

Abstract
Topographically distinct, druggable, allosteric sites may be present on all G protein-coupled receptors (GPCRs). As such, targeting these sites with synthetic small molecules offers an attractive approach to develop receptor-subtype selective chemical leads for the development of novel therapies. A crucial part of drug development is to understand the acute and chronic effects of such allosteric modulators at their corresponding GPCR target. Key regulatory processes including cell-surface delivery, endocytosis, recycling, and down-regulation tightly control the number of receptors at the surface of the cell. As many GPCR therapeutics will be administered chronically, understanding how such ligands modulate these regulatory pathways forms an essential part of the characterization of novel GPCR ligands. This is true for both orthosteric and allosteric ligands. In this Review, we summarize our current understanding of GPCR regulatory processes with a particular focus on the effects and implications of allosteric targeting of GPCRs.
Keywords: G protein-coupled receptor, allosteric ligand, arrestin, endocytosis, functional selectivity
G protein-coupled receptors (GPCRs) represent the largest group of cell surface receptors encoded by the human genome (∼2%). By binding to a broad variety of ligands (ranging from small ions to amines or large peptides), GPCRs play the essential role of transmitting stimuli from the extracellular milieu and transforming them into specific cellular responses. The diverse physiological roles played by GPCRs, together with evidence for aberrant GPCR expression or signaling in various pathological conditions, emphasize the fundamental biological and clinical importance of this family of membrane proteins, and support their prominent position as targets in drug development programs. As such, GPCRs are currently the therapeutic target of more than 30% of marketed drugs.1 Classical approaches to GPCR drug discovery have focused upon developing small molecules that target the site at which endogenous hormones or neurotransmitters bind, the so-called “orthosteric” site. Such molecules can either mimic or inhibit the actions of these endogenous ligands. However, the attrition rate of modern drug discovery is higher than ever and the development of selective compounds as potential drug leads represents a significant challenge. One of the key issues in this regard is the fact that many GPCRs share high sequence homology within the orthosteric site across receptor subtypes. As a consequence, targeting this site alone is unlikely to yield highly subtype-selective lead compounds. Indeed, during the past decade, the idea of targeting topographically distinct “allosteric sites” as a novel approach to GPCR drug discovery has become a major topic in receptor pharmacology.2,3 The phenomenon of functional selectivity (also called “ligand-directed stimulus bias” or biased agonism) refers to the ability of different ligands that, despite acting via the same receptor and in the same cellular background, differentially activate certain subsets of intracellular signaling pathways to the relative exclusion of the others.4,5 As such, stimulus bias offers a new avenue for attaining “pathway-selective” rather than “receptor-selective” therapeutics. The characterization of the regulatory processes elicited by newly discovered GPCR ligands has often been “secondary” to the main aim of drug discovery and development programs.6 However, the fact that most of the therapies that target GPCRs are based on chronic exposure of the receptor to its ligand raises the important issue of understanding and investigating the long-term regulatory processes of this family of cell surface receptors. Such mechanisms of GPCR regulation also need to be considered for drugs that target allosteric sites on GPCRs. Furthermore, the concept of stimulus bias suggests that measurements of drug action at multiple signaling end points, including those related to receptor regulation, are required to gain a more complete description of ligand efficacy. In this Review, we summarize studies to date that have investigated the action of allosteric modulators upon receptor regulation. In addition, we discuss the implications that the paradigm of stimulus bias may have upon our interpretation of such studies.
GPCR Regulatory Processes and the Role of β-Arrestins
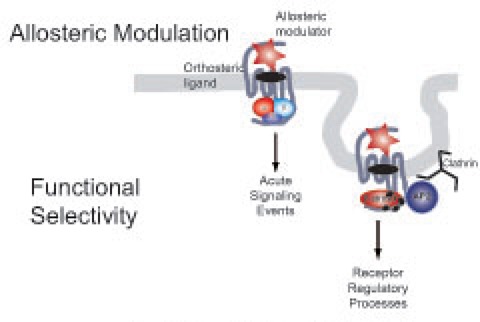
The activation mechanism of a GPCR upon ligand binding involves the transmission of a conformational change to the heterotrimeric G protein that promotes the release of GDP, its replacement by GTP and subsequent conformational rearrangements that result in the activation of several effectors (e.g., adenylate cyclases, Phospholipase C), and generation of second messengers (e.g., cAMP, Inositol phosphates). These conformational rearrangements have been confirmed by the recent solution of the high-resolution crystal structure of the ternary complex of the β2-adrenergic receptor and the stimulatory G protein, Gs.7 The intrinsic GTPase activity of Gα leads to hydrolysis of GTP to GDP, the reassociation of Gα-GDP and Gβγ subunits, and the termination of signaling.8 Upon activation, GPCRs rapidly undergo phosphorylation by GPCR kinases (GRKs) or second messenger activated kinases such as PKA or PKC (Figure 1). Selective phosphorylation of the agonist-activated receptor, and subsequent binding of β-arrestins, prevents the sustained interaction of GPCRs with G proteins, effectively terminating the G protein-mediated signal (receptor desensitization).9 The classical role proposed for arrestins is to act as scaffolds, binding to the coat structure of clathrin-coated pits (CCPs), thereby promoting endocytosis of arrestin bound receptors (receptor internalization). Eventually, internalized receptors can recycle back to the cell surface (receptor resensitization) or be targeted for lysosomal degradation (receptor down-regulation). However, an alternative role for β-arrestins has recently been demonstrated for a large group of GPCRs; β-arrestins are also able to recruit diverse signaling proteins to activated receptors at plasma and endosomal membranes and thus to be essential signaling mediators. For example, β-arrestins have been shown to scaffold the formation of multiprotein complexes with intracellular kinases such as the MAPKs ERK or JNK, AKT as well as with phosphatases such PP2A.10,11 It has therefore been proposed that β-arrestins mediate a second wave of GPCR signaling that is distinct from the “classical” G protein-dependent signaling at the plasma membrane (Figure 1).
Figure 1.

Schematic representation of GPCR regulation by β-arrestin mediated endocytosis. Phosphorylation of the activated receptor by GRKs triggers the recruitment of β-arrestin and the scaffolding of the endocytic machinery that results in receptor internalization. Once in endosomes, GPCRs can be dephosphorylated and recycled to the plasma membrane or, alternatively, be targeted for lysosomal degradation via multivesicular bodies (MVB).
Allosteric Targeting of GPCRs
In recent years it has become apparent that potentially all GPCRs contain topographically distinct, druggable, allosteric binding sites and the targeting of such sites presents an approach to achieve greater subtype selectivity. This fact, together with the increasing use of functional rather than ligand binding approaches in drug discovery programs to screen for small molecule leads, has led to an explosion in the identification of allosteric ligands. Allosteric ligands will bind to a receptor selecting a distinct receptor conformation and modulating both orthosteric ligand affinity and/or efficacy and are thus often referred to as allosteric modulators (Figure 2).12 Since allosteric modulators still allow the endogenous agonist to bind to the receptor, allosteric ligands have also been pursued for their ability to “fine tune” the physiological responses linked to a particular receptor, thus accommodating both temporal and spatial rhythms of normal signaling.12 Furthermore, their modulatory effect is limited, saturable, and therefore less prone to overdose. Currently, two GPCR allosteric modulators have achieved FDA approval; maraviroc that targets the chemokine receptor CCR5 for the treatment of HIV13 and cinacalcet that targets the calcium sensing receptor (CaSR) for the treatment of hyperthyroidism.14
Figure 2.
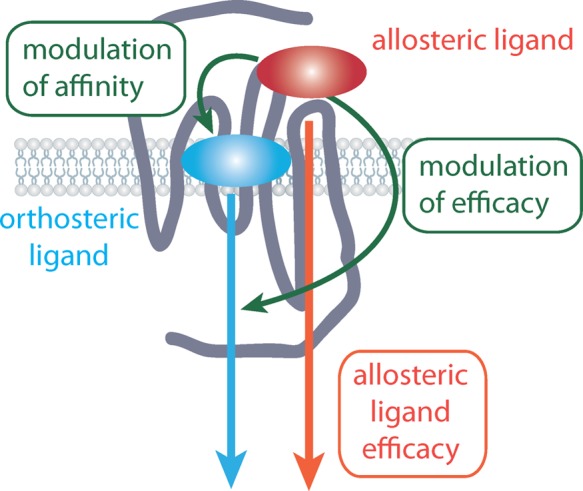
GPCR drug discovery has predominantly focused upon targeting the orthosteric site where the endogenous agonist binds. Allosteric modulators bind to a site on a GPCR that is topographically distinct from this orthosteric binding site. Allosteric modulators will bind to a receptor selecting a distinct receptor conformation but will still allow orthosteric ligands to bind. As such, allosteric ligands can modulate both orthosteric ligand affinity and efficacy. Of particular note, allosteric ligands can also possess their own intrinsic efficacy.
The concept of allosterism was formalized in 1965, by the seminal Monod–Wyman–Changeux (MWC) model, which proposed a conformational selection mechanism to account for ligand actions at bacterial regulatory enzymes.15 Since then, this model has been extended to other protein families. However, in the field of GPCRs, descriptions of allosteric mechanisms have remained largely theoretical or phenomenological. We have recently described an unprecedented example of a GPCR allosteric modulator whose action is entirely consistent with a two-state, MWC mechanism16 and proposed a pharmacological framework for the study and classification of allosteric modulators across different GPCR families. Deviations from the predicted MWC behavior may then suggest the existence of more complex (e.g multistate) or mixed (allosteric/orthosteric) modes of action. An important prediction of a two-state mechanism is that all allosteric ligands should display some agonism or inverse agonism in their own right (depending on their ability to select the active or inactive receptor states respectively).17 In practice, this is not always observed because the bioassay used cannot detect these effects due to signal threshold limitations. However, in an overexpressed receptor system, or an assay monitoring a sensitive or amplified response, low-level allosteric agonism or inverse agonism can be readily unmasked. Indeed, this has been shown to be the case for several GPCR allosteric modulators (Table 1).
Table 1. Examples to Date in Which the Effect of Allosteric Ligands upon Receptor Regulation (Internalization/β-Arrestin Recruitment) Have Been Studied.
| GPCR family | GPCR | allosteric ligand | modulator activitya | intrinisc activity | effect on regulation | ref |
|---|---|---|---|---|---|---|
| Family A | M1 mAChR | TBPB | novel M1 agonist | agonist | slow arrestin recruitment | (28) |
| no effect in internalization | ||||||
| AC260584 | novel M1 agonist | agonist | slow arrestin recruitment | (27, 28) | ||
| no effect in internalization | ||||||
| AC-42 | novel M1 agonist | agonist | promotes internalization and downregulation | (27) | ||
| OR no effect in internalization | (29) | |||||
| 77-LH-28-1 | novel M1 agonist | agonist | promotes internalization but not downregulation | (29) | ||
| M2 mAChR | Gallamine | negative | increase in cell surface exp. | (25) | ||
| Alcuronium | negative | increase in cell surface exp. | (25) | |||
| C7/3-phth | negative | inverse ago? | increase in cell surface exp. | (25) | ||
| M4 mAChR | LY2033298 | positive | agonist | promotes internalization | (24) | |
| A1 AdoR | PD81723 | positive | agonist | no effect in internalization | (60, 61) | |
| LH-R | Org42599 | NR | agonist | pharmacological chaperone: cell surface rescue | (62) | |
| CXCR3 | VUF10661 | NR | agonist | promotes internalization | (63) | |
| CXCR4 | RSVM peptide | NR | agonist | promotes internalization | (64) | |
| ASLW peptide | NR | agonist | no effect in internalization | |||
| CB1 | Org27569 | positive | agonist | promotes internalization | (31) | |
| G protein-independent ERK1/2 phosphorylation | ||||||
| Family B | GLP-1R | compound 2 | positive | agonist | promotes internalization | (32) |
| Family C | mGluR7 | AMN082 | NSA | agonist | promotes internalization | (34) |
| mGluR5 | CDPPB | positive | agonist | no effect in internalization in striatum | (35) | |
| promotes internalization in frontal cortex | ||||||
| CaSR | NPS R-586 | positive | agonist | increase of cell surface expression of wt CaR | (36) | |
| rescue of loss-of-function mutants |
In interactions with endogenous agonist except in Org27569 in which the orthosteric agonist was CP555940. NR: not reported. NSA: Novel selective agonist.
Allosteric Ligands Are Likely to Modulate GPCR Regulatory Processes
The expectation of all allosteric ligands to display efficacy in their own right means that not all allosteric modulators will conform to one of the suggested advantages of allosteric targeting of GPCRs: The effect of an allosteric modulator will only be observed in the presence of the orthosteric endogenous ligand and therefore will allow for the aforementioned temporal and spatial control of signal modulation. Furthermore, it also emphasizes the fact that, having efficacy on their own, allosteric modulators can not only modulate receptor activation but also potentially elicit receptor regulatory processes (Figure 3). Allosteric modulators could potentially induce receptor desensitization/internalization or conversely increase cell surface expression, act as chaperones of receptor cell surface delivery, or prevent the internalization induced by the endogenous/orthosteric ligand. Indeed, examples of all such scenarios have been described in literature (Table 1).
Figure 3.

Both orthosteric and allosteric GPCR ligands acting at the same receptor can engage acute signaling and regulatory pathways by interacting with distinct effector proteins (Eff. A-C) and regulatory proteins such as β-arrestin (β-arr). Both orthosteric and allosteric ligands may select different subsets of these signaling and regulatory pathways by stabilizing distinct receptor conformations, a phenomenon termed functional selectivity or stimulus bias. The subset of these pathways and processes engaged by a ligand–receptor complex will underlie the physiological effect of the ligand.
If we look outside the GPCR family, there is an example where the effect of an allosteric modulator upon receptor regulation may have clinical relevance: the case of the GABAA receptors (GABAARs). GABAARs are multisubunit ion channels allosterically modulated by benzodiazepines, widely used allosteric modulators in clinical practice for their sedative, anxiolytic, anticonvulsant, and muscle relaxant actions.18 Benzodiazepines bind to a high affinity binding site located at the α/γ subunit interface but do not open the channel by themselves. Binding to their recognition site leads to a conformational change that that results in an increase of the apparent affinity of the channel for the neurotransmitter GABA.19
Use-dependence and tolerance observed after long-term administration of benzodiazepines are commonly linked to the modulation of the expression of GABAAR binding sites at the cell surface. Such modulation has been shown to be brain-region-specific.20 Moreover, the prolonged effect of benzodiazepines in receptor regulation has also been shown to be subunit dependent.21 In vivo studies on long-term benzodiazepine treatments reveal downregulation of α1 and β3 subunits in hippocampal and cortical brain regions and γ2 subunits in the cerebral cortex. In contrast, benzodiazepine withdrawal causes upregulation of α4 and γ4 subunits.22 More recently, Jacob and co-workers observed that 24 h treatment of hippocampal neurons with flurazepam dramatically decreased α2 subunit-containing GABAAR surface and total levels without comparable changes in levels of the α1 subunit. They suggest that flurazepam exposure enhances degradation of α2 subtype GABAARs after endocytosis, leading to a reduction in inhibitory synapse size and number along with a decrease in the efficacy of synaptic inhibition.23
Despite the ever-increasing number of allosteric ligands targeting GPCRs, a surprisingly small number of studies have investigated the effects of such ligands upon receptor regulation. However, examples exist at all three major receptor family classes: Family A (e.g., muscarinic, adenosine, and luteinizing hormone -LH- receptors), Family B (e.g., glucagon-like peptide-1 -GLP-1- receptors), and Family C (e.g., mGluR5, mGluR7, and CaSR). Muscarinic acetylcholine receptors (mAChRs) are perhaps the most studied Family A GPCRs in terms of targeting by small molecule allosteric modulators. Many of the properties of allosteric modulators have been investigated using the mAChR family as a model GPCR system, and several studies have reported the effects of allosteric modulators on mAChR regulation. In agreement with its allosteric agonism, the M4 mAChR modulator LY2033298 has been shown to induce receptor internalization.24 In contrast, at the M2 mAChR, gallamine, alcuronium, and C7/3-phth, negative allosteric modulators of the endogenous agonist acetylcholine- enhanced cell surface receptor expression.25,26 More recently, several studies have focused in the regulation of the M1 mAChR by putative allosteric agonists (AC-42, TBPB) and obtained contradictory results.27−29 However, it should be noted that it is still unclear if these ligands represent “pure” allosteric modulators or, instead, represent a different and potentially more complex bitopic (i.e., dual allosteric/orthosteric) binding mode, hence their classification as novel M1mAChR agonists. Interestingly, some studies have also focused on the ability of these ligands to promote β-arrestin recruitment to the receptor. In particular, Ma et al. reported that BQCA (a positive allosteric modulator of M1 mAChR) is able to induce β-arrestin recruitment with significantly higher potencies than TBPB or AC-42 (novel M1 mAChR agonists).30 It is also worth noting that from all these studies, only one of them investigated the modulatory effect on orthosteric ligand-induced internalization, showing that LY2033298 behaves as an allosteric potentiator when internalization is interrogated in an interaction paradigm as an additional functional end point.24 More recently, another interesting example of an allosteric ligand of a Family A receptor that mediates receptor internalization is Org27569 at the cannabinoid CB1 receptor. Ahn et al.31 described the ability of Org27569 not only to mediate receptor internalization but also to engender Gαi/o G protein-independent ERK1/2 phosphorylation.
Receptor regulation by allosteric ligands has also been reported for the prototypical Family B receptor, the GLP-1R. In particular, the small molecule allosteric agonist of GLP-1R, Compound 2, induces receptor internalization in a manner similar to the endogenous peptide, though with slower kinetics.32 Interestingly, β-arrestin-1 mediates GLP-1R signaling to insulin secretion in cultured pancreatic β cells but does not play a role in GLP-1R desensitization/internalization.33 However, the ability of Compound 2 to recruit arrestins to the GLP-1R and/or exert modulatory effects upon the physiological outcomes related to this recruitment still remains to be addressed.
Family C receptors present the most striking separation between defined orthosteric and allosteric sites, located in the extracellular Venus Fly Trap domain and the transmembrane bundle, respectively. Multiple allosteric modulators have been described for Family C receptors, with the mGluRs and CaSR being prototypical examples. For instance, AMN082, an allosteric agonist of mGluR7, was shown to internalize the receptor in hippocampal neurons.34 mGluRs allosteric modulators have also provided interesting examples on differential regulation depending on regions of receptor expression. For example, the effect of prolonged treatment to the mGluR5 positive allosteric modulator, CDPPB, is dependent on the brain region, by which mGluR5 expressed in the cortex is more susceptible to desensitization/internalization than those expressed in the striatum.35
It is also worth noting the possibility of allosteric modulators to act as pharmacological chaperones for cell surface delivery of GPCRs. An example of such a scenario is the CaSR. Loss- or gain-of-function mutations identified in different pathological conditions (familial hypocalciuric hypercalcemia (FHH) and neonatal severe hyperparathyroidism (NSHPT) or autosomal dominant hypocalcemia (ADH), respectively) suggest that signaling changes may result from differences in cell surface expression. Allosteric modulators of CaSR such as the calcimimetic NPS R-568 have been shown to differentially regulate the function of plasma membrane-localized CaSR by regulating receptor turnover.36 NPS R-568, by favoring active conformations, reduces WT CaSR or loss-of-function CaSR mutant ubiquitination, and degradation, hence increasing the levels of functional cell surface CaSR.
The therapeutic relevance of the role of allosteric modulators in GPCR regulation still needs further investigation. One possible research venue is the modulatory effects derived from arrestin recruitment to the receptor and triggering or modulation of arrestin-mediated signals. Concomitantly, an alternative application would rely in the so-called “functional-antagonism”, whereby internalization of receptor would inhibit its function by removing it from the cell surface. This latter mechanism has been suggested to be of value in protecting cells from HIV infection in the presence of chemokines and it has been demonstrated that allosteric targeting of CCR5 affecting its internalization properties offers advantageous characteristics as opposed to competitive antagonists.37
Functional Selectivity and Receptor Regulation: Implications for the Characterization of Allosteric Ligands
The first unequivocal evidence of functional selectivity was the observation that ligands acting at the same receptor can exhibit reversals in the rank order of potency from one pathway to another.38 Such inversion in the rank order of potency cannot be explained by differences in the amplification of the signaling pathways or the detection systems used.5 It follows that these data cannot be easily reconciled with the two-state model classically used to explain agonism, antagonism and inverse agonism, suggesting instead that different ligands can promote distinct receptor active states with preferences toward different signaling pathways. To date, there are numerous examples of receptors for which functionally selective ligands have been described, and it is widely accepted that receptors can exist in multiple conformational states, each eliciting a particular subset of signals. Although many examples of ligand-biased signaling result in relatively modest differences in agonism between individual pathways, some compounds show more extreme pathway bias, characterized by selective agonism toward one pathway and a lack of activity toward another. Many of the best characterized examples of such “extreme” bias refer to drugs with differential activities in G protein-mediated signals versus G protein-independent pathways that often involve the scaffolding protein β-arrestin.6
Functional selectivity has also been described at the level of receptor endocytosis (Figure 3). Indeed, the existence of noninternalizing and internalizing ligands has been reported for several GPCRs, and the mechanisms behind such behaviors are starting to be unraveled. As mentioned previously, agonist occupation of GPCRs results in rapid receptor phosphorylation at sites largely within the third intracellular loop and C-terminal tail. This process not only mediates the uncoupling of the receptor from its cognate G protein but also drives G protein-independent receptor signaling. It is now clear that GPCR phosphorylation is a complex regulatory mechanism that involves mainly, but not only, members of the GPCR kinase family (GRKs). The consequence of this phosphorylation is the recruitment of arrestins that mediate both G protein-independent signaling as well as receptor internalization.9 In line with the idea that GPCRs can adopt multiple conformations that result in differential engagement of signaling proteins, it can be assumed that the phosphorylation patterns adopted by a receptor will be a reflection not only of the complement of kinases and phosphatases expressed in a given cell or tissue but also of the receptor conformation following agonist occupation.39 Several examples have been recently described that show that biased orthosteric ligands can direct receptor signaling by driving receptor phosphorylation profiles, providing “barcodes” that encode for a particular signaling outcome; for example muscarinic M3 mAChR, β2-adrenergic receptor, and the chemokine receptors CXCR4 or CCR7.40−43
Pioneering work with the β2-adrenergic and angiotensin AT1 receptors demonstrated that recruitment of β-arrestins to phosphorylated receptors can be the initial step for G protein-independent signals as they selectively scaffold intracellular kinases and other signaling effectors.44,45 Interestingly, β-arrestins also recruit signaling proteins in the endosomal membranes, raising the possibility for receptors that are essentially regarded as cell surface proteins, such as GPCRs, to elicit intracellular signals, although this is just starting to be understood.11
In the past decade, numerous ligands that differentially bias receptor signals toward G protein-dependent or -independent pathways have been described, although the physiological relevance of such bias has only been established for a few of them. β-Arrestin-biased signaling has been proposed as a potential therapeutic mechanism for drugs targeting the orthosteric site of β2-adrenergic, AT1, PTH, muscarinic M3, GLP-1, dopamine D2, or the nicotinic acid receptors33,46−52 (or ref (6) for review). However, it is worth noting that biased agonism may not always result in a more positive therapeutic profile. It is equally possible that biased agonism may underlie unwanted side effects, although the evidence for this is still very limited.
When receptor internalization is considered as an “end point”, stimulus bias can then be extended to ligands that differentially regulate a given GPCR. The most prominent example of this scenario is the opioid receptors. Different from most of the synthetic and endogenous ligands, the widely used analgesic morphine shows compromised ability to internalize the μ-opioid receptor, and this has been proposed to account for the development of tolerance to this drug.53−55 Recent studies suggest receptor phosphorylation as well as differential recruitment of β-arrestin1 versus β-arrestin2 as a potential mechanism to explain the different regulatory events induced by the different opiates.56 Stimulus bias for receptor regulation has also recently been shown for the somatostatin SST2A receptor. Two somatostatin analogues currently in clinical investigation (SOM230 and KE108) have been shown to display functional selectivity, not only in terms of acute signaling but also in terms of receptor phosphorylation, endocytosis, and recycling.57 Functional selectivity in terms of differential receptor internalization has also been demonstrated at the cannabinoid receptor CB2. Despite both orthosteric ligands promoting ERK phosphorylation as well as β-arrestin recruitment, CP55940 was found to internalize the CB2 receptor while the aminoalkylindole WIN55212–2 failed to do so.58
Given that the binding of an allosteric modulator to a receptor stabilizes a distinct receptor conformation, it follows that such allosteric modulators may themselves display pathway bias both in terms of their own intrinsic efficacy or their modulation of orthosteric ligand effects.59 Therefore, it cannot be assumed that if an allosteric modulator is quiescent in the absence of an orthosteric agonist in terms of one signaling pathway it will also be quiescent in terms of receptor regulation (Figure 3). Moreover, it will not necessarily follow that if a modulator is an enhancer of an acute signaling pathway it will display similar potentiation of receptor regulatory events. Thus, it follows that effects upon receptor regulation should be a key focus in studies to characterize novel and existent allosteric modulators in addition to standard readouts of acute receptor signaling events.
Concluding Remarks
Recent research has given us insight into the complexities of GPCR regulation and trafficking. These processes are likely to be both receptor-dependent and in many cases ligand-dependent. Given that many of these GPCR ligands will be administered chronically, an essential part of understanding GPCR ligand efficacy is to understand their ability to modulate these receptor regulatory processes. This in turn will be central to the ability to understand and predict their clinical efficacy. It has now become clear that the development of allosteric modulators offers an attractive approach to achieve selective targeting of GPCRs. These allosteric ligands, in addition to modulating orthosteric ligand effects, may possess efficacy in their own right. As such, it is essential to understand how such ligands modulate GPCR regulatory processes.
Glossary
Abbreviations
- CCP
clathrin-coated pit
- GPCR
G protein-coupled receptor
- HIV
human immunodeficiency virus
Author Contributions
† Authors J.R.L. and M.C. contributed equally to this work.
Author Contributions
J.R.L. and M.C. wrote the manuscript. A.A. wrote specific parts of the manuscript.
Work in the authors’ laboratory is supported by the National Health and Medical Research Council (NHMRC) of Australia Project Grants APP1011796 and APP1011920. M.C. is a Monash Fellow, and J.R.L. is a Monash University Larkins Fellow and NHMRC Career Development Awardee. J.R.L. acknowledges the financial support of The Netherlands Organization for Scientific Research [NWO VENI Grant 863.09.018].
The authors declare no competing financial interest.
References
- Rask-Andersen M.; Almén M. S.; Schiöth H. B. (2011) Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery 10, 579–590. [DOI] [PubMed] [Google Scholar]
- Christopoulos A. (2002) Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discovery 1, 198–210. [DOI] [PubMed] [Google Scholar]
- Conn P.; Christopoulos A.; Lindsley C. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T.; Miller L. J. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol. Rev. 62, 265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallaert W.; Christopoulos A.; Bouvier M. (2011) Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin. Drug Discovery 6, 811–825. [DOI] [PubMed] [Google Scholar]
- Whalen E. J.; Rajagopal S.; Lefkowitz R. J. (2011) Therapeutic potential of β-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S. G. F.; Devree B. T.; Zou Y.; Kruse A. C.; Chung K. Y.; Kobilka T. S.; Thian F. S.; Chae P. S.; Pardon E.; Calinski D.; Mathiesen J. M.; Shah S. T. A.; Lyons J. A.; Caffrey M.; Gellman S. H.; Steyaert J.; Skiniotis G.; Weis W. I.; Sunahara R. K.; Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Vera T. M.; Vanhauwe J.; Thomas T. O.; Medkova M.; Preininger A.; Mazzoni M. R.; Hamm H. E. (2003) Insights into G protein structure, function, and regulation. Endocr. Rev. 24, 765–781. [DOI] [PubMed] [Google Scholar]
- Lefkowitz R. J.; Whalen E. J. (2004) beta-arrestins: traffic cops of cell signaling. Curr. Opin. Cell Biol. 16, 162–168. [DOI] [PubMed] [Google Scholar]
- DeWire S. M.; Ahn S.; Lefkowitz R. J.; Shenoy S. K. (2007) β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 69, 483–510. [DOI] [PubMed] [Google Scholar]
- Murphy J. E.; Padilla B. E.; Hasdemir B.; Cottrell G. S.; Bunnett N. W. (2009) Endosomes: a legitimate platform for the signaling train. Proc. Natl. Acad. Sci. U.S.A. 106, 17615–17622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May L.; Leach K.; Sexton P.; Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51. [DOI] [PubMed] [Google Scholar]
- Dorr P.; Westby M.; Dobbs S.; Griffin P.; Irvine B.; Macartney M.; Mori J.; Rickett G.; Smith-Burchnell C.; Napier C.; Webster R.; Armour D.; Price D.; Stammen B.; Wood A.; Perros M. (2005) Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49, 4721–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg J. S. (2005) Cinacalcet HCl, an Oral Calcimimetic Agent for the Treatment of Secondary Hyperparathyroidism in Hemodialysis and Peritoneal Dialysis: A Randomized, Double-Blind, Multicenter Study. J. Am. Soc. Nephrol. 16, 800–807. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J.-P. (1965) On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 88–118. [DOI] [PubMed] [Google Scholar]
- Canals M.; Lane J. R.; Wen A.; Scammells P. J.; Sexton P. M.; Christopoulos A. (2012) A Monod-Wyman-Changeux Mechanism Can Explain G Protein-coupled Receptor (GPCR) Allosteric Modulation. J. Biol. Chem. 287, 650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M.; Sexton P. M.; Christopoulos A. (2011) Allostery in GPCRs: “MWC” revisited. Trends Biochem. Sci. 36, 663–672. [DOI] [PubMed] [Google Scholar]
- Tallman J. F.; Thomas J. W.; Gallager D. W. (1978) GABAergic modulation of benzodiazepine binding site sensitivity. Nature 274, 383–385. [DOI] [PubMed] [Google Scholar]
- Sigel E.; Buhr A. (1997) The benzodiazepine binding site of GABAA receptors. Trends Pharmacol. Sci. 18, 425–429. [DOI] [PubMed] [Google Scholar]
- Jacob T. C.; Moss S. J.; Jurd R. (2008) GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M.; Bettler B. (2012) Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat. Rev. Neurosci. 13, 380–394. [DOI] [PubMed] [Google Scholar]
- Uusi-Oukari M.; Korpi E. R. (2010) Regulation of GABA(A) receptor subunit expression by pharmacological agents. Pharmacol. Rev. 62, 97–135. [DOI] [PubMed] [Google Scholar]
- Jacob T. C.; Michels G.; Silayeva L.; Haydon J.; Succol F.; Moss S. J. (2012) Benzodiazepine treatment induces subtype-specific changes in GABAA receptor trafficking and decreases synaptic inhibition. Proc. Natl. Acad. Sci. U.S.A. 109, 18595–18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K.; Loiacono R.; Felder C.; McKinzie D.; Mogg A.; Shaw D.; Sexton P.; Christopoulos A. (2009) Molecular Mechanisms of Action and In Vivo Validation of an M4Muscarinic Acetylcholine Receptor Allosteric Modulator with Potential Antipsychotic Properties. Neuropsychopharmacology 35, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May L. T.; Lin Y.; Sexton P. M.; Christopoulos A. (2005) Regulation of M2 muscarinic acetylcholine receptor expression and signaling by prolonged exposure to allosteric modulators. J. Pharmacol. Exp. Ther. 312, 382–390. [DOI] [PubMed] [Google Scholar]
- Avlani V. A.; Gregory K. J.; Morton C. J.; Parker M. W.; Sexton P. M.; Christopoulos A. (2007) Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 282, 25677–25686. [DOI] [PubMed] [Google Scholar]
- Davis C.; Bradley S.; Schiffer H.; Friberg M.; Koch K.; Tolf B.-R.; Bonhaus D.; Lameh J. (2009) Differential regulation of muscarinic M1 receptors by orthosteric and allosteric ligands. BMC Pharmacol. 9, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A. A.; Heilman C. J.; Brady A. E.; Miller N. R.; Fuerstenau-Sharp M.; Hanson B. J.; Lindsley C. W.; Conn P. J.; Lah J. J.; Levey A. I. (2010) Differential effects of allosteric M(1) muscarinic acetylcholine receptor agonists on receptor activation, arrestin 3 recruitment, and receptor downregulation. ACS. Chem. Neurosci. 1, 542–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. L.; Langmead C. J.; Wood M. D.; Challiss R. A. J. (2009) Contrasting effects of allosteric and orthosteric agonists on m1 muscarinic acetylcholine receptor internalization and down-regulation. J. Pharmacol. Exp. Ther. 331, 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Seager M. A.; Seager M.; Wittmann M.; Jacobson M.; Bickel D.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; Pearson M.; McCampbell A.; Gaspar R.; Shughrue P.; Danziger A.; Regan C.; Flick R.; Pascarella D.; Garson S.; Doran S.; Kreatsoulas C.; Veng L.; Lindsley C. W.; Shipe W.; Kuduk S.; Sur C.; Kinney G.; Seabrook G. R.; Ray W. J. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H.; Mahmoud M. M.; Kendall D. A. (2012) Allosteric modulator ORG27569 induces CB1 Cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi-independent ERK1/2 kinase activation. J. Biol. Chem. 287, 12070–12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman K.; Huang Y.; Johnston N.; Bradley S. J.; Wilkinson G. F.; Willars G. B. (2010) Comparative effects of the endogenous agonist glucagon-like peptide-1 (GLP-1)-(7–36) amide and the small-molecule ago-allosteric agent “compound 2” at the GLP-1 receptor. J. Pharmacol. Exp. Ther. 334, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda N.; Imamura T.; Yoshizaki T.; Babendure J.; Lu J.-C.; Olefsky J. (2008) Î2-Arrestin-1 mediates glucagon-like peptide-1 signaling to insulin secretion in cultured pancreatic Î2 cells. Proc. Natl. Acad. Sci. U.S.A. 105, 6614–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey K. A.; Yuan X.; Lavezzari G.; Roche K. W.; McBain C. J. (2007) mGluR7 undergoes rapid internalization in response to activation by the allosteric agonist AMN082. Neuropharmacology 52, 108–117. [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S.; Obrien J. A.; Doran S.; Nguyen S. J.; Flick R. B.; Uslaner J. M.; Chen H.; Finger E. N.; Williams T. M.; Jacobson M. A.; Hutson P. H. (2012) Differential effects of the mGluR5 positive allosteric modulator CDPPB in the cortex and striatum following repeated administration. Neuropharmacology 62, 1453–1460. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Breitwieser G. E. (2007) Rescue of calcium-sensing receptor mutants by allosteric modulators reveals a conformational checkpoint in receptor biogenesis. J. Biol. Chem. 282, 9517–9525. [DOI] [PubMed] [Google Scholar]
- Muniz-Medina V. M.; Jones S.; Maglich J. M.; Galardi C.; Hollingsworth R. E.; Kazmierski W. M.; Ferris R. G.; Edelstein M. P.; Chiswell K. E.; Kenakin T. P. (2009) The relative activity of “function sparing” HIV-1 entry inhibitors on viral entry and CCR5 internalization: is allosteric functional selectivity a valuable therapeutic property?. Mol. Pharmacol. 75, 490–501. [DOI] [PubMed] [Google Scholar]
- Spengler D.; Waeber C.; Pantaloni C.; Holsboer F.; Bockaert J.; Seeburg P. H.; Journot L. (1993) Differential signal transduction by five splice variants of the PACAP receptor. Nature 365, 170–175. [DOI] [PubMed] [Google Scholar]
- Tobin A. B.; Butcher A. J.; Kong K. C. (2008) Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol. Sci. 29, 413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher A. J.; Prihandoko R.; Kong K. C.; McWilliams P.; Edwards J. M.; Bottrill A.; Mistry S.; Tobin A. B. (2011) Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 286, 11506–11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles K. N.; Xiao K.; Ahn S.; Shukla A. K.; Lam C. M.; Rajagopal S.; Strachan R. T.; Huang T.-Y.; Bressler E. A.; Hara M. R.; Shenoy S. K.; Gygi S. P.; Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signaling 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busillo J.; Armando S.; Sengupta R.; Meucci O.; Bouvier M.; Benovic J. (2010) Site-specific Phosphorylation of CXCR4 Is Dynamically Regulated by Multiple Kinases and Results in Differential Modulation of CXCR4 Signaling. J. Biol. Chem. 285, 7805–7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidar D. A.; Violin J. D.; Whalen E. J.; Lefkowitz R. J. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U.S.A. 106, 9649–9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.; Maudsley S.; Luttrell L. M.; Lefkowitz R. J.; Daaka Y. (1999) Src-mediated tyrosine phosphorylation of dynamin is required for beta2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J. Biol. Chem. 274, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Tohgo A.; Pierce K. L.; Choy E. W.; Lefkowitz R. J.; Luttrell L. M. (2002) beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436. [DOI] [PubMed] [Google Scholar]
- Wisler J. W.; DeWire S. M.; Whalen E. J.; Violin J. D.; Drake M. T.; Ahn S.; Shenoy S. K.; Lefkowitz R. J. (2007) A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Ahn S.; Shenoy S. K.; Karnik S. S.; Hunyady L.; Luttrell L. M.; Lefkowitz R. J. (2003) Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty-Palmer D.; Flannery P.; Yuan L.; Corsino L.; Spurney R.; Lefkowitz R. J.; Luttrell L. M. (2009) A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl. Med. 1, 1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K. C.; Butcher A. J.; McWilliams P.; Jones D.; Wess J.; Hamdan F. F.; Werry T.; Rosethorne E. M.; Charlton S. J.; Munson S. E.; Cragg H. A.; Smart A. D.; Tobin A. B. (2010) M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proc. Natl. Acad. Sci. U.S.A. 107, 21181–21186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quoyer J.; Longuet C.; Broca C.; Linck N.; Costes S.; Varin E.; Bockaert J.; Bertrand G.; Dalle S. (2010) GLP-1 mediates antiapoptotic effect by phosphorylating Bad through a beta-arrestin 1-mediated ERK1/2 activation in pancreatic beta-cells. J. Biol. Chem. 285, 1989–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B.; Salahpour A.; Didriksen M.; Ghisi V.; Beaulieu J.-M.; Gainetdinov R. R.; Caron M. G. (2008) Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U.S.A. 105, 13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters R. W.; Shukla A. K.; Kovacs J. J.; Violin J. D.; DeWire S. M.; Lam C. M.; Chen J. R.; Muehlbauer M. J.; Whalen E. J.; Lefkowitz R. J. (2009) beta-Arrestin1 mediates nicotinic acid-induced flushing, but not its antilipolytic effect, in mice. J. Clin. Invest. 119, 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn L. M.; Lefkowitz R. J.; Caron M. G. (2002) Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J. Neurosci. 22, 10494–10500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang V. C.; Chieng B.; Azriel Y.; Christie M. J. (2011) Cellular Morphine Tolerance Produced by Arrestin-2-Dependent Impairment of -Opioid Receptor Resensitization. J. Neurosci. 31, 7122–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal K. M.; Schmid C. L.; Groer C. E.; Bohn L. M. (2011) Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63, 1001–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groer C. E.; Schmid C. L.; Jaeger A. M.; Bohn L. M. (2011) Agonist-directed interactions with specific arrestins determine μ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 286, 31731–31741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao Y. J.; Ghosh M.; Schonbrunn A. (2011) Ligand-dependent mechanisms of sst2A receptor trafficking: role of site-specific phosphorylation and receptor activation in the actions of biased somatostatin agonists. Mol. Endocrinol. 25, 1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood B. K.; Wager-Miller J.; Haskins C.; Straiker A.; Mackie K. (2012) Functional selectivity in CB(2) cannabinoid receptor signaling and regulation: implications for the therapeutic potential of CB(2) ligands. Mol. Pharmacol. 81, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach K.; Sexton P.; Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol. Sci. 28, 382–389. [DOI] [PubMed] [Google Scholar]
- Klaasse E.; den Hout, van G.; Roerink S.; de Grip W.; Ijzerman A.; Beukers M. (2005) Allosteric modulators affect the internalization of human adenosine A1 receptors. Eur. J. Pharmacol. 522, 1–8. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Linden J. (1996) Effects of long-term treatment with the allosteric enhancer, PD81,723, on Chinese hamster ovary cells expressing recombinant human A1 adenosine receptors. Mol. Pharmacol. 50, 104–111. [PubMed] [Google Scholar]
- Newton C. L.; Whay A. M.; McArdle C. A.; Zhang M.; van Koppen C. J.; van de Lagemaat R.; Segaloff D. L.; Millar R. P. (2011) Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. U.S.A. 108, 7172–7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholten D. J.; Canals M.; Wijtmans M.; de Munnik S.; Nguyen P.; Verzijl D.; de Esch I. J. P.; Vischer H. F.; Smit M. J.; Leurs R. (2012) Pharmacological characterization of a small-molecule agonist for the chemokine receptor CXCR3. Br. J. Pharmacol. 166, 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachpatzidis A.; Benton B. K.; Manfredi J. P.; Wang H.; Hamilton A.; Dohlman H. G.; Lolis E. (2003) Identification of allosteric peptide agonists of CXCR4. J. Biol. Chem. 278, 896–907. [DOI] [PubMed] [Google Scholar]