

Abstract
Chronic inflammation in the central nervous system (CNS) is a central feature of many neurodegenerative and autoimmune diseases. As an immunologically competent cell, the astrocyte plays an important role in CNS inflammation. It is capable of expressing a number of cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β) that promote inflammation directly and through the recruitment of immune cells. Checkpoints are therefore in place to keep tight control over cytokine production. Adenylate/uridylate-rich elements (ARE) in the 3′ untranslated region of cytokine mRNAs serve as a major checkpoint by regulating mRNA stability and translational efficiency. Here, we examined the impact of KH-type splicing regulatory protein (KSRP), an RNA binding protein which destabilizes mRNAs via the ARE, on cytokine expression and paracrine phenotypes of primary astrocytes. We identified a network of inflammatory mediators, including TNF-α and IL-1β, whose expression increased 2 to 4-fold at the RNA level in astrocytes isolated from KSRP−/− mice compared to littermate controls. Upon activation, KSRP−/− astrocytes produced TNF-α and IL-1β at levels that exceeded control cells by 15-fold or more. Conditioned media from KSRP−/− astrocytes induced chemotaxis and neuronal cell death in vitro. Surprisingly, we observed a prolongation of half-life in only a subset of mRNA targets and only after selective astrocyte activation. Luciferase reporter studies indicated that KSRP regulates cytokine gene expression at both transcriptional and post-transcriptional levels. Our results outline a critical role for KSRP in regulating pro-inflammatory mediators and have implications for a wide range of CNS inflammatory and autoimmune diseases.
Keywords: RNA stability, AU-rich element, TNF-α, IL-1β
INTRODUCTION
Astrocytes are the primary glial cells in the central nervous system (CNS) and outnumber neurons by more than 5-fold (Sofroniew and Vinters, 2010). They provide a variety of functions ranging from structural and trophic support of the neurovascular unit to synaptic function and neuronal plasticity (Dong and Benveniste, 2001; Sofroniew and Vinters, 2010). Astrocytes also play a pivotal role in regulating CNS inflammatory responses in pathological states such as multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, and brain cancer. Astrocytes produce a wide range of cytokines and chemokines which can attract, activate, or attenuate other immune cells including microglia and peripherally derived lymphocytes. An important level of control for this molecular diversity is post-transcriptional regulation. Many mRNAs of factors produced by astrocytes, such as tumor necrosis factor alpha (TNF-α) and interleukin 1 beta (IL-1β), contain adenylateuridylate rich elements (ARE) in the 3′ untranslated region (UTR), which directly regulate the stability and translational efficiency of the transcript. KH-type splicing regulatory protein (KSRP) and other adenylate-uridylate–rich RNA-binding proteins (AUBP) such as Hu Antigen R (HuR) interact with the ARE to modulate this level of gene regulation. KSRP, also known as far upstream element binding protein 2 (FBP2), was first characterized as a DNA-binding protein that enhances c-myc transcription (Davis-Smyth et al., 1996; Duncan et al., 1994). Subsequently, KSRP was found to bind AREs in the 3′ untranslated region (UTR) and promote mRNA deadenylation and degradation by recruiting the exosome (Gherzi et al., 2010). Here, we investigated the impact of KSRP−/− on molecular and cellular phenotypes of astrocytes. We found that KSRP−/− astrocytes expressed significantly higher levels of select inflammatory mediators including TNF-α and IL-1β. Luciferase reporter studies indicate that KSRP exerts a suppressive effect at the post-transcriptional, and surprisingly, the transcriptional level. KSRP deletion led to production of soluble factors that induced astrocyte chemotaxis and neuronal apoptosis. Our results indicate that KSRP is an important suppressor of inflammatory cytokine expression in astrocytes.
MATERIALS AND METHODS
Plasmids
Cloning of the pGL2-TNF-3′ UTR and pGL2-HelN1-promoter constructs was described previously (King, 1996; Nabors et al., 2003). To generate pGL2-TNF(−655), pGL2-TNF(−610), pGL2-TNF(−122), and pGL2-TNF(−78), the human TNF-α promoter was amplified by PCR using pXP1-TNF as a template [kindly provided by Dr. Economou (Rhoades et al., 1992)]. The primers used were: TNF655-Fwd, 5′-ATATATACGCGTATGAAAGAAGAAGGCCTGC-3′, TNF610-Fwd, 5′-ATATATACGCGTATTTCACTCCCCGGGGCT-3′, TNF122-Fwd, 5′-ATATATACGCGTTACCGCTTCCTCCAGATGA-3′, TNF78-Fwd, 5′-ATATATACGCGTGTTTTCCGCTGGTTGAATGA-3′, TNF3-Rev, 5′-ATATATCTCGATTGTGCCAACAACTGCCTT-3′. The mouse TREM-1 promoter (+54 to −1,200) (Hosoda et al., 2011), kindly provided by Dr. Nagaoka (Juntendo University, Tokyo, Japan), was cloned into pGL2-Basic vector at MluI/BglII sites. A plasmid containing heterogeneous nuclear ribonucleoprotein C (hnRNP C) was a gift from Dr. Scott Blume (University of Alabama at Birmingham). The pSV-β-galactosidase control vector was purchased from Promega. Construction of pcDNA3-FLAG-KSRP was described previously (Lin et al., 2007).
Cell Culture
Primary astrocytes were isolated from KSRP−/− (Lin et al., 2011) or littermate control cortex as described previously (Qin et al., 2008). Cells were cultured until confluent and then shaken vigorously at room temperature for 3 h. Cells were trypsinized at room temperature and seeded in six-well plates. Purity of astrocytes was confirmed by immunostaining for glial fibrillary acidic protein (GFAP). RAW 264.7 and N2a cells were cultured according to manufacturer’s protocol (ATCC).
Transfection and Luciferase Assays
Astrocytes were transfected by electroporation (Invitrogen) following the manufacturer’s protocol. The amount of luciferase reporter plasmid was kept constant (1 μg) and three doses of expression plasmid were used (1, 2, and 3 μg). DNA for each transfection was kept constant with empty pcDNA vector. Transfections were done in duplicate. Reporter plasmids (0.2 μg for N2A and 0.5 μg for RAW264.7 cells), pcDNA3-FLAG-KSRP or pcDNA3-myc-hnRNP C (0.2–1.5 μg), and a SV40-β-galactosidase plasmid (0.2 μg) were cotransfected using the TransIT-LT kit (Mirus). Luciferase activity was assayed using a kit (Promega) and measured with the Spectrafluor plus machine (Tecan U.S.). Values were normalized to β-galactosidase activity as previously described (Nabors et al., 2003).
Western blot, RNA Immunoprecipitation, and ELISA
Whole cell extracts were prepared using the M-Per kit (Pierce) and quantitated with a bicinchoninic acid protein assay kit. Fifty micrograms of protein were resolved by gel electrophoresis, blotted and probed with antibodies to the following targets: FLAG and α-tubulin (Sigma), MYC, HuR, TIA-1, TIAR (Santa Cruz), TTP (Abcam), and luciferase (Millipore). For RNA immunoprecipitation, WT astrocytes were treated with lipopolysaccharide (LPS) at 1 μg/ml or vehicle for 24 h, and cytoplasmic extracts were harvested using NE-PER (Pierce Endogen). Two hundred micrograms of extract were immunoprecipitated as described elsewhere (Lu et al., 2009). RNA was extracted from the precipitate and subjected to qRT-PCR. RNA input of each target was determined by qRT-PCR. For ELISA, astrocytes were seeded in 6-well plates and treated with LPS (1 μg/mL) or TNF-α (10 ng/mL). TNF-α, IL-1β, and vascular endothelial growth factor (VEGF) were quantified in the culture media by ELISA (R & D system). Values were normalized to volume of growth media. Total RNA was extracted from the cells and quantitated to ensure an equivalent input of cells.
RNA Isolation, Quantitation, Kinetics, and Microarray Analysis
Total RNA was extracted and quantitated with the RiboGreen kit (Invitrogen). One microgram of RNA was reverse transcribed according to the manufacturer’s specifications (Applied Biosystems). Quantitative RT-PCR was carried out as described previously (Nabors et al., 2003) using the 7900HT Fast Real-Time PCR System and commercially available primers and probes (Applied Biosystems). Target mRNA values were normalized to S9 mRNA values. For RNA kinetics, astrocytes were treated with TNF-α (10 ng/mL) or LPS (1 μg/mL) for 24 h. Half-lives were determined as previously described (Lu et al., 2009). For microarray analysis, we performed single chip assays using 300 ng of total RNA from KSRP−/− and wild type (WT) astrocytes. Double strand cDNA was generated by linear amplification using Random Primer-T7 oligomers and reverse transcriptase. Biotin-labeled cRNA was synthesized by in vitro transcription (IVT) using the 3′-Amplification Reagents for IVT labeling (Affymetrix) followed by cRNA fragmentation and preparation of hybridization cocktail. Arrays were hybridized overnight at 45°C, and then washed, stained, and scanned the next day. Gene expression levels were extracted using Affymetrix GeneChip Command Console. GeneChip data was extracted, normalized and summarized using the robust multiaverage method in GeneSpring v10.0.
Migration and Neuronal Death Assays
The migration assay was carried out as described (Ding et al., 2002; Tezel et al., 2001). Briefly, tissue culture inserts with 8 μm diameter pores (Costar) were coated on the lower surface with 10 μg/mL vitronectin (Invitrogen) at 37°C overnight, followed by blocking with 1% BSA in DMEM for 1 h at 37°C. Primary astrocytes were trypsinized, washed and resuspended in migration assay buffer (DMEM with 1% BSA, 2 mM L-glutamine) at 4 × 105 cells/ml. A 100 μL cell suspension was seeded on the insert, placed in 24-well plate with astrocyte conditioned media, and incubated at 37°C. After 48 h, cells on the lower insert surface were fixed with 4% paraformaldehyde (PFA) for 10 min. Cells were washed and stained with 1% crystal violet for 15 min. Migrated cells were counted in 8 random high power fields and averaged from three independent experiments. For the cell death assays, cortical neurons were cultured in 24-well plates with neurobasal medium supplemented with B27 and 0.5 mM L-glutamine. Neurons were maintained for 7 days, after which half of the medium was replaced with fresh medium. Neurons were treated with astrocyte conditioned media at different doses for 24 h. For apoptosis, neurons were fixed in 4% PFA and incubated with cleaved caspase-3 antibody (Cell Signaling) at 1:500 followed by a secondary anti-rabbit Alexa Fuor 488 antibody (Invitrogen). Images were viewed under a Nikon Eclipse TI microscope. Positive cells were expressed as a percentage of total cell nuclei. For nuclear fragmentation, cell chromatin was stained with 4′, 6′-diamidino-2-phenylindole (DAPI) and neuronal viability was quantified by evaluating nuclear morphology. Neurons with condensed nuclei and/or fragmented chromatin were considered nonviable (Kaltschmidt et al., 1999; Morrison et al., 2006; Zhang et al., 2008). Viable and nonviable neurons were counted from eight fields for each treatment.
Statistical Analyses
All statistical analyses were performed with Graphpad Prism v. 5 software using a Student’s t test.
RESULTS
TNF-α and IL-1β Are Upregulated in KSRP−/− Astrocytes
Astrocytes can produce high levels of TNF-α and IL-1β in response to environmental cues such as TNF-α stimulation (Dong and Benveniste, 2001). Since the mRNAs of these inflammatory cytokines are regulated post-transcriptionally through the ARE, we investigated the impact of a KSRP−/− genotype on their expression in astrocytes. Because AUBPs can regulate each other, we first tested for HuR, TIAR, TIA-1, and tristetraprolin (TTP) expression by Western blot in KSRP−/− astrocytes (Fig. 1). KSRP was expectedly absent, but we found no changes in other AUBPs either in basal or activated (TNF-α stimulated) cells compared to littermate controls. We next analyzed TNF-α and IL-1β mRNA expression by qRT-PCR at baseline and after activation (Fig. 2). In control astrocytes, both mRNAs were induced after TNF-α stimulation (2-fold for TNF-α and 10-fold for IL-1β). KSRP−/− astrocytes, however, had significantly higher mRNA levels (2- to 2.5-fold) under basal and stimulated conditions (P < 0.05). KC (CXCL1) mRNA and VEGF, both of which contain AREs in the 3′ UTR, were not affected indicating that KSRP deletion did not have a global effect on ARE-containing mRNAs. We next tested the impact of LPS stimulation on TNF-α and IL-1β expression (Fig. 3). LPS produced a higher overall induction of both mRNAs in control cells, but as with TNF-α stimulation, there was a 2 to 3-fold increase in KSRP−/− cells. We quantitated these cytokines in the culture media 24 and 72 hours after stimulation and found a 15-fold increase in TNF-α production in KSRP−/− versus control cells. With IL-1β, no protein was detected in WT cells whereas KSRP−/− cells produced ~2 pg/mL. Neither TNF-α nor IL-1β was detected in either cell type under basal conditions or after TNF-α stimulation (data not shown). Secreted VEGF showed no change consistent with the mRNA levels (Supp. Info. Fig. 1). We examined astrocytes from heterozygous (KSRP−/+) mice and found intermediate increases of TNF-α (~1.7-fold) and IL-1β (1.5 to 2.1-fold; Supp. Info. Fig. 2) consistent with a KSRP dose effect. Except for the LPS stimulated condition, differences between mutant and wild-type astrocytes remained significant. To determine whether increases of TNF-α and IL-1β expression in KSRP−/− astrocytes were related to changes in RNA kinetics, we measured the half-lives of those targets under basal, TNF-α or LPS-stimulated conditions (Fig. 4). For both mRNAs, no differences in half-life were observed at baseline or with TNF-α stimulation. The degradation curves were essentially superimposable. The short mRNA half-life of TNF-α (~0.3 h) was similar to that reported in other cell types and it did not change after TNF-α stimulation (Nabors et al., 2003; Park et al., 2008). After LPS stimulation, there was a 3-fold increase in TNF-α half-life in control cells (0.9 h), but for KSRP−/− cells, the half-life extended to 1.7 h. The IL-1β half-life did not change substantially in control cells after LPS stimulation whereas in KSRP−/− cells it increased nearly 2-fold (3.0 to 5.8 h). In summary, loss of KSRP led to global increases in TNF-α and IL-1β mRNA levels, but only stimulus-dependent changes in mRNA stability.
Fig. 1.

KSRP−/− genotype does not affect other ARE RNA binding proteins in astrocytes. Western blot of protein extracts prepared from cultured KSRP−/− and littermate control (WT) astrocytes at baseline and after TNF-α stimulation (10 ng/mL for 24 h). Antibodies are shown to the left of the blot.
Fig. 2.

KSRP−/− genotype enhances TNF-α-induced TNF-α and IL-1β mRNA expression in astrocytes. qRT-PCR analysis of TNF-α and IL-1β in KSRP−/− or littermate control (WT) astrocytes treated with vehicle (PBS) or TNF-α (10 ng/mL) for 24 h. All mRNA values were normalized to the housekeeping mRNA, S9, and expressed as a fold-induction over unstimulated WT astrocytes (set at a value of 1). Data points are the mean (±SEM) of at least four independent experiments. *P, < 0.05 (compared with WT).
Fig. 3.
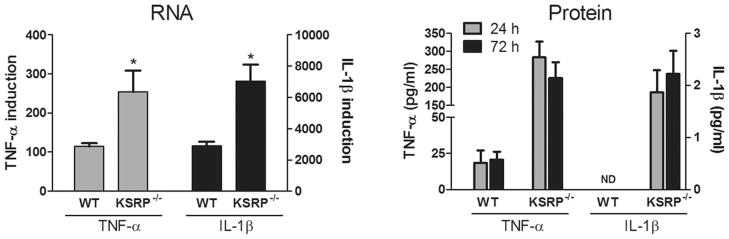
KSRP−/− genotype enhances LPS-induced mRNA and protein expression of TNF-α and IL-1β. Left panel: astrocytes were treated with LPS (1 μg/mL) for 24 h, and analyzed for TNF-α and IL-1β mRNAs by qRT-PCR. Target mRNA expression was normalized to S9 mRNA and values are expressed as a fold-induction over unstimulated WT astrocytes (set at a value of 1). Data points are the mean (±SEM) of at least four independent experiments. *P < 0.05 compared with WT. Right panel: ELISA analysis of culture media from astrocytes stimulated with LPS for the time points designated. Data points are the mean (±SEM) of two independent experiments tested in duplicate.
Fig. 4.

LPS-induced TNF-α and IL-1β mRNA stabilization is enhanced in KSRP−/− astrocytes. WT or KSRP−/− astrocytes were treated with vehicle (PBS), TNF-α (10 ng/mL) or LPS (1 μg/mL) for 24 h, followed by 5 μg/mL ActD to block transcription. RNA was collected at indicated time intervals and analyzed by qRT-PCR. RNA expression was normalized to S9 and expressed as a percentage of the level before ActD was added (time 0). Values are the mean (±SEM) of at least three independent experiments. Estimated half-lives in hours are shown in parentheses.
Other mRNA Targets Are Affected by the KSRP−/− Genotype
To identify other mRNA targets affected by KSRP deletion, we performed a pilot microarray analysis on KSRP−/− and WT astrocytes and screened for mRNAs that changed by more than 2-fold in KSRP−/− versus control (not shown). Three identified targets were further investigated: C-type lectin domain family 5, member A (CLEC5A), triggering receptor expressed on myeloid cells (TREM) and chemokine, C-C motif, ligand 12(CCL12). Each target was validated by qRT-PCR and showed more than a 2-fold increase compared to control cells under basal conditions (Table 1). TNF-α stimulation induced all three targets by an additional 2 to 4-fold, with TREM-1 showing the highest induction. KSRP−/− cells again had a 2 to 3-fold increase in expression of these mRNA targets. LPS produced a 4-fold induction of CCL12 but had no effect on CLEC5A or TREM-1. One gene, inducible nitric oxide synthase (iNOS), whose regulation has been linked to KSRP (Pautz et al., 2010 ), was not identified in the microarray. Quantitative RT-PCR validated this result, showing no significant difference between KSRP−/− and control cells either at baseline or upon induction with astrocyte stimulation (Supp. Info. Fig. 3). Interestingly, fuse binding protein 1 (FBP1), which is closely related to KSRP, was identified in the array and found to be increased by 2-fold in KSRP−/− cells (Supp. Info. Fig. 3). A similar pattern of upregulation was also observed when KSRP was silenced in a hepatoma cell line (Malz et al., 2009). FBP3, on the other hand, which is the third member of the family, was not changed. We next measured the RNA half-lives of TREM-1, CLEC5A and CCL12 under basal and stimulated conditions (Table 1 and Supp. Info. Fig. 4). All three mRNAs had similar degradation curves in KSRP−/− and control astrocytes under basal and TNF-α stimulated conditions, although the half-life of CLEC5A could not be estimated because it exceeded the last time point of 6 h. TNF-α stimulation stabilized CCL12 but only modestly prolonged TREM-1 mRNA half-life (from 2.2 to 3.6 h). LPS induced stabilization of CCL12 in KSRP−/− but not control cells (> 6.0 versus 6.0 h). In summary, there were significant increases in all three target mRNAs in KSRP−/− astrocytes with only selective prolongation of half-lives depending on the stimulus type and the mRNA target.
TABLE 1.
Expression and Kinetics of Three mRNA Targets Identified by Microarray Showing Greater Than 2-fold Increase in KSRP−/− Astrocytes
| Gene | Cell | mRNAa | Half-life (h)b | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Basal | TNF | LPS | Basal | TNF | LPS | ||
| CLEC5A | WTc | 1.0 | 2.2 ± 0.1 | 0.9 ± 0.1 | >6.0 | >6.0 | nd |
| KO | 2.3 ± 0.3** | 5.0 ± 0.5** | 2.0 ± 0.3* | >6.0 | >6.0 | ||
| TREM-1 | WT | 1.0 | 3.9 ± 0.4 | 1.0 ± 0.1 | 2.2 | 3.6 | nd |
| KO | 2.6 ± 0.3** | 10.7 ± 1.4** | 3.2 ± 0.8** | 2.2 | 3.6 | ||
| CCL12 | WT | 1.0 | 2.2 ± 0.8 | 4.1 ± 0.5 | 5.1 | >6.0 | 6.0 |
| KO | 2.3 ± 0.3* | 5.0 ± 0.5* | 9.8 ± 1.4* | 5.1 | >6.0 | >6.0 | |
mRNA values were determined by qRT-PCR and are the mean ± SD of 3–8 independent samples. All values are expressed as fold-induction over wild type astrocytes under basal conditions.
RNA half life values were extrapolated from degradation curves generated from three independent experiments (see Supp. Info. Fig. 4).
WT, wild-type astrocytes from littermate controls; KO, KSRP−/− astrocytes; nd, not done.
P < 0.005;
P < 0.05 comparing KO to WT.
KSRP Negatively Regulates Gene Expression in Astrocytes at the Post-Transcriptional Level
Our findings thus far indicated that KSRP deletion led to increased RNA stability of certain targets but not others (despite an increase in target mRNA levels). To investigate mechanisms of KSRP regulation in astrocytes, we used luciferase reporters to assess the impact of the 3′ UTR (post-transcriptional regulation) and the promoter (transcriptional regulation) on target mRNA expression. For the former, we co-transfected a luciferase reporter fused to the ARE portion of the TNF-α 3′UTR along with a FLAG-KSRP or myc-hnRNP C (control) expression plasmid (Fig. 5A). The antisense orientation of the 3′ UTR fragment served as a second control. When KSRP was expressed ectopically in WT astrocytes, there was a significant and gene dose-dependent decrease in luciferase activity (P <0.0001), but no change with the control 3′ UTR or with hnRNP C (Fig. 5B). Ectopic KSRP also significantly inhibited TNF-α 3′ UTR mediated expression in KSRP−/− astrocytes (Supp. Info. Fig. 5A). To confirm that the reduced luciferase activity was related to reduced protein expression, we assessed reporter activity in the neuronal-like N2a cell line. We observed a significant dose-dependent reduction in luciferase activity in conjunction with luciferase protein, as determined by Western blot, and luciferase mRNA as determined by qRT-PCR (Supp. Info. Fig. 5B–D). While KSRP has been shown to bind TNF-α mRNA in other cell systems (and regulate the mRNA post-transcriptionally), the other four targets have not. So, to begin identifying a link, we did RNA immunoprecipitation with LPS-stimulated wild-type astrocyte extracts using an anti-KSRP antibody (Fig. 6). Bound mRNA was quantitated by qRT-PCR and expressed as a percent of target mRNA in the extract (input). We identified increased binding to each of the target mRNAs (over IgG control) except TREM-1 where no binding was detected. RNA immunoprecipitation of extracts from unstimulated astrocytes displayed the same binding pattern (not shown). Inspection of the 3′ UTRs identified at least one ARE in the 3′ UTR of the bound targets, but none in TREM-1, which may explain the absence of binding. The similar pattern of binding by HuR, another AUBP, is further supportive of this explanation.
Fig. 5.

KSRP negatively regulates gene expression via the TNF-α 3′ UTR. A: Schematic figure of the luciferase reporters used showing the ARE portion of the TNF-α 3′UTR or the antisense control UTR downstream from the luciferase gene. B: The upper panel is a Western blot of WT astrocytes transfected with increasing amounts of FLAG-KSRP or myc-hnRNPC control plasmids (see Materials and Methods). The blot was probed with an anti-FLAG (KSRP) or anti-myc (hnRNP C) antibody. The lower panel shows relative luciferase activities of the reporters following co-transfection with increasing amounts of expression plasmid (1–3 μg) versus reporter plasmid (1 μg). After normalizing to the transfection control (β-galactosidase), all values were expressed as a percent of the luciferase activity when the reporter was co-transfected with pcDNA alone. The 3′ UTR tested is shown in parentheses. All data points represent the mean (±SEM) of three independent experiments. **P < 0.003, **** P <0.0001, comparing TNF-α to control 3′ UTR for KSRP.
Fig. 6.
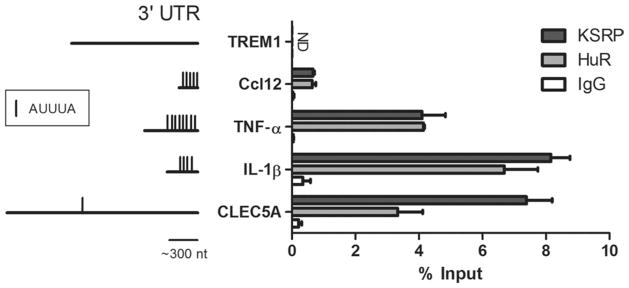
KSRP associates with all mRNA targets except TREM-1. RNA immunoprecipitation was performed on cytosolic extract from LPS-stimulated WT astrocytes using antibodies to KSRP, HuR, or control IgG. RNA from the precipitates was analyzed by qRT-PCR and expressed as a percentage of the input. To the left is a schematic diagram showing the different 3′ UTRs (mouse orthologs) and the presence of AUUUA pentamers. Data represent the mean (±SEM) of two independent assays, each evaluated in duplicate.
KSRP Negatively Regulates Gene Expression Through Transcriptional Pathways
Since differences in RNA expression under basal or TNF-α-stimulated conditions did not appear to be related to changes in RNA half-life, along with the absence of KSRP binding to TREM-1 mRNA, we examined the possibility that KSRP modulates transcription. We used a luciferase reporter downstream from portions of the TNF-α and TREM-1 promoters (Hosoda et al., 2011; Rhoades et al., 1992). Both promoters contain activator protein-1 (AP-1) and nuclear factor kappaB (NF-κB) sites as previously described (Fig. 7A). For a control, we used another mammalian promoter, Hel-N1 (King 1996). The reporters were co-transfected with FLAG-KSRP or myc-hnRNP C expression plasmids and a transfection control into astrocytes (Fig. 7B). We observed a significant and gene dose-dependent decline in luciferase activity for TNF-α and TREM-1 promoters when KSRP was co-transfected. At the highest gene dose, promoter activity was reduced to 36% (TNF-α) and 53% (TREM-1) of baseline activity. The control expression plasmid, hnRNP C, showed no effect on promoter activity at any gene dose. The effect was specific as KSRP failed to inhibit Hel-N1 promoter activity. We determined whether this negative regulation occurs in other cell types by testing TNF-α promoter activity in N2a cells (Fig. 8). KSRP, but not hnRNP C, produced a significant gene-dose dependent reduction in luciferase activity (Fig. 8A) with a concomitant decrease in protein expression (Fig. 8B). To determine the impact of KSRP on TNF-α promoter activity following LPS stimulation, we used RAW264.7, a macrophage cell line which is strongly induced by LPS (Supp. Info. Fig. 6). There was a 20-fold induction of luciferase activity in control (hnRNP C)-transfected cells which was similar to cells transfected with empty pcDNA vector. This induction was blunted by nearly 60% with KSRP co-expression (P < 0.0001) whereas hnRNP C had no effect. To characterize further the cis elements involved in this negative regulation, we performed additional mapping of the TNF-α promoter in N2a cells (Fig. 8C). We observed a significant reduction in luciferase activity when a 45 nucleotide segment at the 5′ end of the promoter region (−611 to −655) was deleted (P < 0.008). This segment included the third NF-κB site. Although additional deletions, which included the other two NF-κB sites, reduced overall luciferase activity, there was no significant difference compared to the control. These results suggest that the furthest upstream NF-kB site contributes to the negative regulation induced by KSRP. In summary, KSRP specifically inhibited TNF-α and TREM-1 promoter activity in primary astrocytes (and other cells) which may contribute to the reduced RNA and protein levels of these inflammatory mediators in the basal and stimulated states.
Fig. 7.

KSRP specifically inhibits TNF-α and TREM-1 promoter activity in astrocytes. A: Schematic diagram of TNF-α, TREM-1, or Hel-N1 promoters used in the reporter assay. B: Luciferase activity of the reporters after co-transfection with FLAG-KSRP or myc-hnRNP C plasmid at three doses (see Materials and Methods). A transfection control plasmid was used to adjust for transfection efficiency. Values are expressed as a percentage of luciferase activity in cells transfected with reporter alone. Data points for all transfection experiments shown in this figure represent the mean (±SEM) of at least three independent experiments performed in duplicate. *P < 0.03, **P < 0.003, ***P < 0.0003 compared with hnRNP C control at the equivalent gene dose.
Fig. 8.

KSRP inhibits TNF-α promoter activity in N2a cells. A: Left panel, Western blot of N2a cells 48 h after co-transfection of a luciferase reporter containing the TNF-α promoter and Flag-KSRP or myc-hnRNP C. Gene dose is expressed as molar ratio of the luciferase reporter to the expression plasmid. The blot was probed with FLAG (KSRP) and myc (hnRNP C) antibodies. Right panel, luciferase activity of transfected N2a cells. B: Western blot of luciferase protein after N2a cells were transfected with KSRP or hnRNP C and the TNF-α reporter plasmid. Antibodies are shown to the right. C: Mapping analysis of TNF-α promoter. N2a cells were transfected with KSRP or pcDNA (control), reporter plasmid and a transfection control plasmid. Data points for all transfections are the mean (±SEM) of at least three independent experiments analyzed in duplicate. **P <0.008; ****P <0.0001.
KSRP−/− Cells Induce Cortical Neuron Toxicity and Astrocyte Migration
Since KSRP−/− astrocytes produce increased amounts of soluble factors (e.g., TNF-α, IL-1β), we postulated that these cells would exert paracrine effects on neighboring cells in the CNS. We tested the phenotypes of neuronal toxicity and astrocyte chemotaxis. We used astrocyte-conditioned media (ACM) from unstimulated cells to avoid the confounding effect of LPS or TNF-α in the media. For neurotoxicity we used rat primary cortical neurons (Fig. 9A). ACM was added to the culture media, and after 24 h cells were immunostained with a cleaved caspase-3 antibody to detect apoptosis. We observed a significant and concentration dependent increase in immunoreactive cells with KSRP−/− ACM versus wild-type, with the highest dose showing a ~60% increase in positive cells (P < 0.0001). The number of non-viable cells, as determined by condensation and/or fragmentation of the nucleus, was also significantly increased with exposure to KSRP−/− ACM at similar doses (P < 0.001; Supp. Info. Fig. 7). For chemotaxis, we tested migration of wild-type astrocytes through an 8 μm pore filter using a dual chamber with ACM in the lower chamber. After 48 h, filters were removed and stained with crystal violet (Fig. 9B). We observed a significant and concentration-dependent increase in migrated astrocytes with KSRP−/− versus control ACM. At the higher dose, there was a 60% increase in migrated cells over control (P < 0.0001). These results suggest that KSRP regulates factors that exert paracrine effects on other cells in the milieu.
Fig. 9.
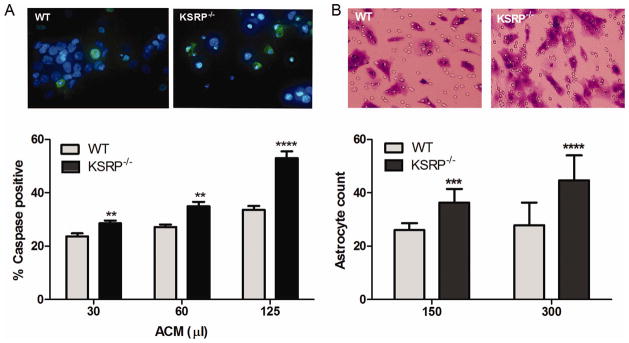
Conditioned media from KSRP−/− astrocytes induces cortical neuron toxicity and increased astrocyte migration. A: Rat cortical neurons were treated with different amounts of astrocyte conditioned media (ACM) from KSRP−/− or littermate control (WT) astrocytes for 24 h. Neurons were stained with a cleaved caspase-3 antibody and DAPI. Immunostained neurons (green) were quantitated. Data points are the mean (±SEM) of eight high-power fields from three independent experiments. **P <0.007; ****P <0.0001 compared with WT. B: WT astrocytes were plated onto tissue culture inserts with 8 μm diameter pores and placed on wells containing KSRP−/− or control ACM. After 48 h, migrated cells on the lower surface were detected with crystal violet (photomicrographs) and quantified. Data points represent the mean (±SEM) of eight high-powered fields from three independent experiments. ***P < 0.003; ****P <0.0001 compared with WT. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
In this study we investigated the role of KSRP in regulating cytokine expression in astrocytes, and its impact on cellular phenotypes relevant to the inflammatory response. We showed that a subset of cytokines, including TNF-α, IL-1β, and TREM-1, is upregulated in astrocytes when KSRP is deleted. This molecular phenotype led to production of soluble factors that enhanced astrocyte chemotaxis and neuronal toxicity. Surprisingly, there were no obvious effects on RNA half-life at baseline for these targets, and only selective effects after LPS stimulation. Luciferase reporter studies suggested that KSRP modulates gene expression in astrocytes at the transcriptional and post-transcriptional levels.
Our findings implicate KSRP as a dampener for the expression of select inflammatory mediators in astrocytes. Remarkably, the attenuating effect of KSRP was relatively uniform across the targets (2–3 fold at the mRNA level). The specificity of regulation was underscored by the absence of an effect on KC and VEGF mRNA. Protein secretion which directly impacts paracrine and autocrine functions showed much greater changes. TNF-α and IL-1β levels in the culture media, for example, were 15-fold or greater versus WT astrocytes after LPS stimulation (Fig. 3). Two possibilities may contribute to this enhancement. First, KSRP modulates translational efficiency in addition to mRNA stability. A recent report showed that KSRP silencing in Hela cells led to relocation of IL-1α and IL-6 mRNA to polysomes with increased protein secretion (Dhamija et al., 2011). Translational silencing of these targets was mediated through the ARE. The second possibility is the increased expression of TREM-1, a cell surface receptor, and CLEC5a, a trans membrane protein, both of which augment release of cytokines such as TNF-α and IL-1β (Aoki et al., 2009; Bakker et al., 1999; Bouchon et al., 2001; Chen et al., 2008). Furthermore, astrocytes express chemokine receptors which can respond to secreted factors leading to further production of inflammatory mediators (Dorf et al., 2000; Tanabe et al., 1997). TNF-α, IL-1β and CC12 are also well known to amplify the inflammatory response by promoting chemotaxis, recruitment, and activation of immune cells (both peripheral and central) (Gabay et al., 2010; Ming et al., 1987; Sarafi et al., 1997). A link to the innate immune system was also recently identified in a study of embryonic fibroblasts where deletion of KSRP led to stabilization of Type I interferon mRNAs and resistance to viral infection (Lin et al., 2011).
The astrocyte is capable of producing many secreted factors, and our pilot microarray data showed additional, albeit not yet validated, targets that changed by 2-fold or more with KSRP deletion (Dong and Benveniste 2001; Lieberman et al., 1989). KSRP knockdown studies in other cell types indicate that a broader group of genes is regulated by KSRP under different cellular conditions (Dhamija et al., 2011; Graham et al., 2010; Ruggiero et al., 2007; Winzen et al., 2007). Interestingly, IL-1β, TREM-1, CCL12, and CLEC-5A were not among the targets identified in those studies suggesting that the regulation is cell-type dependent as well. With such a broad molecular phenotype, the astrocyte, not surprisingly, can have diverse effects on surrounding cells, ranging from supportive to toxic (Renault-Mihara et al., 2008; Sofroniew and Vinters 2010; Whitney et al., 2009; Williams et al., 2007). Increased astrocyte migration with KSRP−/− ACM (Fig. 9) is relevant to glial responses in inflammatory or traumatic insults to the central nervous system. Such migration may be protective by limiting the inflammatory response, or potentially harmful by producing astrogliosis and scarring (Renault-Mihara et al., 2008; Sofroniew and Vinters 2010). While the data indicate that KSRP modulates this phenotype, it is unclear which targets are responsible. KC, a chemokine linked to astrocyte migration (Heesen et al., 1996), was not upregulated in KSRP−/− cells and thus is unlikely to be a candidate (Fig. 2). TNF-α and IL-1β, on the other hand, inhibited astrocyte migration in a scratch wound model (Faber-Elman et al., 1995). The identity of factor(s) producing cortical neuron death (Fig. 9) is also unknown. The neurotoxic role of astrocytes through secretion of toxic factors has been well established in a number of neuroinflammatory and degenerative diseases including multiple sclerosis, amyotrophic lateral sclerosis, Parkinson’s and Alzheimer’s disease (Blackburn et al., 2009; Chuanyu et al., 2011; Williams et al., 2007). TNF-α and IL-1β have both been identified as potential mediators of neuronal injury (Allan et al., 2005; Chuanyu et al., 2011; Sriram and O’Callaghan, 2007), and their upregulation in KSRP−/− astrocytes may have contributed to the neurotoxicity observed here.
The molecular mechanism for the attenuating effect of KSRP appears to be bimodal, involving transcriptional and post-transcriptional pathways. KSRP, like many other RBPs, can bind DNA and RNA and has multiple functions (Dreyfuss et al., 1993). KSRP and its homologues have been linked to transcriptional regulation, RNA splicing, 3′ processing, editing, localization, stability and miRNA maturation (Danckwardt et al., 2011; Gherzi et al., 2010). A number of these functions could potentially suppress mRNA expression, either directly through interaction with the mRNA target or its promoter, or indirectly by modulating other factors that regulate target expression (e.g. a transcriptional factor, miRNA or RBP). The absence of RNA half-life changes for the targets in KSRP−/− astrocytes at baseline or with TNF-α stimulation, despite significant increases in mRNA levels, suggests another regulatory mechanism at play. In another study using Hela cells, more than 400 mRNA targets were enriched by greater than 2-fold after KSRP knockdown yet the vast majority of targets, including TNF-α, were not stabilized (Winzen et al., 2007). Our finding of KSRP-induced inhibition of TNF-α and TREM-1 promoter activity points to transcriptional repression as an alternative mechanism of regulation. While KSRP and other members of the FBP family are known for their transactivation of c-myc, they possess promoter repressor activity (Chung et al., 2006; Duncan et al., 1996; Liu et al., 2000). Although inspection of TNF-α and TREM-1 promoter sequences used here did not reveal classic FUSE binding elements, a more recent study indicates that KSRP binds to a much broader spectrum of DNA loci with no obvious consensus sequence (Benjamin et al., 2008). We were not able to detect an interaction of KSRP by chromatin immunoprecipitation or by electrophoretic mobility shift assay (not shown). Although these negative data do not exclude a direct effect, it is possible that KSRP is modulating promoter activity indirectly by affecting expression of other transcription factors. FBP1 mRNA levels, for example, increased by 2-fold in KSRP−/− astrocytes (Supp. Info. Fig. 3). Our mapping studies suggest that the negative effect of KSRP, in part, involves sequence containing the third NF-κB site in the TNF-α promoter (Fig. 8). Interestingly, all inhibited promoters, including a cytomegalovirus promoter which was used to drive our transfection control in pilot studies (not shown), contain a number of NF-κB sites whereas the unaffected promoters, Hel-N1 and SV40 (used to drive the β-galactosidase transfection control reporter), had none. Additional studies, however, are clearly required for definitive characterization of response element(s) and the involvement of NF-κB. The inhibitory effect is also not restricted to astrocytes, and includes other immune cells, thus broadening the importance of KSRP in cytokine regulation (Supp. Info. Figs. 5 and 6).
We observed differential RNA stabilization of TNF-α, IL-1β, and CCL12 in KSRP−/− cells after LPS stimulation indicating a clear role for KSRP in the post-transcriptional regulation of these targets. This finding was supported by reporter studies with the TNF-α 3′ UTR (Fig. 5 and Supp. Info. Fig. 5) and RNA immunoprecipitation indicating that KSRP bound to these transcripts (Fig. 6). With TREM-1, there was no effect of KSRP deletion on RNA degradation, suggesting that promoter inhibition (Fig. 7) is the predominant mechanism. The absence of any clear-cut ARE in its 3′ UTR coupled with the lack of binding to KSRP (and HuR), is further supportive of that possibility. Interestingly, we observed stress-dependent selective stabilization of target mRNAs in astrocytes. The TNF-α mRNA half-life tripled (from 0.3 h to 0.9 h, Fig. 4) in LPS-stimulated WT astrocytes whereas IL-1β showed no change. This differential stabilization of cytokine mRNAs (containing AREs) has been observed with other targets in other cells (Brown et al., 1996; Chen et al., 2006; Lindsten et al., 1989; Nabors et al., 2003; Schuler and Cole, 1988; Tebo et al., 2003), underscoring the complexities of post-transcriptional regulation.
In summary, we have identified KSRP as a significant checkpoint in regulating a subset of proinflammatory mediators in astrocytes. These findings have implications for CNS autoimmune and degenerative diseases, where a sustained inflammatory response is critical for disease progression.
Supplementary Material
Acknowledgments
The authors wish to thank the tissue culture core for assistance with neuronal cultures (P30 HD038395) and the UAB Microarray Facility (P30 CA013148-39).
Grant sponsor: National Institute of Health; Grant numbers: NS064133, NS057664, GM068758, CA131468; Grant sponsor: Merit Review award from the Department of Veterans Affairs.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat Rev Immunol. 2005;5:629–640. doi: 10.1038/nri1664. [DOI] [PubMed] [Google Scholar]
- Aoki N, Kimura Y, Kimura S, Nagato T, Azumi M, Kobayashi H, Sato K, Tateno M. Expression and functional role of MDL-1 (CLEC5A) in mouse myeloid lineage cells. J Leukocyte Biol. 2009;85:508–517. doi: 10.1189/jlb.0508329. [DOI] [PubMed] [Google Scholar]
- Bakker ABH, Baker E, Sutherland GR, Phillips JH, Lanier LL. Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Natl Acad Sci USA. 1999;96:9792–9796. doi: 10.1073/pnas.96.17.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin LR, Chung H-J, Sanford S, Kouzine F, Liu J, Levens D. Hierarchical mechanisms build the DNA-binding specificity of FUSE binding protein. Proc Natl Acad Sci USA. 2008;105:18296–18301. doi: 10.1073/pnas.0803279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn D, Sargsyan S, Monk PN, Shaw PJ. Astrocyte function and role in motor neuron disease: A future therapeutic target? GLIA. 2009;57:1251–1264. doi: 10.1002/glia.20848. [DOI] [PubMed] [Google Scholar]
- Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- Brown CY, Lagnado CA, Vadas MA, Goodall GJ. Differential regulation of the stability of cytokine mRNAs in lipopolysaccharide-activated blood monocytes in response to interleukin-10. J Biol Chem. 1996;271:20108–20112. doi: 10.1074/jbc.271.33.20108. [DOI] [PubMed] [Google Scholar]
- Chen S-T, Lin Y-L, Huang M-T, Wu M-F, Cheng S-C, Lei H-Y, Lee C-K, Chiou T-W, Wong C-H, Hsieh S-L. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. 2008;453:672–676. doi: 10.1038/nature07013. [DOI] [PubMed] [Google Scholar]
- Chen Y-L, Huang Y-L, Lin N-Y, Chen H-C, Chiu W-C, Chang C-J. Differential regulation of ARE-mediated TNFα and IL-1β mRNA stability by lipopolysaccharide in RAW264.7 cells. Biochem Biophys Res Commun. 2006;346:160–168. doi: 10.1016/j.bbrc.2006.05.093. [DOI] [PubMed] [Google Scholar]
- Chuanyu L, Rui Z, Kai G, Zheng W, Michael Yaoyao Y, Lok Ting L, Dehua C, Albert Cheung Hoi Y. Astrocytes: Implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Liu J, Dundr M, Nie Z, Sanford S, Levens D. FBPs are calibrated molecular tools to adjust gene expression. Mol Cell Biol. 2006;26:6584–6597. doi: 10.1128/MCB.00754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danckwardt S, Gantzert A-S, Macher-Goeppinger S, Probst HC, Gentzel M, Wilm M, Grone H-J, Schirmacher P, Hentze MW, Kulozik AE. p38 MAPK controls prothrombin expression by regulated RNA 3′ end processing. Mol Cell. 2011;41:298–310. doi: 10.1016/j.molcel.2010.12.032. [DOI] [PubMed] [Google Scholar]
- Davis-Smyth T, Duncan RC, Zheng T, Michelotti G, Levens D. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J Biol Chem. 1996;271:31679–31687. doi: 10.1074/jbc.271.49.31679. [DOI] [PubMed] [Google Scholar]
- Dhamija S, Kuehne N, Winzen R, Doerrie A, Dittrich-Breiholz O, Thakur BK, Kracht M, Holtmann H. Interleukin-1 activates synthesis of interleukin-6 by interfering with a KH-type splicing regulatory protein (KSRP)-dependent translational silencing mechanism. J Biol Chem. 2011;286:33279–33288. doi: 10.1074/jbc.M111.264754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Stewart J, Prince CW, Chang P-L, Trikha M, Han X, Grammer JR, Gladson CL. Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain. Cancer Res. 2002;62:5336–5343. doi: 10.1100/tsw.2002.247. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. GLIA. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dorf ME, Berman MA, Tanabe S, Heesen M, Luo Y. Astrocytes express functional chemokine receptors. J Neuroimmunol. 2000;111(1–2):109–121. doi: 10.1016/s0165-5728(00)00371-4. [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem. 1993;62:289–321. doi: 10.1146/annurev.bi.62.070193.001445. [DOI] [PubMed] [Google Scholar]
- Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, Levens D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8:465–480. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- Duncan R, Collins I, Tomonaga T, Zhang T, Levens D. A unique transactivation sequence motif is found in the carboxyl- terminal domain of the single-strand-binding protein FBP. Mol Cell Biol. 1996;16:2274–2282. doi: 10.1128/mcb.16.5.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber-Elman A, Lavie V, Schvartz I, Shaltiel S, Schwartz M. Vitronectin overrides a negative effect of TNF-alpha on astrocyte migration. FASEB J. 1995;9:1605–1613. doi: 10.1096/fasebj.9.15.8529840. [DOI] [PubMed] [Google Scholar]
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- Gherzi R, Chen CY, Trabucchi M, Ramos A, Briata P. The role of KSRP in mRNA decay and microRNA precursor maturation. Wiley Interdisciplinary Reviews-RNA. 2010;1:230–239. doi: 10.1002/wrna.2. [DOI] [PubMed] [Google Scholar]
- Graham JR, Hendershott MC, Terragni J, Cooper GM. mRNA degradation plays a significant role in the program of gene expression regulated by phosphatidylinositol 3-kinase signaling. Mol Cell Biol. 2010;30:5295–5305. doi: 10.1128/MCB.00303-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesen M, Tanabe S, Berman MA, Yoshizawa I, Luo Y, Kim RJ, Post TW, Gerard C, Dorf ME. Mouse astrocytes respond to the chemokines MCP-1 and KC, but reverse transcriptase-polymerase chain reaction does not detect mRNA for the KC or new MCP-1 receptor. J Neurosci Res. 1996;45:382–391. doi: 10.1002/(SICI)1097-4547(19960815)45:4<382::AID-JNR7>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hosoda H, Tamura H, Kida S, Nagaoka I. Transcriptional regulation of mouse TREM-1 gene in RAW264.7 macrophage-like cells. Life Sci. 2011;89(3–4):115–122. doi: 10.1016/j.lfs.2011.05.007. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-κB potentiates amyloid β-mediated neuronal apoptosis. Proc Natl Acad Sci USA. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King PH. Cloning the 5′ flanking sequence of Hel-N1: Evidence for positive regulatory elements governing cell-specific transcription. Brain Res. 1996;723(1,2):141–147. doi: 10.1016/0006-8993(96)00044-3. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Pitha PM, Shin HS, Shin ML. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci USA. 1989;86:6348–6352. doi: 10.1073/pnas.86.16.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-J, Duffy A, Chen C-Y. Localization of AU-rich element-containing mRNA in cytoplasmic granules containing exosome subunits. J Biol Chem. 2007;282:19958–19968. doi: 10.1074/jbc.M702281200. [DOI] [PubMed] [Google Scholar]
- Lin WJ, Zheng X, Lin CC, Tsao J, Zhu X, Cody JJ, Coleman JM, Gherzi R, Luo M, Townes TM, et al. Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol Cell Biol. 2011;31:3196–207. doi: 10.1128/MCB.05073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- Liu J, He L, Collins I, Ge H, Libutti D, Li J, Egly J-M, Levens D. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5:331–341. doi: 10.1016/s1097-2765(00)80428-1. [DOI] [PubMed] [Google Scholar]
- Lu L, Wang S, Zheng L, Li X, Suswam EA, Zhang X, Wheeler CG, Nabors LB, Filippova N, King PH. Amyotrophic lateral sclerosis-linked mutant SOD1 sequesters Hu antigen R (HuR) and TIA-1 related protein (TIAR): Implications for impaired posttranscriptional regulation of vascular endothelial growth factor. J Biol Chem. 2009;284:33989–33998. doi: 10.1074/jbc.M109.067918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malz M, Weber A, Singer S, Riehmer V, Bissinger M, Riener MO, Longerich T, Soll C, Vogel A, Angel P, et al. Overexpression of far upstream element binding proteins: A mechanism regulating proliferation and migration in liver cancer cells. Hepatology. 2009;50:1130–1139. doi: 10.1002/hep.23051. [DOI] [PubMed] [Google Scholar]
- Ming W, Bersani L, Mantovani A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J Immunol. 1987;138:1469–1474. [PubMed] [Google Scholar]
- Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D’Mello SR. Neuroprotection by Histone Deacetylase-Related Protein. Mol Cell Biol. 2006;26:3550–3564. doi: 10.1128/MCB.26.9.3550-3564.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabors LB, Suswam E, Huang Y, Yang X, Johnson MJ, King PH. Tumor necrosis factor-α induces angiogenic factor up-regulation in malignant glioma cells: A role for RNA stabilization and HuR. Cancer Res. 2003;63:4181–4187. [PubMed] [Google Scholar]
- Park PH, Huang H, McMullen MR, Mandal P, Sun L, Nagy LE. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-alpha production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. J Biol Chem. 2008;283:26850–26858. doi: 10.1074/jbc.M802787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide-Biol Ch. 2010;23:75–93. doi: 10.1016/j.niox.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault-Mihara F, Okada S, Shibata S, Nakamura M, Toyama Y, Okano H. Spinal cord injury: Emerging beneficial role of reactive astrocytes’ migration. Int J Biochem Cell Biol. 2008;40:1649–1653. doi: 10.1016/j.biocel.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Rhoades KL, Golub SH, Economou JS. The regulation of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. J Biol Chem. 1992;267:22102–22107. [PubMed] [Google Scholar]
- Ruggiero T, Trabucchi M, Ponassi M, Corte G, Chen C-Y, al-Haj L, Khabar K, Briata P, Gherzi R. Identification of a set of KSRP target transcripts upregulated by PI3K-AKTsignaling. BMC Mol Biol. 2007;8:28. doi: 10.1186/1471-2199-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (MCP)-5: A novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185:99–109. doi: 10.1084/jem.185.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler GD, Cole MD. GM-CSF and oncogene mRNA stabilities are independently regulated in trans in a mouse monocytic tumor. Cell. 1988;55:1115–1122. doi: 10.1016/0092-8674(88)90256-5. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, O’Callaghan JP. Divergent roles for tumor necrosis factor-alpha in the brain. J Neuroimmune Pharmacol. 2007;2:140–153. doi: 10.1007/s11481-007-9070-6. [DOI] [PubMed] [Google Scholar]
- Tanabe S, Heesen M, Berman MA, Fischer MB, Yoshizawa I, Luo Y, Dorf ME. Murine astrocytes express a functional chemokine receptor. J Neurosci. 1997;17:6522–6528. doi: 10.1523/JNEUROSCI.17-17-06522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebo J, Der S, Frevel M, Khabar KSA, Williams BRG, Hamilton TA. Heterogeneity in control of mRNA stability by AU-rich elements. J Biol Chem. 2003;278:12085–12093. doi: 10.1074/jbc.M212992200. [DOI] [PubMed] [Google Scholar]
- Tezel G, Hernandez MR, Wax MB. In vitro evaluation of reactive astrocyte migration, a component of tissue remodeling in glaucomatous optic nerve head. Glia. 2001;34:178–189. doi: 10.1002/glia.1052. [DOI] [PubMed] [Google Scholar]
- Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108:1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Piaton G, Lubetzki C. Astrocytes—Friends or foes in multiple sclerosis? Glia. 2007;55:1300–1312. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- Winzen R, Thakur BK, Dittrich-Breiholz O, Shah M, Redich N, Dhamija S, Kracht M, Holtmann H. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol Cell Biol. 2007;27:8388–8400. doi: 10.1128/MCB.01493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen H-M, Jaramillo E, Wang L, D’Mello SR. Histone deacetylase-related protein inhibits AES-mediated neuronal cell death by direct interaction. J Neurosci Res. 2008;86:2423–2431. doi: 10.1002/jnr.21680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.