, IL) was administered using an agent-specific vaporizer that delivered a set percentage of anesthetic. Midazolam (Sigma-Aldrich Chemical, St. Louis, MO) was dissolved in 10% dimethyl sulfoxide (DMSO) immediately before administration. For control experiments, 10% DMSO was used as a vehicle. Fentanyl, an intravenous narcotic, was purchased from Abbott Laboratories (Chicago, IL); rocuronium, an intravenous muscle relaxant, was purchased from Baxter Pharmaceutical Solutions, LLC (Bloomington, IN). These agents were delivered using a multi-channel infusion pump (Harvard Apparatus, PHD 2000, Holliston, MA).
On the day of the experiment the anesthesia induction of sham and experimental animals was done using isoflurane as needed in ambient air (3 L of flow) delivered via nose cone. Each piglet was placed in the supine position and orotracheally intubated with a polyethylene tube (4 mm internal diameter). Artificial ventilation was immediately initiated with 25% oxygen/nitrogen and isoflurane (as needed while the invasive monitoring lines were placed), using 10–12 mL/kg tidal volume and 20–25 strokes per min delivered via SAR-830 ventilator (CWE, Ardmore, PA). The exhaled gases (e.g., oxygen, carbon dioxide, nitrous oxide, isoflurane) were continuously analyzed using the infrared analyzer (Datex Ultima, Chalfont, St. Giles, UK).
After the induction of general anesthesia, arterial and venous catheters (polyethylene, PE-50) were placed in the right femoral artery for continuous monitoring of blood pressure, heart rate and arterial blood gases, and in the left femoral vein for continuous infusion of intravenous lactate Ringer solution at 4 mL/kg/h. Femoral vascular access was chosen to avoid interference with blood flow to the brain. To facilitate artificial ventilation, piglets were paralyzed using continuous intravenous infusion of a muscle relaxant, rocuronium (15 nmol/kg/min)17 after the placement of an intravenous line; the administration of rocuronium was discontinued 30 min before termination of the desired anesthesia protocol. After placement of the invasive lines, the animals were allowed 15–20 min to stabilize. The anesthesia protocol was then initiated and the minute ventilation was adjusted to maintain stable physiological parameters, which were continuously monitored for 4 h.
Monitoring of vital functions
The adequacy of ventilation and oxygenation was continuously monitored, first by analyzing the composition, in mmHg, of the exhaled end tidal oxygen (EtO2 was kept around 22–23 mmHg) and carbon dioxide (EtCO2) collected from the exhalation limb of the anesthesia breathing circuit. We also analyzed the composition of gases in the arterial blood (pH, partial pressure of oxygen, bicarbonate concentration, oxygen saturation, SaO2 in %) (Nova Biomedical, Inc., Waltham, MA). Arterial blood pressure was continuously monitored using a 23Db blood pressure transducer referred to the mid-thoracic level. This was connected to the cannula inserted into the right femoral artery, which also allowed sampling of the arterial blood as needed. Systolic, diastolic, and mean arterial pressures were obtained using custom software in Pascal computer language and an Intel digital computer. Heart rate was derived from the pressure signal. The animal core temperature was continuously monitored with a rectal temperature probe and maintained using heating pads.
At the termination of an anesthesia protocol, piglets were kept normothermic and inspected continuously for 2 h after anesthesia. As soon as a regular breathing pattern, airway reflexes, and purposeful movements returned, the trachea was extubated. Animals were allowed to feed at will.
Histopathological studies
At 2 h after anesthesia treatment, piglets were deeply anesthetized and perfused with paraformaldehyde (4%) in cacodylate buffer (0.1%), pH 7.4, for histopathology studies of apoptosis. Caspase-3 immunochemical staining (ICC) was used because it is an excellent method for marking neurons in an early stage of apoptosis.3,6,7 Brain tissue was sectioned into 50-μm-thick coronal sections, washed in 0.01 mol/L phosphate-buffered saline (PBS), quenched for 10 min in a solution of methanol containing 3% hydrogen peroxide, then incubated for 1 h in blocking solution (2% BSA/0.2% milk/0.1% triton X-100 in PBS). The tissue was incubated overnight in caspase-specific antibodies, such as rabbit anti-active caspase-3 antiserum, (D175, Cell Signaling Technology, Beverly, MA) diluted 1:1000 in blocking solution. After incubation with D175 primary antibody, sections were incubated for 1 h in secondary antibody (goat anti-rabbit 1:200 in blocking solution), then reacted in the dark with ABC reagents (standard Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA). Sections were preincubated for 10 min in a filtered mixture containing 6 mL of 0.1 mol/L Tris buffer, 2 mg 3,3′-diaminobenzidine (DAB), and 400 mg imidazole, then for 15 min in 6 mL of the same DAB/imidazole/Tris mixture containing 3 μL H2O2.
Quantitative histology
To determine the density of caspase-3-positive neurons in a given brain region, we used the optical disector and fractionator method.18 A counting frame (0.05 × 0.05 mm, disector height, 0.05 mm) and a high numerical aperture objective lens were used to visualize caspase-3-positive neurons. Unbiased sampling of each brain region was achieved by randomly selecting 10–12 viewing fields over which the counting frame was positioned for counting at different focal levels (Stereo Investigator System, MBF Bioscience, Burlington, VT).3–7 The numerical density of caspase-3-stained neurons in any given region was expressed as the number of cells/mm3.
Statistical analyses
To identify anesthesia-induced pathomorphological changes, we assessed the stereological cell count data using ANOVA models that included a between-subject variable, such as treatment (experimental group, sham, and “true” controls). When ANOVAs with repeated measures were done, the Bonferroni correction or Tukey’s test was used to help maintain prescribed alpha levels (e.g., 0.05). The data points are presented as mean ± SEM.
Findings
We used continuous monitoring of cardiovascular and respiratory functions to assure adequate tissue perfusion, as well as oxygenation and ventilation during anesthesia. Vital signs (Fig. 1A) and arterial blood gas values (Fig. 1B) were recorded. Figure 1A illustrates the results obtained from the experimental animal which, for anesthesia, was given a combination of midazolam, N2O, and isoflurane, and sham control group which received a continuous infusion of fentanyl. Figure 1B illustrates the results obtained from the experimental animal only since this was the most toxic anesthesia regimen. All measured physiological parameters were within physiological ranges for piglets (indicated with dashed lines in each graph).
Figure 1.

Anesthesia does not cause significant disturbances in vital signs (A), cardiorespiratory, or metabolic parameters (B) in 5-day-old piglets. Vital signs were monitored continuously and recorded every 30 min during anesthesia exposure (A). All measured parameters in experimental piglet (exposed to isoflurane, 0.55-vol%, + N2O, 75-vol%, + midazolam, 1 mg/kg, i.m.) and sham control piglet (exposed to a continuous fentanyl infusion, 15 μg/kg/h) were within physiologically acceptable ranges (dashed lines).23,24 Arterial blood gas analysis was done every hour during anesthesia exposure (B). All measured parameters in piglets (exposed to isoflurane, 0.55-vol%, + N2O, 75-vol%, + midazolam, 1 mg/kg, i.m.) were within physiologically acceptable ranges (dashed lines).23
To determine the regional distribution and severity of anesthesia-induced neuroapoptosis caused by fentanyl and the anesthesia cocktail used, we have examined piglet brain regions stained with caspase-3 (Figs. 2–4). The developing parietal (Fig. 2) and occipital (Fig. 3) cortices (in particular, the II and IV cortical layers) were the most sensitive to anesthesia-induced damage. Even though the density of caspase-3-positive neurons in “true” control varied between the parietal and occipital cortices, the number of neurons undergoing apoptosis was small. A somewhat higher degree of apoptosis was detected in sham control (fentanyl-treated)19 than in “true” control, although the difference was not statistically significant. The combination of three anesthetics (isoflurane, midazolam, and nitrous oxide) induced a statistically significant (on average 10- to 20-fold) increase in neuroapoptosis in both cortical regions—parietal and occipital—as compared to that in “true” controls (*, P < 0.001 except for parietal cortex-layer II where P < 0.05). Again in both cortical regions, this combination also induced a statistically significant (on average 5- to 7-fold) increase in neuroapoptosis as compared to that in sham control (†, P < 0.001 except for parietal cortex-layer II where P < 0.05).
Figure 2.

Anesthesia induces significant caspase-3 activation in the developing parietal cortex of 5-day-old piglets. In piglet brains, the sensitivity of the parietal cortices to general anesthesia is quantified as the number of caspase-3-positive cells/mm3 in layers II and IV. The density of caspase-3-positive neurons in “true” control was minimal in parietal cortical regions. In comparison, although a somewhat higher degree of caspase-3 labeling occurred in sham control (exposed to a continuous fentanyl infusion, 15 μg/kg/h), the difference was not statistically significant. The combination of isoflurane (0.55-vol%), N2O (75 vol-%), and midazolam (1 mg/kg, i.m.) induced a significant increase in caspase-3 labeling as compared to labeling in both “true” and sham controls (*, P < 0.05, and †, P < 0.05, respectively in layer II; *, P < 0.001, and †, P < 0.001, respectively in layer IV) (n = 1 piglet per condition, n = 5 sections per brain region).
Figure 4.
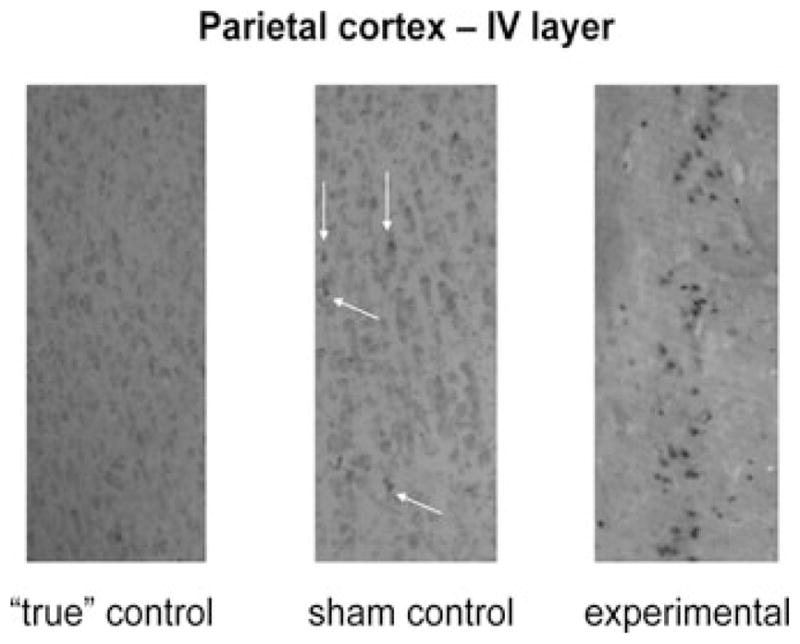
Anesthesia-induced caspase-3 activation in the IV layer of parietal cortex. Exposure of 5-day-old piglet to the triple anesthesia combination (isoflurane, 0.55-vol% + N2O, 75-vol% + midazolam, 1 mg/kg, i.m.) caused significant caspase-3 activation. Only minimal caspase-3 labeling is shown in sham control (magnification, approximately 100×).
Figure 3.

Anesthesia induces significant caspase-3 activation in the developing occipital cortex of 5-day-old piglets. The sensitivity of occipital cortices in piglet brains to general anesthesia is quantified as the number of caspase-3-positive cells/mm3 in layers II and IV. The density of caspase-3-positive neurons in “true” control was minimal in the parietal cortical regions. Although a somewhat higher degree of labeling occurred in sham control (exposed to continuous fentanyl infusion, 15 μg/kg/h), the difference from labeling in “true” control was not statistically significant. The combination of isoflurane (0.55-vol%), N2O (75 vol-%), and midazolam (1 mg/kg, i.m.) caused a significant increase in caspase-3 labeling as compared to that in “true” and sham controls (*, P < 0.001, and †, P < 0.001, respectively) (n = 1 piglet per condition, n = 4–5 sections per brain region).
Figure 4 shows caspase-3 staining of the IV cortical layer of a parietal cortex not exposed to anesthesia (left panel, “true” control), one exposed to fentanyl anesthesia (middle panel, sham control), and one exposed to the triple anesthesia combination (right panel, experimental). In these scenes there was no caspase-3 labeling in “true” controls and minimal labeling in sham controls (marked with white arrows). However, substantial caspase-3 activation was seen in the experimental cortex.
Our piglet experiments indicate that isoflurane, a commonly used general anesthetic, administered in combination with nitrous oxide and midazolam, induces significant apoptotic neurodegeneration in the developing parietal and occipital cortices, specifically targeting the II and IV cortical layers. Although fentanyl, an intravenous narcotic, did not cause a significant increase in apoptotic damage in vulnerable brain regions, the observed caspase-3 activation was at least doubled compared to the amount in “true” control.
Although piglets have a brain-growth spurt that is twice as long as that in guinea pigs and 10 times longer than that in rats, piglets appear to be just as vulnerable as these species to anesthesia-induced developmental neuroapoptosis when exposed to only 4 hrs of general anesthesia.
Primates
Synaptogenesis in primates lasts for about 2 years and, as in humans, occurs both pre- and postnatally.11 Therefore, primates, especially rhesus monkeys, in which brain development closely mimics that in humans, are good animal models for examining anesthesia-induced developmental neuroapoptosis. For the last several years, Slikker and colleagues8 have been studying the neurotoxic effects of ketamine, an intravenous general anesthetic commonly used in the pediatric population. They have found that the developing brain of rhesus monkeys is highly sensitive to ketamine anesthesia administered during an early stage of synaptogenesis. For example, a continuous ketamine infusion that is required to achieve and maintain a surgical plane of anesthesia for 24 h causes in both the fetal rhesus brain cortex (122 days of gestation) and neonatal rhesus brain cortex (5 postnatal days) a significant increase in the number of caspase-3, Fluoro-Jade C, and silver-stain-positive cells when examined 6 h after the cessation of ketamine infusion.8 Interestingly, like rodent and guinea pig brains, the rhesus brain at 35 postnatal days was insensitive to the same anesthesia treatment.8
Discussion
The timing, rather than the duration of anesthesia, determines the vulnerability of the developing brain to anesthesia-induced neuroapoptosis
Collectively, the animal studies published so far have confirmed that the duration of synaptogenesis is not a fundamental determinant of the severity of anesthesia-induced neuronal damage. This is true regardless of the duration of anesthesia exposure. For instance, anesthesia of fairly short duration, such as 4 h in guinea pigs or piglets and 24 h in primates, relative to the 10 and 20 weeks of synaptogenesis, respectively, in guinea pigs and piglets and the 2 years in primates, promotes significant activation of the apoptotic machinery in developing neurons.5,8 Even in rats, despite the fact that histopathological signs of apoptosis could be captured after 6 h of anesthesia, the warning signs of activation of the apoptotic pathways (especially the intrinsic one) could be detected as early as within the first couple of hours of general anesthesia.6 This indicates that, although the final stages of neuronal demise may take some time to evolve, the commitment to neuronal suicide occurs early during general anesthesia.
Guinea pig, piglet, and primate studies clearly confirm that, despite the maintenance of homeostasis and tight control of all vital parameters, significant neuronal apoptosis occurs in the immature brain. This suggests that general anesthetics rather than cardiovascular and respiratory abnormalities, such as hypoxia, hypercarbia, metabolic or respiratory acidosis, hypotension, or hypoglycemia, are the main culprit.4,8
The use of animal species in which brain development follows different schedules (e.g., pre- versus postnatal versus both pre- and postnatal) has enabled us to address the question of whether vulnerability to anesthesia neurotoxicity depends on when synaptogenesis occurs. The answer is that regardless of whether the peak of synaptogenesis occurs in utero or postnatally, the immature brain is at risk. Rodents, in which brain development occurs almost completely postnatally, were just as susceptible1–3,5–7 as were guinea pigs,4 in which brain development almost completely occurs postnatally, and just as susceptible as were piglets and primates,8 in which brain development occurs both before and after birth.
So what seems to matter most when it comes to the vulnerability of the developing brain to anesthesia toxicity? Based on currently available data, the most important determinant appears to be the timing of the exposure to anesthesia relative to the time point in synaptogenesis. Specifically, brain regions that are at the peak of synaptogenesis are most vulnerable.1,2,4,6,8 In all of the mammalian species studied, these are the brain regions that are most severely damaged, regardless of the duration or type of general anesthesia. The precise timing of regional brain development and the exact peak of synaptogenesis for each brain region has not been well established. Nevertheless, it is reasonable to propose that the region-specific vulnerability to general anesthesia demonstrated in the presently available studies is a consequence of the fact that these regions, at the time of anesthesia exposure, are at the peak of synaptogenesis. For example, when rat anterior thalamic nuclei and cerebral cortex are exposed at 7 days of age, they happen to be extremely susceptible1,2,6; when a piglet’s thalamus is exposed at 5 days of age, it is not as sensitive (data not shown), but the cerebral cortex is very vulnerable. Although it was not conceived for that purpose, examination of anesthesia-induced neurodegenerative patterns during brain development could serve as a mapping tool to decipher the specific times when each brain region in different species is at the peak of synaptogenesis.
In conclusion, general anesthesia causes severe neurodegeneration in the mammalian developing brain. The main determinant of the severity of that neurodegeneration appears to be the timing, rather than the duration of the anesthesia exposure; brain regions are most sensitive at the peak of their synaptogenesis. This suggests that general anesthetics cause severe disturbance of the key elements of synaptogenesis by modulating the fine neurotransmitter balance and highly programmed neuronal communication20 at the time when this communication is most vital for the survival and proper functioning of immature neurons. Indeed, anesthesia-induced activation of apoptotic neurodegeneration can lead to permanent harm, manifested as significant neuronal loss in susceptible brain regions4,21 and behavioral impairments (e.g., cognitive deficits) later in life.3,22
Acknowledgments
This study was supported by the NIH/NICHD HD 44517 (to V.J-T.) and Harold Carron endowment (to V.J-T.). V.J-T. is an Established Investigator of the American Heart Association. The authors wish to thank Lisa Carter for technical assistance.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Ikonomidou C, Bosch F, Miksa M, et al. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 2.Ikonomidou C, Bittigau P, Ishimaru MJ, et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 3.Jevtovic-Todorovic V, Hartman RE, Izumi Y, et al. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizzi S, Carter LB, Ori C, et al. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008;18:198–210. doi: 10.1111/j.1750-3639.2007.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu LX, Yon JH, Carter LB, et al. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–1615. doi: 10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 6.Yon JH, Daniel-Johnson J, Carter LB, et al. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;35:815–827. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 7.Yon JH, Carter LB, Reiter RJ, et al. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–530. doi: 10.1016/j.nbd.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 8.Slikker W, Jr, Zou X, Hotchkiss CE, et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–158. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 9.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–808. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 10.Young C, Jevtovic-Todorovic V, Qin YQ, et al. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Brit J Pharmacol. 2005;146:189–197. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 12.Jones DG, Dittmer MM, Reading LC. Synaptogenesis in guinea-pig cerebral cortex: a glutaraldehyde-PTA study. Brain Res. 1974;70:245–259. doi: 10.1016/0006-8993(74)90315-1. [DOI] [PubMed] [Google Scholar]
- 13.Lennon AM, Francon J, Fellous A, et al. Rat, mouse, and guinea pig brain development and microtubule assembly. J Neurochem. 1980;35:804–813. doi: 10.1111/j.1471-4159.1980.tb07076.x. [DOI] [PubMed] [Google Scholar]
- 14.Satas S, Haaland K, Thoresen M, et al. MAC for halothane and isoflurane during normothermia and hypothermia in the newborn piglet. Acta Anaesthesiol Scand. 1996;40:452–456. doi: 10.1111/j.1399-6576.1996.tb04468.x. [DOI] [PubMed] [Google Scholar]
- 15.Hornbein TF, Eger EI, II, Winter PM, et al. The minimum alveolar concentration of nitrous oxide in man. Anest Analg. 1982;61:553–556. [PubMed] [Google Scholar]
- 16.Mahmoudi NW, Cole DJ, Shapiro HM. In-sufficient anesthetic potency of nitrous oxide in the rat. Anesthesiology. 1989;70:345–349. doi: 10.1097/00000542-198902000-00027. [DOI] [PubMed] [Google Scholar]
- 17.Epemolu O, Bom A, Hope F, et al. Reversal of neuromuscular blockade and simultaneous increase in plasma rocuronium concentration after the intravenous infusion of the novel reversal agent Org 25969. Anesthesiology. 2003;99:632–637. doi: 10.1097/00000542-200309000-00018. [DOI] [PubMed] [Google Scholar]
- 18.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. TINS. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- 19.Rajan V, Beharry KDA, Williams P, et al. Pharmacodynamic effects and pharmacokinetic profile of continuous infusion fentanyl in newborn piglets. Biol Neonate. 1998;74:39–47. doi: 10.1159/000014009. [DOI] [PubMed] [Google Scholar]
- 20.Lunardi N, Joksovic P, Todorovic SM, et al. Soc Neurosci Abst Program 10.13. 2008. General anesthetics cause structural and functional perturbations of the developing synapses in rat brain. [Google Scholar]
- 21.Nikizad H, Yon JH, Carter LB, et al. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann NY Acad Sci. 2007;1122:69–82. doi: 10.1196/annals.1403.005. [DOI] [PubMed] [Google Scholar]
- 22.Fredriksson A, Archer T, Alm H, et al. Neuro-functional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–376. doi: 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 23.Chandler CJ, Ong BY, Sitar DS. Kinetic disposition of ethanol in the neonatal piglet and hemodynamic effects in the presence and absence of 4-methylpyrazole. Toxicol Appl Pharmacol. 1989;99:185–192. doi: 10.1016/0041-008x(89)90001-x. [DOI] [PubMed] [Google Scholar]
- 24.Chandler CJ, Ong BY, Sitar DS. Haemo-dynamic alterations in anaesthetized and acutely intoxicated newborn piglets. Br J Pharmacol. 1990;101:227–231. doi: 10.1111/j.1476-5381.1990.tb12118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]