

Abstract
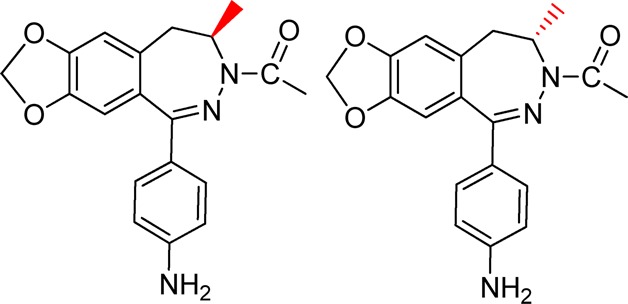
Stereoselectivity of 2,3-benzodiazepine compounds provides a unique way for the design of stereoisomers as more selective and more potent inhibitors as drug candidates for treatment of the neurological diseases involving excessive activity of AMPA receptors. Here we investigate a pair of enantiomers known as Talampanel and its (+) counterpart about their mechanism of inhibition and selectivity toward four AMPA receptor subunits or GluA1–4. We show that Talampanel is the eutomer with the endismic ratio being 14 for the closed-channel and 10 for the open-channel state of GluA2. Kinetic evidence supports that Talampanel is a noncompetitive inhibitor and it binds to the same site for those 2,3-benzodiazepine compounds with the C-4 methyl group on the diazepine ring. This site, which we term as the “M” site, recognizes preferentially those 2,3-benzodiazepine compounds with the C-4 methyl group being in the R configuration, as in the chemical structure of Talampanel. Given that Talampanel inhibits GluA1 and GluA2, but is virtually ineffective on the GluA3 and GluA4 AMPA receptor subunits, we hypothesize that the “M” site(s) on GluA1 and GluA2 to which Talampanel binds is different from that on GluA3 and GluA4. If the molecular properties of the AMPA receptors and Talampanel are used for selecting an inhibitor as a single drug candidate for controlling the activity of all AMPA receptors in vivo, Talampanel is not ideal. Our results further suggest that addition of longer acyl groups to the N-3 position should produce more potent 2,3-benzodiazepine inhibitors for the “M” site.
Keywords: AMPA receptors; stereoselectivity; 2,3-benzodiazepine compounds; Talampanel; structure−activity relationship
Developing selective agents that are capable of differentiating between subunits or subtypes of a protein or receptor, by either affinity or activity, is important for improving our understanding of the functional role of each subunit and subtype and for enhancing our ability to control the target function with minimal or no side effect. If the target is involved in a disease, these selective agents are generally better candidates for drug discovery. For developing selective agents, the chemical structure of a prototypic template may yield useful clues. For instance, if an inhibitor is a small compound and contains a stereogenic center in its chemical structure, such a chiral molecule can be exploited for synthesis of two enantiomers that are mirror images to each other. More often, the two enantiomers exhibit significant difference in their target selectivity and potency, in addition to difference in bioavailability, rate of metabolism, excretion and toxicity.1,2 Therefore, a mechanistic characterization of the two enantiomers provides useful insights into developing more selective and more potent agents. Here we describe a mechanistic study of the enantiomers of the C-4-methyl 2,3-benzodiazepine derivatives, as part of our investigation of the structure–activity relationship of these compounds (Figure 1). 2,3-Benzodiazepine derivatives, also known as GYKI compounds, are a group of compounds synthesized to be potential inhibitors of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors.
Figure 1.

Chemical structures of BDZ-d (Talampanel, GYKI 53773, (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-acetyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine), BDZ-e (GYKI 53774, (S)-(+)-1-(4-aminophenyl)-3,4-dihydro-3-N-acetyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine), BDZ-f (GYKI 53784, (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-methylcarbamyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine), and the prototypic compound GYKI 52466 (1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine).
AMPA receptors are one of the three subtypes of the glutamate ion channel receptor family, and they mediate fast synaptic neurotransmission in the central nervous system.3,4 AMPA receptors are indispensable for brain activities, such as memory and learning.3,4 Excessive channel activity, however, causes abnormal calcium influx, thereby leading to neuronal injury and cell death. AMPA receptor-mediated cell injury is thought to underlie a number of neurological diseases such as epilepsy, amyotrophic lateral sclerosis (ALS), and Parkinson’s disease.5−9 Inhibitors of AMPA receptors are thus potential drugs for a treatment of these neurological diseases. Among the inhibitors developed to date, 2,3-benzodiazepine derivatives are promising drug candidates, because some have shown potent anticonvulsive and neuroprotective effects in cellular and animal models.10−12 The prototypic compound in this family, GYKI 52466 (1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine, see Figure 1 for structure), was discovered in the 1980s and has been used as a template for the synthesis and the activity evaluation of hundreds of new compounds.10−12
The C-4 methyl group in the structure of GYKI 52466 (Figure 1) is important in several ways. First, removing the C-4 methyl group (i.e., replacing it with a hydrogen atom) or substituting it with a longer alkyl group reduces the biological activity, as seen in the inhibition of field potential in hippocampal slices and maximal electroshock seizures in mice.11 Second, although the C-4 methyl group is important in activity, it can be replaced, for example, with a carbonyl group.13,14 This replacement is in fact the change of the azomethine moiety with a ε-lactam moiety. Such a replacement retains biological activities, such as anticonvulsant properties, but changes the binding site.13,14 In other words, 2,3-benzodiazepine compounds with a C-4 carbonyl moiety bind to one site on the receptor, while those with a C-4 methyl group bind to a different site, as shown in our previous studies.15,16
The importance of the C-4 methyl group is further reflected by its stereochemistry, as the C-4 methyl group can adopt two stereoisomeric configurations, provided that the 3,4-double bond is saturated (see the chemical structure of GYKI 52466 in Figure 1). However, the 3,4-dihydro analogue of GYKI 52466, known as GYKI 52895, turns out to be a selective dopamine uptake inhibitor.17 In contrast, acylation at the N-3 position restores AMPA receptor antagonism. Consequently, a large number of acylated dihydro-2,3-benzodiazepines compounds have been synthesized.10,11 Each of these compounds correlates to a pair of enantiomers due to the C-4 stereogenic center. These enantiomers tend to have different biological activities. For example, two (−) stereoisomers, that is, (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-acetyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine (Talampanel or GYKI 53773) and (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-methylcarbamyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine, protect AMPA-receptor mediated cell death in a primary hippocampal culture18 and inhibit AMPA receptors in various cells,19 whereas their (+) stereoisomers are effective. To date, however, the detailed mechanism of how the stereochemistry of the C-4 methyl group affects the inhibitory property of the resulting enantiomers on AMPA receptors is not well understood, nor the selectivity of these two enantiomers for each of the AMPA receptor subunits.
Here we investigate the mechanism of inhibition of the GluA2 AMPA receptor channel opening by a pair of enantiomers that differ from the configuration of the C-4 methyl group. To measure the effect of an inhibitor on the rate of channel opening, we use a laser-pulse photolysis technique that provides a ∼60 μs time resolution. We also characterize whether the change of the C-4 configuration affects (a) the selectivity of the two enantiomers toward each of the four AMPA receptor subunits and (b) the site of binding, as compared with GYKI 52466. The enantiomer pair we chose for this study is Talampanel (Figure 1) and its (+) enantiomer.19−22
Talampanel is perhaps the best known among all 2,3-benzodiazepine compounds.19−22 Animal and electrophysiological studies suggest a strong potential of this compound as a neuroprotective agent.23−27 Thus far, Talampanel has already undergone phase I and II trials for refractory epilepsy,24,28 amyotrophic lateral sclerosis (ALS),29,30 as well as recurrent malignant gliomas.31,32 Recently, however, a much anticipated phase II clinical trial of this compound against ALS (sponsored by Teva Pharmaceutical Industries Ltd.; http://ir.tevapharm.com/phoenix.zhtml?c=73925&p=irol-newsArticle&ID=1555496) ended without appreciable clinical benefit. At the molecular level, very few mechanistic details for Talampanel are known.11,20 Therefore, the study we report here is useful to shed some light on its mechanism of action on AMPA receptors and its subunit selectivity. For simplicity in describing our study, here we name Talampanel and its (+) enantiomer as BDZ-d and BDZ-e (Figure 1), respectively.
Results and Discussion
BDZ-d and BDZ-e Inhibited the Whole-cell Current of GluA2 AMPA Receptors
We used whole-cell current recording and assayed if BDZ-d and BDZ-e inhibited the AMPA receptor activity. As shown, BDZ-d inhibited the homomeric GluA2Qflip channel expressed in human embryonic kidney (HEK-293) cells (Figure 2a). Under the same condition, however, BDZ-e was barely ineffective as an inhibitor (Figure 2a). To quantitatively measure the difference in potency between BDZ-d and BDZ-e, we characterized the inhibition constant for the two compounds by the ratio of the whole-cell current amplitude in the absence and presence of an inhibitor (i.e., A/AI) as a function of inhibitor concentration. For each compound, we further measured the inhibition constant for both the closed-channel (Figure 2b) and open-channel state (Figure 2c) of GluA2Qflip by setting the glutamate concentration at 100 μM and 3 mM, respectively.15,16,33,34 As such, the inhibition constant of BDZ-d was found to be 15 ± 1 μM for the closed-channel state (KI) and 30 ± 4 μM for the open-channel state (K̅I), by the use of eq 1 (all equations, including eq 1, are listed and described in the Supporting Information). Likewise, a KI of 201 ± 27 μM and a K̅I of 304 ± 32 μM were determined for BDZ-e (see also the summary of these data in Table 1). The comparison of the two sets of inhibition constants clearly shows that BDZ-d is at least 10-fold more potent in inhibiting GluA2Qflip than BDZ-e. It is therefore apparent that BDZ-d is the eutomer whereas BDZ-e is the distomer. The endismic ratio, defined as the ratio of the inhibition constant of the distomer to that of the eutomer, is 14 for the closed-channel state and 10 for the open-channel state of GluA2Qflip.
Figure 2.

(a) Representative whole-cell current traces from HEK-293 cells expressing GluA2Qflip receptors, obtained from solution flow experiments. The bar above each current trace represents a pulse of 100 μM glutamate used to evoke receptor response recorded at −60 mV, pH 7.4, and 22 °C. The pair on the left was from the same cell showing that at 20 μM BDZ-d inhibited the current response as compared with the control; the pair on the right was from another cell showing that BDZ-e at the same concentration, that is, 20 μM, only weakly inhibited the receptor response. (b) Effect of BDZ-d on the whole-cell current amplitude of GluA2Qflip receptors; A and AI represent the current amplitude in the absence and presence of an inhibitor, respectively, and I represents inhibitor. A KI of 15 ± 1 μM was determined, by using eq 1 (see eq 1 is in the Supporting Information), for the closed-channel state (100 μM glutamate, ●); a K̅I of 30 ± 4 μM was obtained for the open-channel state (3 mM glutamate, ○). (c) Effect of BDZ-e on the whole-cell current amplitude of GluA2Qflip receptors obtained from the flow measurement. Similarly, a KI of 201 ± 27 μM was determined for the closed-channel state (100 μM glutamate, ●); a K̅I of 304 ± 32 μM was obtained for the open-channel state (3 mM glutamate, ○).
Table 1. Inhibition Constants of 2,3-Benzodiazepines Obtained from Rate and Amplitude Measurements for the Closed-Channel and Open-Channel States of GluA2Qflip.
| rate measurementa |
amplitude measurement |
|||||
|---|---|---|---|---|---|---|
| inhibitor | KI* (μM)be(closed channel) | K̅I* (μM)b,d(open channel) | KI (μM)bd | KI (μM)be | KI (μM)cd(closed channel) | K̅I(μM)cf(open channel) |
| BDZ-d | 66 ± 20 | 57 ± 5 | 18 ± 2 | 16 ± 1 | 15 ± 1 | 30 ± 4 |
| BDZ-e | 201 ± 27 | 304 ± 32 | ||||
| GYKI 52466g | 61 ± 11 | 128 ± 30 | 15 ± 1 | 16 ± 1 | 14 ± 1 | 30 ± 2 |
| BDZ-fh | 22 ± 1 | 20 ± 3 | 4 ± 1 | 5 ± 1 | 4 ± 1 | 5 ± 1 |
The constants obtained from rate measurements represent those in the first step of inhibition as in Figure 5, whereas those obtained from the amplitude measurements represent the overall inhibition constants.
Laser-pulse photolysis measurement.
Flow measurement.
Measurements at 100 μM glutamate for the closed-channel state.
Measurements at ∼300 μM glutamate.
Measurements at 3 mM glutamate.
Ritz et al.15
Wang et al.16
We further examined the inhibition of both compounds on the flop isoform of GluA2 or GluA2Qflop. Using the same method, we found that BDZ-d and BDZ-e showed similar magnitude of inhibition as they did on the flip isoform (Supporting Information Figure 1). Because BDZ-d exhibited a higher potency, we also determined, using eq 1, its inhibition constant (KI) to be 15 ± 1 μM for the closed-channel and K̅I to be 22 ± 1 μM for the open-channel state (Supporting Information Figure 2). Based on the fact that BDZ-d showed similar inhibition constants for the closed-channel and the open-channel state of the flip and flop isoforms of GluA2, we concluded that BDZ-d did not discriminate between the flip and flop isoforms of GluA2. This result is consistent with our observation from the study of other 2,3-benzodiazepine compounds for the flip and flop of GluA2,15,16,33,34 and suggests that the flip/flop sequence cassette most likely is not involved in the site of binding.15,16,33,34 Apparently, these compounds cannot be used to control the difference in various functional properties between the flip and flop isoforms of GluA2, such as desensitization35−39 and channel opening reaction.40 Together, these data show that the single stereoisomeric difference at the C-4 position gives rise to the difference in biological activity of BDZ-d and BDZ-e in that the S configuration significantly diminishes the activity of the enantiomer on the same receptor.
Inhibitory Selectivity of BDZ-d and BDZ-e to AMPA Receptor Subunits
Did BDZ-d and BDZ-e show a similar differential potency on other AMPA receptor subunits? To answer this question, we tested the inhibitory activity of BDZ-d and BDZ-e with the remaining AMPA receptor subunits, GluA1, GluA3, and GluA4. Specifically, a homomeric receptor channel formed by a single subunit was expressed in HEK-239 cells, and was tested with each of the two compounds by the use of whole-cell recording. The inhibitor concentration was kept at 20 μM throughout. By the magnitude of A/AI, BDZ-d exhibited strong inhibitory activity on GluA1 (Figure 3) in addition to GluA2 (Figure 2c), but showed weak activity on both GluA3 and GluA4. In contrast, BDZ-e was virtually ineffective on every AMPA receptor channels (Figure 3). It should be noted that because BDZ-d had low potency against GluA3 and GluA4, the inhibition ratios for both GluA3 and GluA4 were measured at 100 μM and calibrated to the ratios of 20 μM; the same experiment was done with BDZ-e as well. Even at the higher concentration, neither compound showed appreciable inhibition on either GluA3 or GluA4. On GluA1, BDZ-d displayed higher inhibition potency than BDZ-e, similar to GluA2 (Figure 3). On all AMPA receptor subunits, BDZ-d seemed to inhibit preferably the closed-channel over the open-channel state, although both the potency and the conformation selectivity of BDZ-d on GluA3 and GluA4 were weak (Figure 3).
Figure 3.

Selectivity of BDZ-d and BDZ-e for the four AMPA receptor subunits, GluA1, GluA2, GluA3, and GluA4 (all in the flip isoform). Each subunit was expressed in HEK-293 cells separately, and the inhibition was shown as the A/AI ratio for both the closed-channel and the open-channel for each receptor. For GluA1 and 2, 20 μM BDZ-d and BDZ-e were used for the assay. For GluA3 and GluA4, 100 μM BDZ-d and BDZ-e were used for the assay, because both compounds showed diminished inhibition. For the plot, the A/AI ratio was normalized to that of 20 μM inhibitor response.
BDZ-d Inhibited the Channel-Opening Rate Process of GluA2Qflip
We next investigated the mechanism of action of BDZ-d on the GluA2 AMPA receptor. We chose BDZ-d, instead of BDZ-e, because BDZ-d is the eutomer. We further chose GluA2 because it is one of the main receptor targets of action for BDZ-d (Figures 2 and 3). To investigate the mechanism, we used a laser-pulse photolysis technique, together with a photolabile precursor of glutamate or caged glutamate.41 In this experiment, a single laser pulse was used to photolyze caged glutamate (see detail in the Methods) to release free glutamate with an ∼60 μs time resolution.41 The time resolution, represented by the time constant of the photolysis reaction, was sufficient to enable us to measure the channel-opening reaction of the GluA2 receptor42 and the effect of an inhibitor on the channel-opening rate process.15,16,33,34
Shown in Figure 4a are representative whole-cell current traces obtained from the laser-pulse photolysis measurement. As a control (upper trace in Figure 4a), the rise of the glutamate-evoked whole-cell current reflected the opening of the GluA2 channels from a single HEK-293 cell.42 In the presence of BDZ-d (lower trace), the rate of the current rise on the same cell was slowed and the amplitude of the whole-cell current was reduced. Furthermore, that the channel-opening rate remained single exponential at any concentration of glutamate (i.e., between 100 ± 20 μM and 300 ± 50 μM) with and without inhibitor (for up to 60 μM) was consistent with the assumption that the binding of the inhibitor/glutamate was fast relative to the channel opening.15,16,33,34,42 These results suggested that BDZ-d inhibited the GluA2 channel-opening rate, rather than the rate process of ligand binding. To investigate the mechanism of action for BDZ-d, we further characterized the effect of BDZ-d on both channel-closing and the channel-opening rate constants or kcl and kop, respectively (Figure 4b and c) (see a detailed rationale in the Supporting Information). We found BDZ-d affected both kcl and kop, and further determined a K̅I* of 57 ± 5 μM for the open-channel state (Figure 4b), using eq 2, and a KI* of 66 ± 20 μM for the closed-channel state (Figure 4c), using eq 3 (see also the summary of these data in Table 1). The fact that BDZ-d inhibited both kcl and kop was consistent with a noncompetitive mechanism of inhibition for BDZ-d acting on the GluA2 AMPA receptor.15,16,33,34 Together, both the amplitude and rate data from our experiments further clarified that BDZ-d directly inhibits GluA2 AMPA receptor by binding to a noncompetitive site, contrary to what has been perceived.11,20
Figure 4.

(a) BDZ-d inhibited both the rate and the amplitude of the opening of the GluA2Qflip channels, shown in the representative whole-cell current traces from an HEK-293 cell obtained from the laser-pulse photolysis experiment. The upper trace (○) was the control (kobs = 2079 s–1; A = 0.74 nA), and the lower trace (□) contained 20 μM BDZ-d (kobs = 1591 s–1; A = 0.38 nA). The concentration of the photolytically released glutamate was estimated to be ∼100 μM in both cases. (b) Effect of BDZ-d on kcl obtained at 100 μM of photolytically released glutamate and as a function of BDZ-d concentration. A K̅I* of 57 ± 5 μM was determined using eq 2. (c) Effect of BDZ-d on kop obtained at 300 μM of photolytically released glutamate and as a function of BDZ-d concentration. A KI* of 66 ± 20 μM was determined using eq 3. (d) Effect of BDZ-d on the whole-cell current amplitude obtained from the laser-pulse photolysis measurement. A KI of 18 ± 2 μM was determined at 100 μM of photolytically released glutamate (●); a KI of 16 ± 1 μM was obtained at 300 μM of photolytically released glutamate (○).
BDZ-d Inhibited the Channel Opening of GluA2Qflip by a Two-Step Process
It was interesting to note that the inhibition constants of BDZ-d for both the closed-channel and the open-channel state of GluA2Qflip obtained from the amplitude measurement were smaller than the corresponding values from the rate measurement. Specifically, KI values of 18 ± 2 μM and 16 ± 1 μM were determined at 100 and 300 μM glutamate, respectively, from the effect of the inhibition of BDZ-d on the current amplitude in the laser experiment (Figure 4d). However, these values were 3.6-fold smaller than those obtained from the rate measurements (Figure 4b and c; Table 1). Using a solution flow technique, we determined the KI to be 15 ± 1 μM for BDZ-d for the closed-channel state, measured at 100 μM glutamate (Figure 2a, upper line).15,16,33,34,42 These comparisons therefore suggested that a smaller inhibition constant or a stronger inhibition observed from the amplitude measurement was real and was not due to a specific method or technique we used.
To determine the inhibition constant for the open-channel and closed-channel states for BDZ-d, we varied the concentration of glutamate to titrate the open-channel fraction (see eq 1, and eqs a and b in the Supporting Information). For GluA2 AMPA receptor, glutamate concentrations of 100 μM and 3 mM corresponded to the fraction of the open-channel form being ∼4% and ∼95%, respectively (note that the channel opening probability for GluA2 is ∼96%).42 In other words, at a low glutamate concentration the majority of the receptors were in the closed-channel state, whereas at a saturating glutamate concentration the majority of the receptors were in the open-channel state. As such, the use of the two glutamate concentrations allowed us to estimate the inhibition constants for BDZ-d (and BDZ-e) for the two receptor states of GluA2 (Figure 2b and c; Table 1). Based on this rationale, it would be expected that at 100 μM glutamate concentration, the inhibition constant for BDZ-d calculated from the amplitude measurement for the closed-channel state by the solution flow technique was the same as that by the laser-pulse photolysis technique. It would be further expected that at 300 μM glutamate, the inhibition constant of 16 μM calculated from the amplitude (Figure 4d; open circle) from the laser experiment was similar to 15 μM obtained from the solution flow measurement at 100 μm glutamate concentration (Figure 2b, closed symbol) and 18 μM obtained from the laser measurement also at 100 μM glutamate concentration (Figure 4d, closed circle). This was not surprising because a glutamate concentration of 300 μM correlated to ∼10% of the fraction of the open channel population, which was much closer to ∼4% at 100 μM glutamate than ∼95% at 3 mM glutamate. In fact, at 3 mM glutamate, the inhibition constant of 30 μM was determined for BDZ-d for the open-channel state of GluA2Qflip (Figure 2b, open symbol; Table 1).
The discrepancy between the inhibition constants obtained from the amplitude measurement and from the rate measurement can be accounted for by a mechanism we have previously proposed in the study of other 2,3-benzodiazepine compounds.15,16,33,34 By this mechanism, the inhibition of GluA2Qflip channels by BDZ-d involves two steps, and each step contributes to the overall inhibition of the receptor (Figure 5). The first step involves the formation of an inhibitor-receptor intermediate, producing partial inhibition; and the second step involves a rapid isomerization from the loose inhibitor-receptor complex into a tighter, nonconducting complex that yields additional inhibition.15,16,33,34 Furthermore, the first step is slow as compared to the second step. As such, from the rate measurement, we observed only the partial inhibition from the first step or the slow step, that is, a K̅I* of 57 ± 5 μM for the open-channel state (Figure 4b) and a KI* of 66 ± 20 μM for the closed-channel state (Figure 4c). In contrast, the overall inhibition constants determined from the amplitude data, which included additional inhibition from the second step, are smaller: 15 ± 1 μM for the closed-channel state and 30 ± 4 μM for the open-channel state (Table 1).
Figure 5.

A minimal mechanism of inhibition for BDZ-d. The upper row shows the channel-opening reaction of the AMPA receptor. A represents the active, unliganded form of the receptor; L, the ligand (glutamate); AL and AL2, the ligand-bound closed-channel forms; AL2, the open-channel state of the receptor (all the species with a bar sign refer to open-channel state). Furthermore, kop and kcl are the channel-opening and channel-closing rate constants. For simplicity and without contrary evidence, it is assumed that glutamate binds to the receptor in the two steps with equal affinity, represented by the same intrinsic equilibrium dissociation constant, K1. The initial binding of BDZ-d to the receptor is assumed to form a loosely bound, partially conducting intermediate (e.g., IAL2*) (middle row). In the second step (the lower row), the receptor:inhibitor intermediate rapidly isomerizes into a more tightly bound, inhibitory complex (IAL2). The inhibition constants pertinent to various steps in this mechanistic scheme are shown in Table 1. Specifically, KI represents the overall inhibition constant associated with the closed-channel state of the receptor (i.e., column 5 in Table 1); K̅I, the overall inhibition constant associated with open-channel state (i.e., column 6 in Table 1). The values for K̅I* and KI* for step 1 can be found from columns 1 and 2 in Table 1. In addition, kop* and kcl* are the channel-opening and channel-closing rate constant, respectively, of the partially inhibited AMPA receptor.
BDZ-d and GYKI 52466 Bind to the Same Site on GluA2Qflip, and This Site is Stereoselective
We hypothesized that BDZ-d would bind to the same site to which GYKI 52466 binds on GluA2, because both compounds contain a C-4 methyl group15,16 and a 7,8-methylenedioxy moiety.34 To test this hypothesis, we carried out a double inhibition experiment (see the Supporting Information). In this experiment, BDZ-d and GYKI 52466 were applied on GluA2Qflip simultaneously. The concentration of GYKI 52466 was kept at 20 μM, while the concentration of BDZ-d was varied from 5 to 60 μM. The apparent, double-inhibition constant was found to be 14 ± 1 μM (Figure 6) by the use of eq 4, which was identical to the KI of 15 ± 1 μM for BDZ-d alone. This result was consistent with the hypothesis that BDZ-d competed for binding with GYKI 52466 to the same site on the GluA2Qflip receptor. If the two inhibitors bound to two different sites, a double-inhibition constant of ∼7 μM or a much stronger inhibition would be expected (the dashed line in Figure 6, simulated by eq 5 in the Supporting Information).
Figure 6.

Double-inhibition experiment for GYKI 52466 and BDZ-d on the closed-channel state of GluA2Qflip. The concentration of GYKI 52466 was fixed at 20 μM while that of BDZ-d varied from 5 to 60 μM. The apparent double-inhibition constant was determined to be 14 ± 1 μM (○), as compared with a KI of 15 ± 1 μM for BDZ-d alone (●). The dashed line is the simulation of the A/AI ratio by assuming that the two inhibitors bound to two different sites with an apparent, double-inhibition constant of ∼7 μM (when GYKI 52466 was fixed at 20 μM).
There are several key properties of this particular noncompetitive site on the GluA2 AMPA receptor where GYKI 52466 and BDZ-d bind. First, as we reported previously, the presence of the C-4 methyl group in the 2,3-benzodiazepine structures defines a noncompetitive site but only for those compounds that have the 7,8-methylenedioxy moiety (34) (for the brevity of writing, we name this site as the “M” site). The 7,8-methylenedioxy ring moiety (Figure 1) is important because enlarging the 7,8-methylenedioxy ring into even the 7,8-ethylenedioxy one renders the resulting compound binding to a different noncompetitive site34 (we name this site simply as the “E” site”). Both sites, however, are distinctly different from the third noncompetitive site. The third site accommodates those 2,3-benzodiazepine compounds whose C-4 methyl group is replaced with a C-4 carbonyl group (here we name it the “O” site). It should be noted that for replacing the 4-methyl group of GYKI 52466 with a carbonyl group, the double bond between N-3 and C-4 of the diazepine ring has to be saturated as well. As such, the structural change from GYKI 52466 to the resulting compound or 1-(4-aminophenyl)-3,5-dihydro-7,8-methylenedioxy-4H-2,3-benzodiazepin-4-one is in fact a replacement of the azomethine moiety with a ε-lactam moiety.15 By this structure–activity relationship, it is therefore not surprising that both GYKI 52466 and BDZ-d compete for binding to the same noncompetitive site or the “M” site on the GluA2Qflip receptor. All of these sites are schematically shown in Figure 7.
Figure 7.
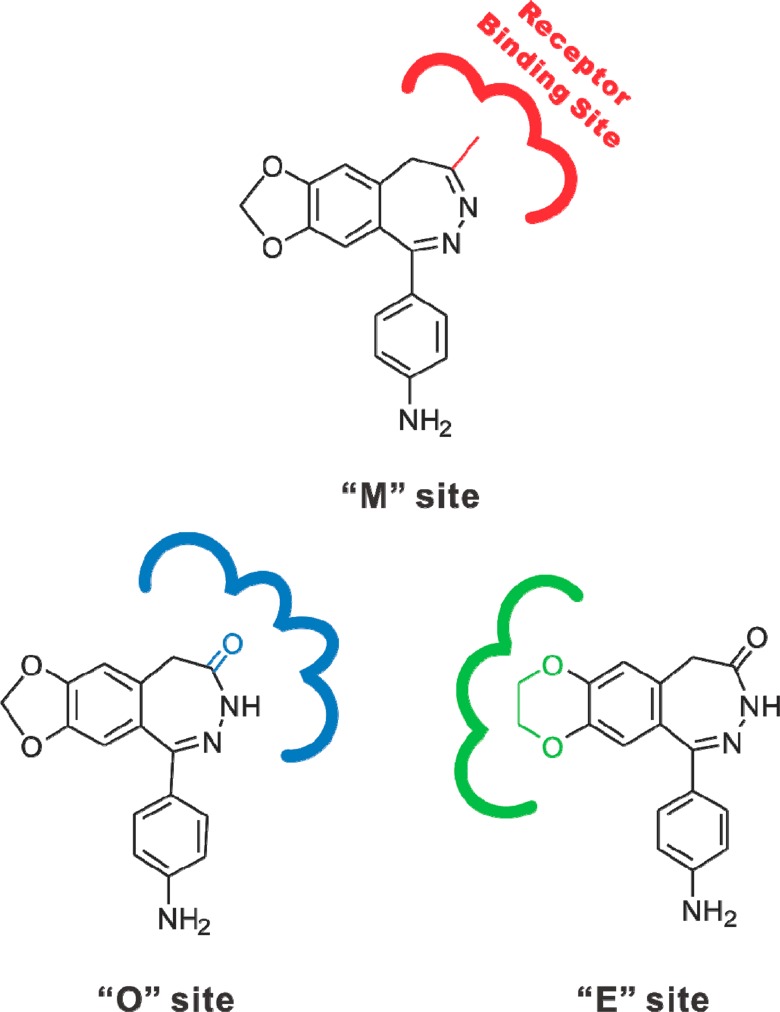
Schematic (and only partial) representation of the three noncompetitive binding sites on the GluA2Qflip AMPA receptor for 2,3-benzodiazepine compounds. The presence of both the C-4 methyl group and the 7,8-methylenedioxy moiety in the 2,3-benzodiazepine structures defines the “M” site (in red color).15,34 Shown here is GYKI 52466 bound to the “M” site. The interaction between the receptor and an inhibitor at the “M” site is stereoselective in that the “M” site preferentially recognizes and accommodates those 2,3-benzodiazepine compounds with a C-4 methyl group in the R configuration. Replacing the C-4 methyl group with a C-4 carbonyl group results in 2,3-benzodiazepin-4-ones that bind to the “O” site (in blue color).15 Shown here is 1-(4-aminophenyl)-3,5-dihydro-7,8-methylenedioxy-4H-2,3-benzodiazepin-4-one bound to the “O” site. However, enlarging the 7,8-methylenedioxy ring into the 7,8-ethylenedioxy one renders the resulting compound binding to the “E” site (in green color).34 Shown here is 1-(4-aminophenyl)-3,5-dihydro-7,8-ethylenedioxy-4H-2,3-benzodiazepin-4-one bound to the “E” site.
Our results demonstrate that BDZ-d binds to and inhibits the GluA2Q AMPA receptors, and it does so much more strongly than BDZ-e. This stereoselectivity is consistent with the one previously reported for 2,3-benzodiazepine compounds.18,19 Our results also establish that the interaction of the receptor through the “M” site with inhibitors like BDZ-d is stereoselective in that the “M” site preferentially recognizes the same 2,3-benzodiazepine compound whose C-4 methyl group is in the R configuration. For an inhibitor with one stereogenic center like the C-4 position, stereoselectivity refers to a receptor’s preferential recognition of one out of two possible stereoisomers.43 At the same site, BDZ-e with the S configuration is not as effective as an inhibitor, most likely due to its less favorable recognition by the receptor and thus a weakened ability to bind to the “M” site. It is further expected that a racemic mixture of BDZ-d and BDZ-e shows a diminished activity as compared to the eutomer BDZ-d alone but improved activity as compared to the distomer BDZ-e alone, which was exactly what we found (see Supporting Information Figure 3).
Assuming that this “M” site is not located on the surface of the GluA2 receptor but in either a cleft or on protruding residues of the receptor, then four simultaneous bonding interactions are involved topologically in order for the receptor to distinguish between the two enantiomers at the receptor site.44 It should be noted that the stereoselectivity at the “M” site is in fact ascribed to the amino acid residues that make up the pocket to bind to the C-4 methyl group of those 2,3-benzodiazepine compounds, such as BDZ-d. At the present, the location and the structure of the “M” site or any other noncompetitive sites on any AMPA receptor subunits is not yet known.
The Effect of N-3 Acylation on the Potency of 2,3-Benzodiazepine Compounds that Bind to the “M” Site
As we have previously reported,16 the “M” site possesses a series of properties for compounds that are structurally similar to GYKI 52466 but more so to those that are derivatized at the N-3 position such as BDZ-d, and BDZ-f (i.e., (−)-1-(4-aminophenyl)-3,4-dihydro-3-N-methylcarbamyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine) (see the chemical structure of BDZ-f in Figure 1). GYKI 524466, BDZ-d, and BDZ-f share the following inhibitory properties. Through this site, (i) these inhibitors show higher potency for the closed-channel than the open-channel state; (ii) these inhibitors are nonselective between the flip and flop variants of GluA2Q or they inhibit both isoforms equally well; (iii) like other 2,3-benzodiazepine compounds we have characterized, BDZ-d inhibits GluA2Qflip by forming an initial, loose intermediate that is still partially conducting, yet this intermediate rapidly isomerizes into a tighter, fully inhibited receptor-inhibitor complex.16
The comparison of the inhibitory potency for all three compounds, that is, GYKI 52466, BDZ-d, and BDZ-f, suggests that acylation at the N-3 position of the 2,3-diazepine ring strengthen the potency of the resulting compounds that bind to the “M” site (see the last two columns in Table 1). The magnitude of the inhibition constants for both the closed-channel and the open-channel state for all three compounds shows that the longer the acyl chain is, the stronger the potency of a resulting compound gets. A derivatization at the N-3 position starts from the GYKI 52466 template (Figure 1). However, the 3,4-dihydro analogue of GYKI 52466, known as GYKI 52895, which is generated by the saturation of the 3,4-double bond, turns out to be a selective dopamine uptake inhibitor.17 In contrast, acylation at the N-3 position by adding an acetyl group, which yields BDZ-d (and BDZ-e), produces the shortest among all N-3 acylated 2,3-benzodiazepine derivatives.11 However, even the eutomer or BDZ-d does not show an improvement in its potency as compared with GYKI 52466 (see Table 1, the last two columns). On the other hand, BDZ-f, which contains a methylcarbamoyl group at the N-3 position, does show an improved potency for both the closed-channel and the open-channel state of GluA2 (Table 1). The fact that BDZ-f is a stronger inhibitor than BDZ-d could be explained by the existence of a large side pocket on the receptor for the “M” site, which can accommodate favorably a large N-3 derivative. Besides the size of an N-3 derivative, other factors could be also involved in strengthening the potency of the resulting compound, such as shape and additional stereochemical arrangement of the same size.16
Based on this hypothesis, we predict that a 2,3-benzodiazepine compound with a longer or larger N-3 derivatization than BDZ-f will possess even a stronger potency. Obviously, this hypothesis requires further testing. It should be noted, however, that our prediction only applies to those compounds that bind to the “M” site on the GluA2 receptor. That means that the compounds must possess a C-4 a methyl group with the R configuration. If the C-4 group is not methyl, but a carbonyl group, the same acylation as described above actually has the opposite effect: the longer the N-3 acyl group, the weaker the potency of resulting compounds.33 This is because these compounds bind to the “O” site, different from the “M” site to which the compounds with the C-4 methyl group such as BDZ-d and BDZ-f bind.
We also predict that the change of the chemical nature of the C-4 methyl group may improve the inhibitory property of a resulting compound. In this case, the CH3– group at the C-4 position may be replaced with a number of perturbations, such as a CH2F– group or a CF3– group. Alternatively, the C-4 CH3– group can be replaced with isosteres, such as a NH2– or OH– group, based on the hydride displacement law.45 A replacement of the C-4 methyl group on the diazepine ring with anyone of these isosteres tests the relevance of the electronegativity without changing the size of a methyl group at the same position. It should be noted that these isosteres must be in the R configuration. Furthermore, the reason by which the size of a methyl group should be preserved is that the replacement of a methyl with a longer alkyl group at the C-4 position is known to reduce potency.11
Selectivity of BDZ-d and BDZ-e to the Four AMPA Receptor Subunits
We also characterized the selectivity profile for both BDZ-d and BDZ-e with each of the four homomeric AMPA receptor channels. Our result shows that BDZ-d exhibits strong inhibition on both GluA1 and GluA2 subunits but only weak inhibition on GluA3 and GluA4 (Figure 3). BDZ-e has the same selectivity profile, although it is a much weaker inhibitor overall (Figure 3). These results therefore allow us to draw several conclusions and implications.
First, that BDZ-d and BDZ-e show the same stereoselectivity on GluA1 as it does on GluA2 suggests that the “M” site also exists on GluA1. Second, the “M” sites to which BDZ-d binds on GluA1 and GluA2 are similar; yet these sites are different from the noncompetitive sites to which BDZ-d binds on either GluA3 or GluA4. We therefore speculate that the weak inhibition of both GluA3 and GluA4 by BDZ-d is due to its weak binding. This speculation is based on the assumption that BDZ-d does not change its conformation when it is bound to different receptor subunits. Furthermore, because BDZ-d does not bind to either GluA3 or GluA4 as strongly as it does to either GluA1 or GluA2, the enantiomeric configuration of the C-4 methyl group has less impact on inhibiting GluA3 and GluA4. In other words, the stereoselectivity of 2,3-benzodiazepine compounds with the C-4 methyl moiety would be less significant on both GluA3 and GluA4 as compared with the GluA1 and GluA2 AMPA receptor subunits.
Mechanistic Evaluation of Talampanel as a Drug Candidate Targeting AMPA Receptors
The results from our study may shed some light to potential problems of using BDZ-d or Talampanel as a single drug for controlling excessive activity of AMPA receptors in vivo, despite the fact that Talampanel is one of the best known 2,3-benzodiazepine compounds.19−22 In terms of potency, Talampanel is not a strong inhibitor of AMPA receptors. It is no better than the prototypic compound GYKI 52466. Even BDZ-f is better than Talampanel or BDZ-d (Table 1).16 If our prediction is correct, a longer N-3 derivitazation with the same C-4 methyl group would be even stronger than BDZ-f and BDZ-d or Talampanel. Surprisingly, Talampanel is ineffective in inhibiting GluA3 and GluA4. This deficiency may explain the failure of a recent, phase II clinical trial where Talampanel was found safe but not efficacious as an ALS drug (see http://ir.tevapharm.com/phoenix.zhtml?c=73925&p=irol-newsArticle&ID=1555496). Evidence suggests that the expression of the GluA3 mRNA (but not the other AMPA receptor subunits) is specifically elevated in motor neurons, after kainic acid infusion but before motor neuron death46 (note that motor neurons can be selectively killed by a continuous low dose of kainic acid infused into animal models). An increase in the GluA3 expression is also observed in the motor neurons of the human superoxide dismutase 1 G93A (DOD1G93A) transgenic mice,47 an ALS animal model, and these motor neurons display an increased vulnerability to glutamate.47 Conversely, the use of a GluA3 antisense peptide nucleic acid or a noncompetitive AMPA receptor inhibitor prolongs the animal survival.48,49 These results suggest that inhibiting the GluA3 subunit should be therapeutically beneficial for ALS treatment. It is therefore not unexpected that the use of Talampanel as a single drug candidate, which does not inhibit the GluA3 subunit, is inefficacious in an ALS clinical test. It should be noted, however, that AMPA receptors play an important role in motor neuron degeneration in ALS,50,51 and the use of AMPA receptor inhibitors remains a promising therapeutic approach. For instance, as shown in experiments with spinal cord slices, AMPA receptor antagonists protect motor neuron degeneration induced by chronic blockade of glutamate uptake as a way to elevate glutamate concentration in order to induce glutamate toxicity.51,52
Conclusion
Chirality can introduce higher selectivity, and possibly specificity as well, for controlling functions of biological molecules in vivo. For instance, the mammalian olfactory system can distinguish the (+)- and (−)-carvone by a caraway-like and a spearmint-like reaction, respectively, although the only difference between the two enantiomers is the configuration of a methylethenyl group.53 For activating glutamate receptors, d-glutamate has lower affinity, and its binding causes less receptor activation.54−56 Chirality can also introduce higher selectivity and perhaps specificity in inhibitor and drug design. This is especially beneficial for developing better 2,3-benzodiazepine compounds for a tighter control of AMPA receptor activity in vivo. The results from this study demonstrate that any effective 2,3-benzodiazepine inhibitors that contain a C-4 methyl group on the diazepine ring, like BDZ-d or Talampanel, must have the R configuration to bind favorably to the noncompetitive receptor site or the “M” site. Our study further suggests that addition of a longer group to the N-3 position should produce a more potent inhibitor for the “M” site. Based on the fact that BDZ-d inhibits GluA1 and GluA2, but is virtually ineffective on GluA3 and GluA4, we hypothesize that the “M” site(s) on GluA1 and GluA2 is different from that of GluA3 and GluA4, most likely reflecting different sets of amino acid residues that make up the “M” site. If the molecular properties of both the AMPA receptors and BDZ-d or Talampanel are used for selecting a single inhibitor as a drug candidate for ALS clinical tests, Talampanel is not ideal.
Methods
Cell Culture and Receptor Expression
HEK-293S cells were cultured in a 37 °C, 5% CO2, humidified incubator, and in the Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen), 100 units of penicillin/mL, and 0.1 mg streptomycin/mL (Sigma-Aldrich, St. Louis, MO). The DNA plasmids encoding all AMPA receptor subunits were prepared as previously described.42,57−59 The HEK-293S cells were transiently transfected to express each AMPA receptor by following a standard calcium phosphate method.60 The cells were also cotransfected with a plasmid encoding green fluorescent protein (GFP) as a transfection marker and a separate plasmid encoding large T-antigen to enhance the receptor expression at the single cell level.61 The weight ratio of the plasmid for GFP and the large T-antigen to that for an AMPA receptor was 1:1:10, respectively. The plasmid used for transient transfection of an AMPA receptor ranged from 5 to 15 μg per 35 mm dish. In general, the cells were used for recording 48 h after transfection.
Whole-Cell Current Recording
Glutamate-induced whole-cell current from transfected HEK-293S cells was recorded on an Axopatch 200B instrument (Molecular Devices, Sunnyvale, CA) at a cutoff frequency of 2–20 kHz by a built-in, four-pole low-pass Bessel filter. The whole-cell current traces were digitized at a 5–50 kHz sampling frequency using a Digidata 1322A (Molecular Devices). All recordings were at −60 mV and room temperature. The pClamp 8 software (Molecular Devices) was used for data acquisition. A recording electrode was made from glass capillary (World Precision Instruments, Sarasota, FL) and had a resistance of ∼3 MΩ when filled with the electrode solution. The electrode solution was composed of (in mM) 110 CsF, 30 CsCl, 4 NaCl, 0.5 CaCl2, 5 EGTA, and 10 HEPES (pH 7.4 adjusted by CsOH). The external solution contained (in mM) 150 NaCl, 3 KCl, 1 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4 adjusted by NaOH). All chemicals were from commercial sources. The GYKI compounds were kindly provided by Dr. Sandor Solyom (from IVAX in Hungary).
Laser-Pulse Photolysis Measurement
The use of the laser-pulse photolysis technique to measure the channel-opening kinetics has been described previously.16 In this experiment, a caged glutamate (e.g., 4-methoxy-7-nitroindolinyl-caged-l-glutamate from Tocris Bioscience) was dissolved in the external buffer and applied to a cell using a flow device.16,57,62 A single laser pulse at 355 nm generated from a pulsed Q-switched Nd:YAG laser (Continuum, Santa Clara, CA), with a pulse length of 8 ns and energy output in the range of 200–1000 μJ, was applied to an HEK-293S cell via optical fiber. To determine the concentration of glutamate generated photolytically by laser photolysis, we calibrated the receptor response in the same cell by applying two solutions of free glutamate with known concentrations before and after laser flash, with reference to the dose–response relation.16 These measurements also allowed us to monitor any damage to the receptors and/or the cell for successive laser experiments with the same cell. A flow device57,62 was used to deliver free glutamate and/or caged glutamate solutions in the absence and presence of inhibitor. The time resolution of this flow device, determined by the rise time of the whole-cell current response (10–90%) to saturating glutamate concentrations, was 1.0 ± 0.2 ms, an average of the measurement from >100 cells expressing the same receptor.63 Furthermore, we used an 8 s time protocol in preincubating all of the 2,3-benzodiazepine compounds in both flow and laser photolysis experiments in order to observe and record full inhibition by these inhibitors, a phenomenon we previously observed with other 2,3-benzodiazepine compounds.15,16,33,34
Experimental Design and Data Analysis
The design of the experiments for determining the inhibition constants for the closed-channel and the open-channel state from the amplitude by varying glutamate concentration has been described in the text. The design of the experiment for measuring the effect of an inhibitor on kop and kcl as a function of inhibitor concentration and at two glutamate concentrations is described in detail in the Supporting Information. All equations used for data analysis are also in the Supporting Information. Furthermore, when a glutamate solution with or without an inhibitor was used to evoke the receptor response, by a solution flow device,16,57,62 the amplitude of the whole-cell current was corrected for receptor desensitization during the rise time.62 The corrected current amplitude was then used for data analysis. Origin 7 (Origin Lab, Northampton, MA) was used for both linear and nonlinear regressions (Levenberg–Marquardt and simplex algorithms). Unless otherwise noted, each data point shown in a plot was an average of at least three measurements collected from three different cells. The error reported refers to the standard error of the fits.
Acknowledgments
We thank Dr. Sandor Solyom from IVAX in Hungary for supplying the GYKI compounds used in this study.
Glossary
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- BDZ
2,3-benzodiazepine derivatives
- ALS
amyotrophic lateral sclerosis
- GYKI 52466
1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- BDZ-d
Talampanel, GYKI 53773, (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-acetyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- BDZ-e
GYKI 53774, (S)-(+)-1-(4-aminophenyl)-3,4-dihydro-3-N-acetyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- BDZ-f
GYKI 53784, (R)-(−)-1-(4-aminophenyl)-3,4-dihydro-3-N-methylcarbamyl-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine
- HEK-293 cells
human embryonic kidney 293 cells
Supporting Information Available
Figures showing the inhibition of BDZ-d and BDZ-e on GluA2Qflop and the inhibition of the mixture of BDZ-d/BDZ-e on GluA2Qflip, and an appendix listing all equations for data analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
C.W. conducted the experiments, produced the plots and table, and wrote the draft; L.N. directed the research and wrote the paper based on the draft written by C.W.
This work was supported by grants from NIH/NINDS (R01 NS060812) and the Muscular Dystrophy Association.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Huang Q.; He R.; Kozikowski A. P. (2011) Stereochemistry at the forefront in the design and discovery of novel anti-tuberculosis agents. Curr. Top. Med. Chem. 11, 810–818. [DOI] [PubMed] [Google Scholar]
- McConnell O.; Bach A. 2nd; Balibar C.; Byrne N.; Cai Y.; Carter G.; Chlenov M.; Di L.; Fan K.; Goljer I.; He Y.; Herold D.; Kagan M.; Kerns E.; Koehn F.; Kraml C.; Marathias V.; Marquez B.; McDonald L.; Nogle L.; Petucci C.; Schlingmann G.; Tawa G.; Tischler M.; Williamson R. T.; Sutherland A.; Watts W.; Young M.; Zhang M. Y.; Zhang Y.; Zhou D.; Ho D. (2007) Enantiomeric separation and determination of absolute stereochemistry of asymmetric molecules in drug discovery: building chiral technology toolboxes. Chirality 19, 658–682. [DOI] [PubMed] [Google Scholar]
- Dingledine R.; Borges K.; Bowie D.; Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61. [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman A. G. (1998) Glutamate receptors in epilepsy. Prog. Brain Res. 116, 371–383. [DOI] [PubMed] [Google Scholar]
- Meldrum B. S.; Akbar M. T.; Chapman A. G. (1999) Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res. 36, 189–204. [DOI] [PubMed] [Google Scholar]
- Kawahara Y.; Kwak S. (2005) Excitotoxicity and ALS: what is unique about the AMPA receptors expressed on spinal motor neurons?. Amyotrophic Lateral Scler. Other Mot. Neuron Disord. 6, 131–144. [DOI] [PubMed] [Google Scholar]
- Kwak S.; Hideyama T.; Yamashita T.; Aizawa H. (2010) AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 30, 182–188. [DOI] [PubMed] [Google Scholar]
- Klockgether T.; Turski L.; Honore T.; Zhang Z. M.; Gash D. M.; Kurlan R.; Greenamyre J. T. (1991) The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine-depleted rats and MPTP-treated monkeys. Ann. Neurol. 30, 717–723. [DOI] [PubMed] [Google Scholar]
- Tarnawa I.; Farkas S.; Berzsenyi P.; Pataki A.; Andrasi F. (1989) Electrophysiological studies with a 2,3-benzodiazepine muscle relaxant: GYKI 52466. Eur. J. Pharmacol. 167, 193–199. [DOI] [PubMed] [Google Scholar]
- Solyom S.; Tarnawa I. (2002) Non-competitive AMPA antagonists of 2,3-benzodiazepine type. Curr. Pharm. Des. 8, 913–939. [DOI] [PubMed] [Google Scholar]
- Szenasi G.; Vegh M.; Szabo G.; Kertesz S.; Kapus G.; Albert M.; Greff Z.; Ling I.; Barkoczy J.; Simig G.; Spedding M.; Harsing L. G. Jr. (2008) 2,3-Benzodiazepine-type AMPA receptor antagonists and their neuroprotective effects. Neurochem. Int. 52, 166–183. [DOI] [PubMed] [Google Scholar]
- De Sarro A.; De Sarro G.; Gitto R.; Grasso S.; Micale N.; Quartarone S.; Zappala M. (1998) 7,8-Methylenedioxy-4H-2,3-benzodiazepin-4-ones as novel AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 8, 971–976. [DOI] [PubMed] [Google Scholar]
- Grasso S.; De Sarro G.; De Sarro A.; Micale N.; Zappala M.; Puia G.; Baraldi M.; De Micheli C. (1999) Synthesis and anticonvulsant activity of novel and potent 2,3-benzodiazepine AMPA/kainate receptor antagonists. J. Med. Chem. 42, 4414–4421. [DOI] [PubMed] [Google Scholar]
- Ritz M.; Wang C.; Micale N.; Ettari R.; Niu L. (2011) Mechanism of Inhibition of the GluA2 AMPA Receptor Channel Opening: The Role of 4-Methyl versus 4-Carbonyl Group on the Diazepine Ring of 2,3-Benzodiazepine Derivatives. ACS Chem. Neurosci. 2, 506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Sheng Z.; Niu L. (2011) Mechanism of Inhibition of the GluA2 AMPA Receptor Channel Opening: Consequences of Adding an N-3 Methylcarbamoyl Group to the Diazepine Ring of 2,3-Benzodiazepine Derivatives. Biochemistry 50, 7284–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth K.; Szabó H.; Pátfalusi M.; Berzsenyi P.; Andrási F. (1990) Pharmacological effects of GYMI 52 895, a new selective dopamine uptake Inhibitor. Eur. J. Pharmacol. 183, 1416–1417. [Google Scholar]
- May P. C.; Robison P. M.; Fuson K. S. (1999) Stereoselective neuroprotection by novel 2,3-benzodiazepine non-competitive AMPA antagonist against non-NMDA receptor-mediated excitotoxicity in primary rat hippocampal cultures. Neurosci. Lett. 262, 219–221. [DOI] [PubMed] [Google Scholar]
- Bleakman D.; Ballyk B. A.; Schoepp D. D.; Palmer A. J.; Bath C. P.; Sharpe E. F.; Woolley M. L.; Bufton H. R.; Kamboj R. K.; Tarnawa I.; Lodge D. (1996) Activity of 2,3-benzodiazepines at native rat and recombinant human glutamate receptors in vitro: stereospecificity and selectivity profiles. Neuropharmacology 35, 1689–1702. [DOI] [PubMed] [Google Scholar]
- Howes J. F.; Bell C. (2007) Talampanel. Neurotherapeutics 4, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G.; Henley J. M. (1993) Binding of the AMPA receptor antagonist [3H]GYKI 53405 to Xenopus brain membranes. NeuroReport 5, 93–94. [DOI] [PubMed] [Google Scholar]
- Donevan S. D.; Yamaguchi S.; Rogawski M. A. (1994) Non-N-methyl-D-aspartate receptor antagonism by 3-N-substituted 2,3-benzodiazepines: relationship to anticonvulsant activity. J. Pharmacol. Exp. Ther. 271, 25–29. [PubMed] [Google Scholar]
- Lodge D.; Bond A.; O’Neill M. J.; Hicks C. A.; Jones M. G. (1996) Stereoselective effects-of 2,3-benzodiazepines in vivo: electrophysiology and neuroprotection studies. Neuropharmacology 35, 1681–1688. [DOI] [PubMed] [Google Scholar]
- Chappell A. S.; Sander J. W.; Brodie M. J.; Chadwick D.; Lledo A.; Zhang D.; Bjerke J.; Kiesler G. M.; Arroyo S. (2002) A crossover, add-on trial of talampanel in patients with refractory partial seizures. Neurology 58, 1680–1682. [DOI] [PubMed] [Google Scholar]
- Denes L.; Szilagyi G.; Gal A.; Nagy Z. (2006) Talampanel a non-competitive AMPA-antagonist attenuates caspase-3 dependent apoptosis in mouse brain after transient focal cerebral ischemia. Brain Res. Bull. 70, 260–262. [DOI] [PubMed] [Google Scholar]
- Erdo F.; Berzsenyi P.; Andrasi F. (2005) The AMPA-antagonist talampanel is neuroprotective in rodent models of focal cerebral ischemia. Brain Res. Bull. 66, 43–49. [DOI] [PubMed] [Google Scholar]
- Aujla P. K.; Fetell M. R.; Jensen F. E. (2009) Talampanel suppresses the acute and chronic effects of seizures in a rodent neonatal seizure model. Epilepsia 50, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langan Y. M.; Lucas R.; Jewell H.; Toublanc N.; Schaefer H.; Sander J. W.; Patsalos P. N. (2003) Talampanel, a new antiepileptic drug: single- and multiple-dose pharmacokinetics and initial 1-week experience in patients with chronic intractable epilepsy. Epilepsia 44, 46–53. [DOI] [PubMed] [Google Scholar]
- Traynor B. J.; Bruijn L.; Conwit R.; Beal F.; O’Neill G.; Fagan S. C.; Cudkowicz M. E. (2006) Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology 67, 20–27. [DOI] [PubMed] [Google Scholar]
- Pascuzzi R. M.; Shefner J.; Chappell A. S.; Bjerke J. S.; Tamura R.; Chaudhry V.; Clawson L.; Haas L.; Rothstein J. D. (2010) A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 11, 266–271. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S.; Tsuzuki K.; Yoshida Y.; Yamada N.; Hagimura N.; Okado H.; Miwa A.; Kurihara H.; Nakazato Y.; Tamura M.; Sasaki T.; Ozawa S. (2002) Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 8, 971–978. [DOI] [PubMed] [Google Scholar]
- Iwamoto F. M.; Kreisl T. N.; Kim L.; Duic J. P.; Butman J. A.; Albert P. S.; Fine H. A. (2010) Phase 2 trial of talampanel, a glutamate receptor inhibitor, for adults with recurrent malignant gliomas. Cancer 116, 1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz M.; Micale N.; Grasso S.; Niu L. (2008) Mechanism of inhibition of the GluR2 AMPA receptor channel opening by 2,3-benzodiazepine derivatives. Biochemistry 47, 1061–1069. [DOI] [PubMed] [Google Scholar]
- Qneibi M. S.; Micale N.; Grasso S.; Niu L. (2012) Mechanism of Inhibition of the GluA2 AMPA Receptor Channel Opening by 2,3-Benzodiazepine Derivatives: Functional Consequences of Replacing 7,8-Methylenedioxy with 7,8-Ethylenedioxy Moiety. Biochemistry 51, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B.; Keinanen K.; Verdoorn T. A.; Wisden W.; Burnashev N.; Herb A.; Kohler M.; Takagi T.; Sakmann B.; Seeburg P. H. (1990) Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science 249, 1580–1585. [DOI] [PubMed] [Google Scholar]
- Mosbacher J.; Schoepfer R.; Monyer H.; Burnashev N.; Seeburg P. H.; Ruppersberg J. P. (1994) A molecular determinant for submillisecond desensitization in glutamate receptors. Science 266, 1059–1062. [DOI] [PubMed] [Google Scholar]
- Koike M.; Tsukada S.; Tsuzuki K.; Kijima H.; Ozawa S. (2000) Regulation of kinetic properties of GluR2 AMPA receptor channels by alternative splicing. J. Neurosci. 20, 2166–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin K. M.; Patneau D. K.; Mayer M. L. (1994) Cyclothiazide differentially modulates desensitization of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor splice variants. Mol. Pharmacol. 46, 129–138. [PubMed] [Google Scholar]
- Kessler M.; Rogers G.; Arai A. (2000) The norbornenyl moiety of cyclothiazide determines the preference for flip-flop variants of AMPA receptor subunits. Neurosci. Lett. 287, 161–165. [DOI] [PubMed] [Google Scholar]
- Pei W.; Huang Z.; Wang C.; Han Y.; Park J. S.; Niu L. (2009) Flip and flop: a molecular determinant for AMPA receptor channel opening. Biochemistry 48, 3767–3777. [DOI] [PubMed] [Google Scholar]
- Wieboldt R.; Gee K. R.; Niu L.; Ramesh D.; Carpenter B. K.; Hess G. P. (1994) Photolabile precursors of glutamate: synthesis, photochemical properties, and activation of glutamate receptors on a microsecond time scale. Proc. Natl. Acad. Sci. U.S.A. 91, 8752–8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Pei W.; Niu L. (2003) Channel-opening kinetics of GluR2Q(flip) AMPA receptor: a laser-pulse photolysis study. Biochemistry 42, 12358–12366. [DOI] [PubMed] [Google Scholar]
- Sundaresan V.; Abrol R. (2002) Towards a general model for protein-substrate stereoselectivity. Protein Sci. 11, 1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecar A. D.; Koshland D. E. Jr. (2000) A new model for protein stereospecificity. Nature 403, 614–615. [DOI] [PubMed] [Google Scholar]
- Grimm H. G. (1925) Structure and Size of the Non-metallic Hydrides Z. Electrochemistry 474–480. [Google Scholar]
- Sun H.; Kawahara Y.; Ito K.; Kanazawa I.; Kwak S. (2006) Slow and selective death of spinal motor neurons in vivo by intrathecal infusion of kainic acid: implications for AMPA receptor-mediated excitotoxicity in ALS. J. Neurochem. 98, 782–791. [DOI] [PubMed] [Google Scholar]
- Spalloni A.; Pascucci T.; Albo F.; Ferrari F.; Puglisi-Allegra S.; Zona C.; Bernardi G.; Longone P. (2004) Altered vulnerability to kainate excitotoxicity of transgenic-Cu/Zn SOD1 neurones. NeuroReport 15, 2477–2480. [DOI] [PubMed] [Google Scholar]
- Rembach A.; Turner B. J.; Bruce S.; Cheah I. K.; Scott R. L.; Lopes E. C.; Zagami C. J.; Beart P. M.; Cheung N. S.; Langford S. J.; Cheema S. S. (2004) Antisense peptide nucleic acid targeting GluR3 delays disease onset and progression in the SOD1 G93A mouse model of familial ALS. J. Neurosci. Res. 77, 573–582. [DOI] [PubMed] [Google Scholar]
- Tortarolo M.; Grignaschi G.; Calvaresi N.; Zennaro E.; Spaltro G.; Colovic M.; Fracasso C.; Guiso G.; Elger B.; Schneider H.; Seilheimer B.; Caccia S.; Bendotti C. (2006) Glutamate AMPA receptors change in motor neurons of SOD1G93A transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J. Neurosci. Res. 83, 134–146. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L.; Van Damme P.; Bogaert E.; Robberecht W. (2006) The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. [DOI] [PubMed] [Google Scholar]
- Van Damme P.; Leyssen M.; Callewaert G.; Robberecht W.; Van Den Bosch L. (2003) The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 343, 81–84. [DOI] [PubMed] [Google Scholar]
- Rothstein J. D.; Jin L.; Dykes-Hoberg M.; Kuncl R. W. (1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 90, 6591–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitereg T. J.; Guadagni D. G.; Harris J.; Mon T. R.; Teranishi R. (1971) Chemical and sensory data supporting the difference between the odors of the enantiomeric carvones. J. Agric. Food Chem. 19, 3. [Google Scholar]
- Gray S. R.; Batstone F. R.; Santiapillai N. F.; Richardson P. J. (1991) Solubilization and purification of a putative quisqualate-sensitive glutamate receptor from crustacean muscle. Biochem. J. 273(Pt 1), 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James V. A.; Walker R. J.; Wheal H. V. (1980) Structure-activity studies on an excitatory receptor for glutamate on leech Retzius neurones. Br. J. Pharmacol. 68, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. H.; Michaelis E. K. (1981) L-glutamate stimulation of Na+ efflux from brain synaptic membrane vesicles. J. Biol. Chem. 256, 10084–10087. [PubMed] [Google Scholar]
- Li G.; Niu L. (2004) How fast does the GluR1Qflip channel open?. J. Biol. Chem. 279, 3990–3997. [DOI] [PubMed] [Google Scholar]
- Pei W.; Huang Z.; Niu L. (2007) GluR3 Flip and Flop: Differences in Channel-Opening Kinetics. Biochemistry 46, 2027–2036. [DOI] [PubMed] [Google Scholar]
- Li G.; Sheng Z.; Huang Z.; Niu L. (2005) Kinetic mechanism of channel opening of the GluRDflip AMPA receptor. Biochemistry 44, 5835–5841. [DOI] [PubMed] [Google Scholar]
- Chen C.; Okayama H. (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Li G.; Pei W.; Sosa L. A.; Niu L. (2005) Enhancing protein expression in single HEK 293 cells. J. Neurosci. Methods 142, 159–166. [DOI] [PubMed] [Google Scholar]
- Udgaonkar J. B.; Hess G. P. (1987) Chemical kinetic measurements of a mammalian acetylcholine receptor by a fast-reaction technique. Proc. Natl. Acad. Sci. U.S.A. 84, 8758–8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Wang C.; Park J. S.; Niu L. (2012) Channel-Opening Kinetic Mechanism of Wild-Type GluK1 Kainate Receptors and a C-Terminal Mutant. Biochemistry 51, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.