

Abstract
Caloric restriction is associated with a reduction in body weight and temperature but a reduction in trabecular bone volume and paradoxically an increase in adipocytes within the bone marrow. The nature of these adipocytes is uncertain, although there is emerging evidence of a direct relationship between bone remodeling and brown adipocytes. For example, in heterotropic ossification, brown adipocytes set up a hypoxic gradient that leads to vascular invasion, chondrocyte differentiation and subsequent bone formation. Additionally, deletion of retinoblastoma protein in an osteosarcoma model leads to increased hibernosarcoma (brown fat tumor). Interestingly, brown adipose tissue (BAT) senescences with age at a time when thermoregulation becomes altered,, bone loss becomes apparent and sympathetic activity increases. Interestingly, heart rate is an unexpected but good predictor of fracture risk in elderly individuals, pointing to a key role for the sympathetic nervous system in senile osteoporosis. Hence the possibility exists that BAT could play an indirect role in age-related bone loss. However, evidence of an indirect effect from thermogenic dysfunction on bone loss is currently limited. Here, we present current evidence for a relationship between brown adipose tissue and bone as well as provide novel insights into the effects of thermoregulation on bone mineral density.
INTRODUCTION
Homeostasis is the process by which an organism maintains life through regulation of biological processes, including balancing energy expenditure and energy intake. Organisms have evolved mechanisms of trading energy-consuming processes for others that may increase their chances of survival. For example, when the body is responding to stress, the sympathetic nervous system shuts down certain processes like digestion and prepares the body for “fight or flight” by elevating heart rate and mobilizing energy. Therefore, it is essential to consider evolutionary adaptations when investigating the relationship between tissues and their homeostatic responses . Bone is a dynamic tissue that not only provides structure and stability, but also serves as a sink for calcium and a physical compartment for hematopoiesis and stem cell recruitment.
The skeleton constantly remodels itself such that every ten years humans generate a new skeleton. This occurs because there is coupling of bone remodeling in a three month cycle whereby bone resorption (by osteoclasts) is coupled to bone formation (by osteoblasts). Activation of these remodeling cycles occurs in response to physical and hormonal signals and requires a significant amount of energy. Hence it is not surprising that bone has emerged as an endocrine organ tied to fuel homeostasis. For example studies have shown that leptin, an adipokine released by fat cells and a sensor for fuel status, activates hypothalamic signals that regulate not only appetite and reproductive status, but also bone turnover and temperature (Barnes et al., 2010). Similarly it is now apparent from recent work that insulin signaling in osteoblasts increases osteoclast recruitment, which in turn releases uncarboxylated osteocalcin (ucOC) into the circulation (Ferron et al., 2010; Fulzele et al.). In a feed-forward mechanism, ucOC then promotes insulin sensitivity in adipose tissue and increases pancreatic β-cell insulin production and secretion (Clemens & Karsenty). Thus, bone is not only important in regulation of calcium levels and providing physical structure, but it is also an endocrine organ regulating energy homeostasis (Clemens & Karsenty; Lee et al., 2007).
Bone remodeling can uncouple during physiologic and pathologic conditions. For example, in the former as peak bone mass is acquired during adolescence, formation exceeds resorption and there is a net gain in bone. Similarly, during lactation, when calcium needs are enhanced and energy demands are immense, resorption far exceeds formation. In both these homeostatic processes, a new steady state returns such that resorption and formation become recoupled and bone mass is restored. On the other hand, pathologic uncoupled bone remodeling leads to a loss of bone mineral density as resorption exceeds formation. The end result is a state of osteoporosis in which bone micro architecture is altered leading to enhanced skeletal fragility. Bone loss can be dramatic during states of calcium deficiency, nutritional deficiencies, skeletal unloading, (e.g. bed rest or micro gravity), menopause in women or advancing age in both men and women, (Syed & Hoey, 2010). However in the latter, it is now clear that bone loss actually starts early after peak bone acquisition and continues its slow but inexorable process long before other signs of aging are apparent. An unresolved question is not whether loss of bone during these states is tied to energy status but rather what mechanisms are operative that tie remodeling status to fuel economy.
A wide variety of animal models have been employed to address this question. The hibernating black bear is a provocative model in which energy intake rises dramatically during the fall season leading to increased body weight, enhanced leptin production and greater bone turnover (Donahue et al., 2006; McGee-Lawrence et al., 2009). High calorie intake initiates hibernation which in turn leads to reduced energy demands associated with relative immobility. However, skeletal loss does not occur and bone remodeling remains coupled albeit at a lower set point in response to lower energy requirements. However, bears are not ideal experimental models for many reasons. Hence, most investigators have focused on,rodents, and in particular mice. For example, we know that In C57BL/6 mice, the most common laboratory strain, that peak bone mass is acquired in the trabecular compartment by 6 weeks of age, and in the cortical compartment by 6 months. Not surprisingly, and similar to humans, trabecular bone loss begins, albeit slowly, as early as 6 weeks of age, as resorption increases while formation remains relatively stable. (Glatt et al., 2007; Syed & Hoey, 2010). In most strains of mice, lactation of multiple pups leads to a state of uncoupled remodeling with excess bone resorption over formation. However, like humans, the remodeling state is re-calibrated post lactation, and bone mass is restored to a new steady state.
As noted, a fundamental question in bone biology relates to the relationship of bone turnover to energy status. During aging in humans, catabolic activities are enhanced, and there is evidence that sympathetic tone increases. Within the bone marrow space, osteoblast number decreases, resorption increases, and there is gradual replacement of bone cell precursors with adipocytes. A similar aging phenotype occurs in mice, with the osteogenic potential of isolated bone marrow stromal cells (BMSCs) markedly decreasing with age (Zhang et al., 2008).Ultimately, there is a loss of osteoblast number and function with age which occurs in tandem with a gain of marrow adipocytes and enhanced expression of PPARγ, a critical factor for fat cell differentiation and energy storage (Lazarenko et al., 2007). This reciprocal relationship has also been observed in other models of secondary osteoporosis including anorexia nervosa, immobility, and type 1 diabetes. (Botolin et al., 2005; Bredella et al., 2009; Das et al., 1975; Devlin et al., 2010). Marrow adiposity can also be induced with the PPARγ agonist rosiglitazone in mice, and this results in reduced osteoblast differentiation and subsequent bone loss. Thus adiposity has been suggested to be an important contributor to age-related changes in bone (Ali et al., 2005; Lazarenko et al., 2007). Because adipocytes and osteoblasts are derived from the same progenitors (mesenchymal stem cells (MSCs)), promotion of the adipocyte lineage could occur at the expense of the osteoblast lineage. Additionally, adipocytes could have direct deleterious effects on bone through secretion of adipokines and pro-inflammatory cytokines that can impair osteoblastogenesis and promote osteoclastogenesis (Magni et al., 2010; Pino et al., 2010).
Adipocytes in the bone marrow may provide important clues as to the relationship between energy sufficiency and bone remodeling. For example, bone marrow fat cells often respond to physiologic changes differently from those in other white adipose depots. In Type 1 diabetes, marrow fat is markedly increased despite a reduction in total fat mass (Botolin et al., 2005). In anorexia nervosa, or calorie deprivation subcutaneous fat almost totally disappears while marrow adiposity becomes pronounced. These scenarios share a common feature; i.e. loss of substrate availability. On the other hand, with high fat feeding in mice, or obesity in humans, there are few marrow adipocytes.
The classification of marrow adipocytes as white, brown or something in between is in question (Kawai et al., 2010). Brown fat is important for thermoregulation in neonates and hibernating animals. Unlike white fat, brown adipocytes divert protons from the generation of ATP in mitochondria towards the production of heat. This function is essential during early life to protect newborns from environmental changes, and is a characteristic feature of hibernating animals. Preformed brown adipocytes are derived from the same mesenchymal precursors that also give rise to osteoblasts, white adipocytes and muscle. Recently retinoblastoma protein has been identified as one regulator of the choice between brown adipocyte and osteoblast (Calo et al.). Brown like adipocytes have also been described in traditional white fat depots; these cells are induced by changes in homeostasis and may be an important counter regulatory mechanism to defend the body against dramatic environmental changes..
DIRECT INTERACTION BETWEEN BROWN ADIPOCYTES AND BONE
Brown adipocytes are derived from mesenchymal stem cells, which can also differentiate into white adipocytes, muscle and osteoblasts. Differentiation into brown adipocytes requires PPARγ, as well as other transcription factors not necessary for white adipogenesis. Work by Spiegelman and colleagues have produced a better understanding of the differentiation of brown adipocytes in recent years. Myf5 expressing progenitor cells can differentiate into both brown adipocytes and muscle, but the switch to brown adipocytes in particular is dependent on the transcription factor PRDM16, which is expressed both during early and late brown adipocyte differentiation (Seale et al., 2008). Kajimura et al. later demonstrated that PRDM16 binds to the active form of the transcription factor C/EBPβ and that overexpression of both PRDM16 and C/EBPβ in fibroblasts is sufficient to induce brown adipocyte differentiation (Kajimura et al., 2009). In their latest work, Seale et al. have demonstrated that PRDM16 and markers of brown adipose tissue (BAT) function (ie UCP-1) are more highly expressed in subcutaneous adipose depots than in intra-abdominal depots after cold challenge (Seale et al.). The role for PRDM16 in mediation of this white to brown adipose phenotype switch was further confirmed when PRDM16 was overexpressed in fat using the aP2 promoter and subcutaneous depots expressed UCP-1, Cidea, and PGC1α while other WAT depots did not (Seale et al.).
Since there is evidence for muscle to brown adipocyte and white to brown adipocyte switching, could there be an osteoblast to brown adipocyte switch? Retinoblastoma protein (Rb) is a tumor suppressor that is commonly mutated in human tumors and is also a key transcriptional regulator of differentiation. When bound to the transcriptional repressor E2F, pRb suppresses activity of the PPARγ promoter, thus inhibiting adipogenic differentiation (Fajas et al., 2002). Interestingly, pRb can also enhance Runx2 activity and subsequent osteoblast maturation (Thomas et al., 2001). However, Calo et al. recently discovered that knockout of Rb in combination with the ablation of the tumor suppressor p53 resulted in a switch from predominantly osteosarcoma (bone tumor) to hibernosarcoma, which is a tumor composed of differentiated brown adipose tissue (Calo et al.). The authors then went on to examine the phenotype of deletion of Rb alone after embryonic day 6.5 (Meox2 expressing cells). Calvarial osteoblasts from these embryos were able to differentiate into adipocyte-like cells after culture with adipogenic media and brown adipose tissue mass was increased in Rb null embryos (e18.5). While brown adipogenesis increased, whole body mineralization as well as calvarial expression of later osteoblast differentiation markers (alkaline phosphatase and bone sialoprotein) were reduced in e18.5 embryos. These are the first studies to indicate that in certain situations, brown adipogenesis could occur at the expense of osteoblastogenesis.
Alternatively, bone formation could be assisted by the presences of brown adipocytes as during heterotropic ossification, i.e. the formation of bone in soft tissue. In mice, heterotropic ossification is modeled by BMP-2 injection in muscle. Olmstead-Davis et al. found that soon after BMP-2 injection, brown adipocytes appear and generate a hypoxic gradient, which triggers vascular invasion, chondrocyte development and subsequent bone formation (Olmsted-Davis et al., 2007). In Misty mice, which have reduced brown adipose tissue, white adipocytes switched to a brown adipocyte-like phenotype after BMP-2 injection (Olmsted-Davis et al., 2007). Although there is no solid evidence of brown adipocytes in bone, it is plausible that brown-like adipocytes could transiently exist and could foster bone formation. Several rodent models provide insight into this possible scenario. C3H/HeJ mice are an inbred strain that has high bone mass. Surprisingly, these animals have an abundance of marrow adiposity. Gene expression demonstrated the presence of markers of brown adipogenesis within the marrow population (C.J. Rosen, unpublished data). These mice also show an exaggerated response to rosiglitazone (Ackert-Bicknell et al., 2009). On the other hand, in mice without brown fat, bone mass is extremely low (C.J. Rosen, unpublished data). Whether brown adipocytes are present in bone marrow and how they could regulate osteogenesis remains to be determined,
INTERACTION BETWEEN THERMOGENESIS AND BONE REMODELING
Here, we will introduce basic principles of thermogenesis, which have been extensively reviewed elsewhere (Morrison et al., 2008; Van Someren et al., 2002). Thermogenesis is the obligatory (result of normal metabolic processes) or facultative production of heat. Facultative thermogenesis occurs with the purpose of generating heat to maintain temperature homeostasis and includes physical activity, shivering, and humoral thermogenesis. Heat production in humoral thermogenesis occurs through brown adipose tissue (BAT) and/or as a result of certain hormones (thyroid, glucagon, epinephrine, growth hormone (GH) and adrenocorticotropic hormone (ACTH)). The oxidation of lipids in brown adipose is regulated by direct sympathetic nervous system (SNS) stimulation via norepinephrine and is accomplished by expression of uncoupling protein 1 (UCP1). UCP-1 facilitates a proton leak across the mitochondrial membrane, resulting in energy released as heat rather than stored as chemical energy. The degree of UCP1 expression and oxidation of lipids is dependent upon the amount of norepinephrine binding to β3-adrenergic receptors.
Preformedinterscapular BAT (iBAT) is present in rodents, human infants and other mammals. Evidence from rodents indicates that when challenged with cold, brown-like adipocytes form in classical white adipose tissue depots and generate heat (wBAT). This process catabolizes lipid stores in white adipose tissue and is the basis of the hypothesis that thermogenic challenge may be used to fight obesity (Kozak et al., 2010). BAT thermogenesis was previously thought to be negligible/absent in adult humans as iBAT reduces in size and function with age. However, images obtained from positron-emission tomography (PET) have recently indicated subscapular and peri-vertebral regions of glucose uptake and heat production in adult humans given cold challenge (Kozak et al., 2010; Virtanen et al., 2009). Additionally, UCP-1 expression has been found in human white adipose tissue biopsies and human preadipocytes isolated from subcutaneous WAT depots have more potential to become brown adipocytes that those isolated from omental or mesenteric depots (Schulz et al., 2011) but there remains no concrete evidence to date of inducible wBAT in humans (Kozak et al., 2010).
As we introduced, cold exposure leads to the sympathetic nervous system stimulation of UCP-1 expression and heat generation in brown adipose. Additionally, brown adipocytes can form in subcutaneous white adipose tissue depots in rodents, and this becomes more prevalent with more severe exposure to cold. Another parallel sympathetic network increases heart rate in response to cold (Morrison et al., 2008). Similarly, we believe that cold could stimulate changes in bone density through activation of the sympathetic nervous system (Figure 1). This is the framework of our hypothesis that brown adipose tissue could have indirect effects on the skeleton. We hypothesize that increased sympathetic activity, due to cold stimulus and/or impaired response to cold by brown adipose tissue, can cause bone loss. As brown adipose tissue function and thermogenesis declines with age in both rodents and humans, we believe it could be a significantly contributing factor to age-related bone loss.
Figure 1.
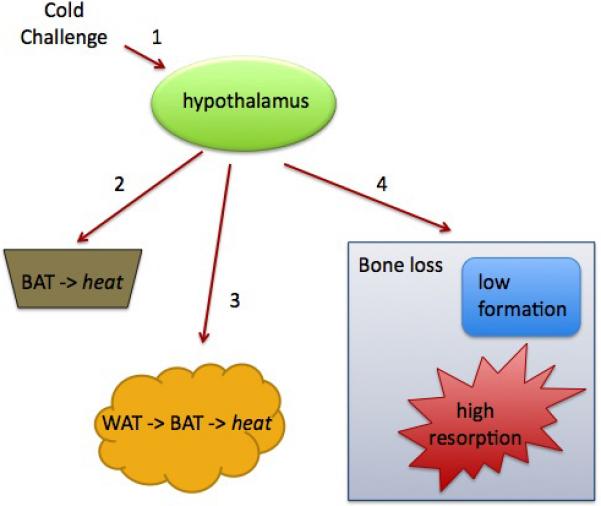
Influence of thermogenesis on bone remodeling. 1, Cold activates sympathetic output from the hypothalamus. Sympathetic nervous system activity stimulates heat production in 2, brown adipose tissue (BAT) and 3, white adipose tissue (WAT). A parallel sympathetic network activates heart rate and could 4, alter bone remodeling. Impaired thermogenesis increases sympathetic activity, which could lead to bone loss.
Bone is innervated by sensory and autonomic (sympathetic and parasympathetic) nerve fibers and hypothalamic relay via leptin and serotonin stimulates sympathetic effects on bone density (Ducy et al., 2000; Takeda et al., 2002; Yadav et al., 2009). Osteoblasts express β2-adrenergic receptors (β2-AR) and deletion of β2-AR increases bone density while overexpression of β2-AR causes bone loss (Bouxsein et al., 2009; Elefteriou et al., 2005). Similarly, treatment with isoproterenol, a beta-agonist, causes bone loss and beta-blockers increase bone density. β2-AR signaling in osteoblasts reduces bone formation and increases expression of RANKL, which in turn increases osteoclast activity (Elefteriou et al., 2005). SNS tone has been implicated in a number of conditions of bone loss, including ovariectomy, stress and unloading (Elefteriou et al., 2005). Despite these effects, the role of the SNS in age-related osteoporosis is not well characterized, and this is in part due to limitations in measuring sympathetic activity.
SNS activity can be measured by assaying sympathetic endpoints (ideally more endpoints measured will provide a clearer picture). Aged 344/N Fischer rats have high circulating catecholamines, including norepinephrine and epinephrine, compared to young rats (Cizza et al., 1995). Elderly humans have increased blood pressure and increased norepinephrine levels that could be due in part to impaired norepinephrine reuptake (Seals & Esler, 2000; Uchino et al., 2010). Despite the evidence of increased SNS activity in humans, not all studues agree and sympathetic activity is likely confounded by other factors (Piazza et al., 2010). Although no direct relationship has been demonstrated between sympathetic activity and bone density in elderly humans, relationships have been drawn between heart rate and bone. A study of elderly women found that elevated resting heart rate (>80 bpm) was a significant risk factor for hip, pelvis, rib and vertebral fractures and this relationship could not be attributed to other risk factors (Kado et al., 2002).
Could impaired thermogenesis lead to some of the age-related changes in sympathetic activity observed in humans? Although cutaneous and neocortical temperature sensing nerve endings remain intact, thermal perception is attenuated with advanced age (Van Someren et al., 2002) and this is augmented by diabetes (even without clinical neuropathy present) (Jensen et al., 1991). Additionally, variability in thermal perception increases with age (Van Someren et al., 2002). When exposed to cold, elderly animals and humans have a larger decrease in core body temperature and take longer to recover, suggesting mechanisms for thermogenesis are less effective (Van Someren et al., 2002). Whether impaired sympathetic output or response to sympathetic stimulus is responsible for reduced response to cold remains unclear.
The interaction between thermoregulation and bone remodeling is even less evident. In fact, the only evidence for an effect of temperature on bone is that cold exposure (1.5 hours to 3 weeks) reduced plasma osteocalcin levels in rats (Patterson-Buckendahl et al., 1995). If one considers osteocalcin to be a marker of bone formation, this study would suggest cold exposure reduces osteoblast function. However, recent evidence indicates osteocalcin levels could increase with increased bone resorption, although this relationship is not always observed (Ferron et al., 2010). Clearly, more research into the direct effects of cold exposure on bone metabolism are necessary to determine if thermoregulation can influence bone metabolism.
There is a mouse model of defective thermoregulation that has altered bone metabolism. Mice deficient in the transcription factor C/EBPβ mice have impaired thermoregulation as a result of reduced capacity for brown adipose tissue to store fatty acids and are unable to maintain body temperature in response to cold exposure (Carmona et al., 2005). These mice had higher induction of UCP1 expression in response to long term cold exposure, likely to compensate for reduced fuel for BAT thermogenesis, which is indicative of increased sympathetic input, although sympathetic activity was not measured. In separate studies, global deletion in C/EBPβ also results in bone loss due to reduced osteoblast activity and increased resorption, depending on the age of the animals (Zanotti et al., 2009). Although C/EBPβ itself is important for osteoblast and adipocyte differentiation (Iyer et al., 2004) and appears to be a negative regulator of osteoclast differentiation, the relationship between bone density and thermogenesis in this model is intriguing and warrants further investigation.
CONCLUSION
Here we present intriguing evidence of a direct relationship (positive and negative) between brown adipose tissue and bone. Both of these are exemplified by specific gene deletions or disease models. Therefore it remains to be determined if brown adipocytes could have an effect on normal bone remodeling or in fracture healing. There is less evidence of a direct effect of thermoregulation on skeletal remodeling, although it is remarkable that during peak bone acquisition in humans, a time of very active bone formation, adipocytes infiltrate long bone marrow. Temperatures in the appendicular skeleton are usually three to four degrees lower than in the abdominal cavity, and it is conceivable that a specific temperature threshold is necessary during high rates of bone formation. There is also circumstantial evidence of defective thermoregulation and increased sympathetic activity in the elderly, as well as an unexplained correlation between heart rate and fracture risk. Thus further investigation into the effects of thermoregulation and brown adipose tissue on the skeleton are justified and may result in development of novel therapeutics.
ACKNOWLEDGEMENTS
C. J. Rosen is supported by NIH grants R24DK084970 and AR45433.
Footnotes
DISCLOSURE STATEMENT
The authors have nothing to disclose.
REFERENCES
- Ackert-Bicknell Cl, Shockley Kr, Horton Lg, Lecka-Czernik B, Churchill Ga, Rosen Cj. Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulinlike growth factor-I. Endocrinology. 2009;150(3):1330–1340. doi: 10.1210/en.2008-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Aa, Weinstein Rs, Stewart Sa, Parfitt Am, Manolagas Sc, Jilka Rl. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146(3):1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- Barnes Mj, Rogers Rc, Van Meter Mj, Hermann Ge. Co-localization of TRHR1 and LepRb receptors on neurons in the hindbrain of the rat. Brain Res. 2010;1355:70–85. doi: 10.1016/j.brainres.2010.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, Faugere Mc, Malluche H, Orth M, Meyer R, Mccabe Lr. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146(8):3622–3631. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein Ml, Devlin Mj, Glatt V, Dhillon H, Pierroz Dd, Ferrari Sl. Mice lacking betaadrenergic receptors have increased bone mass but are not protected from deleterious skeletal effects of ovariectomy. Endocrinology. 2009;150(1):144–152. doi: 10.1210/en.2008-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackert-Bicknell Cl, Shockley Kr, Horton Lg, Lecka-Czernik B, Churchill Ga, Rosen Cj. Strainspecific effects of rosiglitazone on bone mass, body composition, and serum insulinlike growth factor-I. Endocrinology. 2009;150(3):1330–1340. doi: 10.1210/en.2008-0936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali Aa, Weinstein Rs, Stewart Sa, Parfitt Am, Manolagas Sc, Jilka Rl. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146(3):1226–1235. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- Barnes Mj, Rogers Rc, Van Meter Mj, Hermann Ge. Co-localization of TRHR1 and LepRb receptors on neurons in the hindbrain of the rat. Brain Res. 2010;1355:70–85. doi: 10.1016/j.brainres.2010.07.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botolin S, Faugere Mc, Malluche H, Orth M, Meyer R, Mccabe Lr. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146(8):3622–3631. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein Ml, Devlin Mj, Glatt V, Dhillon H, Pierroz Dd, Ferrari Sl. Mice lacking beta-adrenergic receptors have increased bone mass but are not protected from deleterious skeletal effects of ovariectomy. Endocrinology. 2009;150(1):144–152. doi: 10.1210/en.2008-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredella Ma, Fazeli Pk, Miller Kk, Misra M, Torriani M, Thomas Bj, Ghomi Rh, Rosen Cj, Klibanski A. Increased bone marrow fat in anorexia nervosa. J Clin Endocrinol Metab. 2009;94(6):2129–2136. doi: 10.1210/jc.2008-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Quintero-Estades Ja, Danielian Ps, Nedelcu S, Berman Sd, Lees Ja. Rb regulates fate choice and lineage commitment in vivo. Nature. 466(7310):1110–1114. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona Mc, Hondares E, Rodriguez De La Concepcion Ml, Rodriguez-Sureda V, Peinado-Onsurbe J, Poli V, Iglesias R, Villarroya F, Giralt M. Defective thermoregulation, impaired lipid metabolism, but preserved adrenergic induction of gene expression in brown fat of mice lacking C/EBPbeta. Biochem J. 2005;389(Pt 1):47–56. doi: 10.1042/BJ20050009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizza G, Pacak K, Kvetnansky R, Palkovits M, Goldstein Ds, Brady Ls, Fukuhara K, Bergamini E, Kopin Ij, Blackman Mr, et al. Decreased stress responsivity of central and peripheral catecholaminergic systems in aged 344/N Fischer rats. J Clin Invest. 1995;95(3):1217–1224. doi: 10.1172/JCI117771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens Tl, Karsenty G. The osteoblast: An insulin target cell controlling glucose homeostasis. J Bone Miner Res. doi: 10.1002/jbmr.321. [DOI] [PubMed] [Google Scholar]
- Das Sk, Scott Mt, Adhikary Pk. Effect of the nature and amount of dietary energy on lipid composition of rat bone marrow. Lipids. 1975;10(10):584–590. doi: 10.1007/BF02532721. [DOI] [PubMed] [Google Scholar]
- Devlin Mj, Cloutier Am, Thomas Na, Panus Da, Lotinun S, Pinz I, Baron R, Rosen Cj, Bouxsein Ml. Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res. 2010;25(9):2078–2088. doi: 10.1002/jbmr.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue Sw, Galley Sa, Vaughan Mr, Patterson-Buckendahl P, Demers Lm, Vance Jl, Mcgee Me. Parathyroid hormone may maintain bone formation in hibernating black bears (Ursus americanus) to prevent disuse osteoporosis. J Exp Biol. 2006;209(Pt 9):1630–1638. doi: 10.1242/jeb.02185. [DOI] [PubMed] [Google Scholar]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling Af, Beil Ft, Shen J, Vinson C, Rueger Jm, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000;100(2):197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- Elefteriou F, Ahn Jd, Takeda S, Starbuck M, Yang X, Liu X, Kondo H, Richards Wg, Bannon Tw, Noda M, Clement K, Vaisse C, Karsenty G. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434(7032):514–520. doi: 10.1038/nature03398. [DOI] [PubMed] [Google Scholar]
- Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril Mb, Miard S, Auwerx J. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3(6):903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- Ferron M, Wei J, Yoshizawa T, Del Fattore A, Depinho Ra, Teti A, Ducy P, Karsenty G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142(2):296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulzele K, Riddle Rc, Digirolamo Dj, Cao X, Wan C, Chen D, Faugere Mc, Aja S, Hussain Ma, Bruning Jc, Clemens Tl. Insulin receptor signaling in osteoblasts regulates postnatal bone acquisition and body composition. Cell. 142(2):309–319. doi: 10.1016/j.cell.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt V, Canalis E, Stadmeyer L, Bouxsein Ml. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22(8):1197–1207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- Iyer Vv, Kadakia Tb, Mccabe Lr, Schwartz Rc. CCAAT/enhancer-binding protein-beta has a role in osteoblast proliferation and differentiation. Exp Cell Res. 2004;295(1):128–137. doi: 10.1016/j.yexcr.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Jensen Ts, Bach Fw, Kastrup J, Dejgaard A, Brennum J. Vibratory and thermal thresholds in diabetics with and without clinical neuropathy. Acta Neurol Scand. 1991;84(4):326–333. doi: 10.1111/j.1600-0404.1991.tb04963.x. [DOI] [PubMed] [Google Scholar]
- Kado Dm, Lui Ly, Cummings Sr. Rapid resting heart rate: a simple and powerful predictor of osteoporotic fractures and mortality in older women. J Am Geriatr Soc. 2002;50(3):455–460. doi: 10.1046/j.1532-5415.2002.50110.x. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni Jv, Gygi Sp, Spiegelman Bm. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009;460(7259):1154–1158. doi: 10.1038/nature08262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai M, Sousa Km, Macdougald Oa, Rosen Cj. The many facets of PPARgamma: novel insights for the skeleton. Am J Physiol Endocrinol Metab. 2010;299(1):E3–9. doi: 10.1152/ajpendo.00157.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak Lp, Koza Ra, Anunciado-Koza R. Brown fat thermogenesis and body weight regulation in mice: relevance to humans. Int J Obes (Lond) 2010;34(Suppl 1):S23–27. doi: 10.1038/ijo.2010.179. [DOI] [PubMed] [Google Scholar]
- Lazarenko Op, Rzonca So, Hogue Wr, Swain Fl, Suva Lj, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148(6):2669–2680. doi: 10.1210/en.2006-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Nk, Sowa H, Hinoi E, Ferron M, Ahn Jd, Confavreux C, Dacquin R, Mee Pj, Mckee Md, Jung Dy, Zhang Z, Kim Jk, Mauvais-Jarvis F, Ducy P, Karsenty G. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni P, Dozio E, Galliera E, Ruscica M, Corsi Mm. Molecular aspects of adipokine-bone interactions. Curr Mol Med. 2010;10(6):522–532. doi: 10.2174/1566524011009060522. [DOI] [PubMed] [Google Scholar]
- Mcgee-Lawrence Me, Wojda Sj, Barlow Ln, Drummer Td, Castillo Ab, Kennedy O, Condon Kw, Auger J, Black Hl, Nelson Ol, Robbins Ct, Donahue Sw. Grizzly bears (Ursus arctos horribilis) and black bears (Ursus americanus) prevent trabecular bone loss during disuse (hibernation). Bone. 2009;45(6):1186–1191. doi: 10.1016/j.bone.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison Sf, Nakamura K, Madden Cj. Central control of thermogenesis in mammals. Exp Physiol. 2008;93(7):773–797. doi: 10.1113/expphysiol.2007.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmsted-Davis E, Gannon Fh, Ozen M, Ittmann Mm, Gugala Z, Hipp Ja, Moran Km, Fouletier-Dilling Cm, Schumara-Martin S, Lindsey Rw, Heggeness Mh, Brenner Mk, Davis Ar. Hypoxic adipocytes pattern early heterotopic bone formation. Am J Pathol. 2007;170(2):620–632. doi: 10.2353/ajpath.2007.060692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Buckendahl P, Kvetnansky R, Fukuhara K, Cizza G, Cann C. Regulation of plasma osteocalcin by corticosterone and norepinephrine during restraint stress. Bone. 1995;17(5):467–472. doi: 10.1016/8756-3282(95)00281-x. [DOI] [PubMed] [Google Scholar]
- Piazza Jr, Almeida Dm, Dmitrieva No, Klein Lc. Frontiers in the use of biomarkers of health in research on stress and aging. J Gerontol B Psychol Sci Soc Sci. 2010;65(5):513–525. doi: 10.1093/geronb/gbq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino Am, Rios S, Astudillo P, Fernandez M, Figueroa P, Seitz G, Rodriguez Jp. Concentration of adipogenic and proinflammatory cytokines in the bone marrow supernatant fluid of osteoporotic women. J Bone Miner Res. 2010;25(3):492–498. doi: 10.1359/jbmr.090802. [DOI] [PubMed] [Google Scholar]
- Schulz Tj, Huang Tl, Tran Tt, Zhang H, Townsend Kl, Shadrach Jl, Cerletti M, Mcdougall Le, Giorgadze N, Tchkonia T, Schrier D, Falb D, Kirkland Jl, Wagers Aj, Tseng Yh. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A. 2011;108(1):143–148. doi: 10.1073/pnas.1010929108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe Hm, Erdjument-Bromage H, Tempst P, Rudnicki Ma, Beier Dr, Spiegelman Bm. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe Hm, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, Spiegelman Bm. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 121(1):96–105. doi: 10.1172/JCI44271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals Dr, Esler Md. Human ageing and the sympathoadrenal system. J Physiol. 2000;528(Pt 3):407–417. doi: 10.1111/j.1469-7793.2000.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed Fa, Hoey Ka. Integrative physiology of the aging bone: insights from animal and cellular models. Ann N Y Acad Sci. 2010;1211:95–106. doi: 10.1111/j.1749-6632.2010.05813.x. [DOI] [PubMed] [Google Scholar]
- Takeda S, Elefteriou F, Levasseur R, Liu X, Zhao L, Parker Kl, Armstrong D, Ducy P, Karsenty G. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–317. doi: 10.1016/s0092-8674(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Thomas Dm, Carty Sa, Piscopo Dm, Lee Js, Wang Wf, Forrester Wc, Hinds Pw. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8(2):303–316. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- Uchino Bn, Birmingham W, Berg Ca. Are older adults less or more physiologically reactive? A meta-analysis of age-related differences in cardiovascular reactivity to laboratory tasks. J Gerontol B Psychol Sci Soc Sci. 2010;65B(2):154–162. doi: 10.1093/geronb/gbp127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Someren Ej, Raymann Rj, Scherder Ej, Daanen Ha, Swaab Df. Circadian and age-related modulation of thermoreception and temperature regulation: mechanisms and functional implications. Ageing Res Rev. 2002;1(4):721–778. doi: 10.1016/s1568-1637(02)00030-2. [DOI] [PubMed] [Google Scholar]
- Virtanen Ka, Lidell Me, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto Nj, Enerback S, Nuutila P. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360(15):1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- Yadav Vk, Oury F, Suda N, Liu Zw, Gao Xb, Confavreux C, Klemenhagen Kc, Tanaka Kf, Gingrich Ja, Guo Xe, Tecott Lh, Mann Jj, Hen R, Horvath Tl, Karsenty G. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138(5):976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Stadmeyer L, Smerdel-Ramoya A, Durant D, Canalis E. Misexpression of CCAAT/enhancer binding protein beta causes osteopenia. J Endocrinol. 2009;201(2):263–274. doi: 10.1677/JOE-08-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ou G, Hamrick M, Hill W, Borke J, Wenger K, Chutkan N, Yu J, Mi Qs, Isales Cm, Shi Xm. Age-related changes in the osteogenic differentiation potential of mouse bone marrow stromal cells. J Bone Miner Res. 2008;23(7):1118–1128. doi: 10.1359/JBMR.080304. [DOI] [PMC free article] [PubMed] [Google Scholar]