

Abstract

The transient receptor potential vanilloid subfamily member 1 (TRPV1) cation channel is known to be involved in pain nociception and neurogenic inflammation, and accumulating evidence suggests that it plays an important role in several central nervous system (CNS)-related disorders. TRPV1-specific positron emission tomography (PET) radioligands can serve as powerful tools in TRPV1-related (pre)clinical research and drug design. We have synthesized several potent TRPV1 antagonists and accompanying precursors for radiolabeling with carbon-11 or fluorine-18. The cinnamic acid derivative [11C]DVV24 and the aminoquinazoline [18F]DVV54 were successfully synthesized, and their biological behavior was studied. In addition, the in vivo behavior of a 123I-labeled analogue of iodo-resiniferatoxin (I-RTX), a well-known TRPV1 antagonist, was evaluated. The binding affinities of DVV24 and DVV54 for human TRPV1 were 163 ± 28 and 171 ± 48 nM, respectively. [11C]DVV24, but not [18F]DVV54 or 123I-RTX, showed retention in the trigeminal nerve, known to abundantly express TRPV1. Nevertheless, it appears that ligands with higher binding affinities will be required to allow in vivo imaging of TRPV1 via PET.
Keywords: TRPV1, positron emission tomography, carbon-11, fluorine-18, 123I-RTX
The transient receptor potential vanilloid subfamily member 1 (TRPV1) receptor is a nonselective, Ca2+ permeable cation channel belonging to the TRP ion channel family.1 TRPV1 is mainly expressed on a subpopulation of primary sensory neurons of the dorsal root and trigeminal ganglia and their nociceptive terminals. Expression of functional TRPV1 receptors has also been demonstrated in various non-neuronal cell types, such as human skin cells,2 urothelial cells,3 and pancreatic β-cells,4 and evidence of an active population of TRPV1 receptors in the central nervous system (CNS) is emerging. However, the extent to which these central and non-neuronal TRPV1 channels are expressed is still a subject of controversy, and their exact (patho)physiological role remains elusive.5−9
TRPV1 is pursued as a target for the development of a new class of anti-inflammatory and analgesic drugs. Opening of TRPV1 channels expressed on sensory neurons, by agonists [e.g., capsaicin, resiniferatoxin (RTX), or anandamide], heat, or acidic pH,10 leads to depolarization of the cell membrane via calcium influx whereby painful stimuli are transmitted and pro-inflammatory neuropeptides such as substance P and calcitonin gene-related peptide are released.11,12 Therefore, TRPV1 has been suggested to play an essential role in the pathogenesis of various pain conditions and chronic inflammatory disorders.13 In the CNS, TRPV1 regulates multiple functions in response to stress14 and mediates synaptic plasticity, which highlights its potential role in the control of emotional responses, learning, and epileptic activity.15 Furthermore, accumulating evidence suggests that TRPV1 channels may contribute to the pathogenesis of febrile seizures, stroke, and neurodegenerative brain disorders such as Parkinson’s disease.16,17 These CNS-related observations could indicate new potential therapeutic applications, yet more research is needed to elucidate the exact (pathological) role of these central TRPV1 receptors.
Positron emission tomography (PET) tracers can serve as powerful tools in studying the efficacy of therapeutics that target TRPV1 and in investigating altered TRPV1 levels under pathophysiological conditions. Previously, we have synthesized and evaluated [11C]SB366791, a specific TRPV1 PET radioligand. However, its clinical use is doubtful because of its relatively low binding affinity (280 ± 56 nM) for human TRPV1 (hTRPV1).18 Therefore, we aimed to develop PET radioligands that display higher binding affinities. After a literature survey, several potent TRPV1 antagonists belonging to different structural classes were selected, including cinnamic acid derivatives,19 aminoquinazolines,20 and urea derivatives.21 To allow radiolabeling, some of these molecules required small structural changes. Moreover, the chlorine atom in SB366791 was substituted with a trifluoromethyl group. It has been shown that the nature of the 4-position ring substituent in other structurally related compounds plays an important role in determining the activity for TRPV1. More hydrophobic substituents such as a trifluoromethyl group were reported to increase the TRPV1 inhibitory activity.10 Suitable precursor and authentic reference compounds were synthesized, and their TRPV1 binding and activity profile was studied. Radiolabeling of the precursor compounds with carbon-11 or fluorine-18 provided two new TRPV1 PET radioligands, [11C]DVV24 and [18F]DVV54. Their pharmacokinetic properties were studied by means of biodistribution studies and radiometabolite analysis. Additionally, the in vivo behavior of a 123I-labeled analogue of iodo-resiniferatoxin (I-RTX), a highly potent TRPV1 antagonist,22 was studied.
Results and Discussion
Chemistry
The synthesis of the cinnamic acid derivative 1 was described previously.18 Compound 2 was synthesized in four steps, starting from 3-acetamidophenol following the procedure reported for the synthesis of 1, but using 4-trifluoromethylcinnamic acid instead of 4-chlorocinnamic acid. Alkylation of 1 with 2-fluoroethyl tosylate yielded compound 3 (Figure 1). DVV24 was obtained in one step via the formation of a peptide bond between 4-(trifluoromethyl)cinnamic acid and 3-methoxyaniline using EDCI and HOBt as described by Gunthorpe et al. (Figure 2).19
Figure 1.

Cinnamic acid derivatives. Structure of compounds 1 and 2 and synthesis of compound 3. (i) 2-Fluoroethyl tosylate, Cs2CO3, 5 h at 120 °C.
Figure 2.
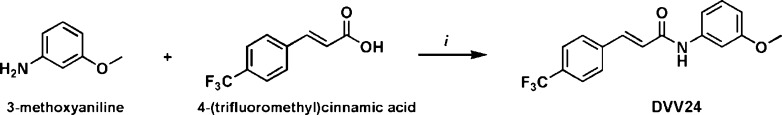
Synthesis of cinnamic acid derivative DVV24. (i) EDCI, HOBt, 16 h at room temperature.
The urea derivatives were synthesized in two steps according to a method described in the patent literature.23 In the first step, intermediates 4 and 5 were obtained by alkylation of the amine of N-methyl-p-toluidine and p-toluidine, respectively, through a nucleophilic substitution using 2-bromoethylamine hydrobromide. Next, ureas 6 and 7 were formed by reaction of the nucleophilic amine moiety of 4 and 5, respectively, with 2-bromophenyl isocyanate (Figure 3).
Figure 3.

Synthesis of urea derivatives 6 and 7. (i) Toluene, reflux for 24 h. (ii) CH2Cl2, 1 h at room temperature.
The synthesis of four aminoquinazolines was achieved via a multistep procedure based on previously described methods.20,24,25 Compound 16 was obtained via a nucleophilic substitution reaction on the chlorine of 15 using sodium methanolate, and DVV54 was formed via a nucleophilic fluorine for chlorine exchange by reacting a solution of 15 with tetrabutylammonium fluoride (TBAF) in THF under high pressure. Compound 20 was obtained by debenzylation of 19 using H2 in the presence of palladium on activated carbon (Pd/C, 10 wt % loading) (Figure 4).
Figure 4.

Synthesis of aminoquinazolines 16, 20, and DVV54. (i) Na2CO3, Pd(PPh3)4, 16 h at 80 °C. (ii) H2SO4, HNO3, 1 h at room temperature. (iii) KMnO4, 4 h at 110 °C. (iv) (a) SOCl2, 4 h at 100 °C; (b) NH3 gas, 30 min at 0 °C. (v) SnCl2, 2 h at 80 °C. (vi) 2-Chloro-1,1,1-trimethoxyethane, 4 h at 130 °C. (vii) POCl3, 16 h at 80 °C. (viii and xiii) 4-(Trifluoromethyl)aniline, 4 h at 75 °C. (ix) (a) NaOCH3, 1 h at 60 °C; (b) metallic Na, 16 h at 60 °C. (x) TBAF, 5 min at 150 °C. (xi) (a) Benzyloxy acetic acid, oxalyl chloride, 1 h at 0 °C; (b) 17, benzyloxy acetic acid, oxalyl chloride, 1 h at room temperature; (c) NaOH, 1 h at room temperature. (xii) POCl3, 18 h at 70 °C. (xiv) Pd/C (10 wt %), hydrogenation for 62 h.
Radiolabeling
To radiolabel urea precursor 7 with carbon-11, multiple experimental conditions were tested. The type of labeling reagent [[11C]CH3I or [11C]methyl triflate ([11C]CH3OTf)], solvent, amount of precursor, base, reaction time and temperature, and type of oven were changed, nevertheless, without success. Under neutral conditions, the reaction did not proceed, whereas addition of a base to activate the nucleophile resulted in degradation of 7. The different radiolabeling conditions are summarized in Table 1.
Table 1. Summary of the Experimental Conditions for Radiolabeling of [11C]-6, [11C]-16, [11C]DVV24, and [18F]DVV54a.
| precursor (μg) | reagent | solvent | base | reaction time, temperature | tracer | yield and isolated activities |
|---|---|---|---|---|---|---|
| 2 (200) | [11C]CH3I | DMF | Cs2CO3 | 5 min at 70 °C | [11C]DVV24 | 55–75%,c1443–3404 MBq |
| 15 (1000) | [18F]F– | DMF | K222/K2CO3 | 15 min at 150 °C | [18F]DVV54 | 10–44%c1258–2257 MBq |
| 20 (200) | [11C]CH3I | DMF | dry NaH | 5 min at 90 °C | [11C]-16 | NYd |
| 20 (200) | [11C]CH3OTf | THF | dry NaH | 5 min at 65 °C | [11C]-16 | NYd |
| 7 (200) | [11C]CH3OTf | CH3CN or DMF | Cs2CO3 | 5 min at room temperature or 70 °C | [11C]-6 | NYd |
| 7 (200) | [11C]CH3I | CH3CN | 0.01 M NaOH | 5 min at 70 or 90 °C | [11C]-6 | NYd |
| 7 (200) | [11C]CH3I | DMF | no base | 5 min at 130 °C | [11C]-6 | NYd |
| 7 (1000) | [11C]CH3I | DMF | no base or Cs2CO3 | 4 min at 130 °C | [11C]-6 | NYd |
Cinnamic acid derivatives, 2 and [11C]DVV24; urea derivatives, 7 and [11C]-6; aminoquinazolines, 15, 20, [18F]DVV54, and [11C]-16.
Microwave oven 60 W.
Radiochemical yield based on HPLC-recovered radioactivity relative to [11C]CH3I or [18F]F–.
No yield, meaning no product was formed under these conditions.
Unlike the ureas, the radiolabeled cinnamic acid derivative [11C]DVV24 (Figure 5) was efficiently synthesized in one step by methylation of phenolic precursor 2 using [11C]CH3I (Table 1). Attempts to radiolabel 20 with carbon-11 using [11C]CH3I or [11C]CH3OTf to obtain [11C]-16 were unsuccessful because the reactions did not proceed, yet [18F]DVV54 (Figure 5) was obtained by replacement of the chlorine atom of precursor 15 with 18F using [18F]F– in the presence of kryptofix 222 (K222)/K2CO3 (Table 1). The tracers were purified by reversed phase high-performance liquid chromatography (RP-HPLC), and their identity was confirmed by co-elution with the nonradioactive reference compounds after co-injection on an analytical RP-HPLC system (Figures A.1 and A.2 of the Supporting Information). [11C]DVV24 and [18F]DVV54 were obtained within 45 and 60 min, respectively. Both tracers showed high radiochemical purity (RCP > 99%) and good specific activity (SA of [11C]DVV24 = 50–225 GBq/μmol; SA of [18F]DVV54 = 16–44 GBq/μmol).
Figure 5.

TRPV1-targeting radioligands [11C]DVV24 (cinnamic acid derivative), [18F]DVV54 (aminoquinazoline), and 123I-RTX.
RTX was successfully radiolabeled with iodine-123 using the iodogen method.26 Iodine-123 is a radioisotope suitable for single-photon emission-computed tomography (SPECT) imaging. It was used instead of the PET radionuclide iodine-124 to perform the preliminary biological evaluation because of its shorter half-life (13 hours for iodine-123 vs 4.2 days for iodine-124). The desired 123I-RTX (Figure 5) was isolated using RP-HPLC; its identity was confirmed, and the RCP was >99%.
Competition Binding and 45Ca2+ Uptake Experiments
The binding assays and 45Ca2+ uptake experiments were performed according to a previously described method.18 Briefly, the binding affinities (expressed as Kd) of the nonradioactive reference and precursor compounds were evaluated using a competitive binding assay with [3H]RTX as the radioligand. Functional 45Ca2+ uptake experiments were performed to study their antagonism and agonism (expressed as Ki and EC50, respectively) profile. The assays were performed on Chinese hamster ovary (CHO) cells transfected with hTRPV1 or rat TRPV1 (rTRPV1) in the presence of a fixed concentration of [3H]RTX (binding assays) or capsaicin (antagonism assays) and various concentrations of the competing ligands. The results of the binding and functional assays are listed in Table 2 together with the calculated logD (clogD) and polar surface area (PSA) values.
Table 2. Overview of the Kd, Ki, clogD, and PSA Values of the Nonradioactive Reference and Precursor Compoundsa.
| [3H]RTX binding [Kd (nM)]b | ant. [Ki (nM)]b | clogD (pH 7.4) | PSA (Å2) | |
|---|---|---|---|---|
| SB366791 | 780 ± 140 (rTRPV1) | 334 ± 17 (rTRPV1) | 4.02 | 38.33 |
| 280 ± 56 (hTRPV1) | 197 ± 17 (hTRPV1) | |||
| 1 | 900 ± 140 (rTRPV1) | 128 ± 34 (rTRPV1) | 3.78 | 49.33 |
| 2 | 215 ± 11 (rTRPV1) | 55 ± 10 (rTRPV1) | 4.06 | 49.33 |
| 3 | 4600 ± 1700 (rTRPV1) | 5600 ± 1600 (rTRPV1) | 4.14 | 38.33 |
| DVV24 | 227 ± 10 (rTRPV1) | 171 ± 17 (rTRPV1) | 4.29 | 38.33 |
| 163 ± 28 (hTRPV1) | 125 ± 33 (hTRPV1) | |||
| 6 | 173 ± 33 (rTRPV1) | 37 ± 12 (rTRPV1) | 4.29 | 47.53 |
| 7 | 2800 ± 280 (rTRPV1) | 1486 ± 16 (rTRPV1) | 3.66 | 56.32 |
| 16 | 11.3 ± 1.5 (rTRPV1) | 1.61 ± 0.57 (rTRPV1) | 6.21 | 59.93 |
| 42.3 ± 5.7 (hTRPV1) | 2.58 ± 0.26 (hTRPV1) | |||
| DVV54 | 32 ± 11 (rTRPV1) | 3.13 ± 0.78 (rTRPV1) | 6.51 | 50.70 |
| 171 ± 48 (hTRPV1) | 8.3 ± 1.0 (hTRPV1) | |||
| I-RTX | 0.61 ± 0.08 (rTRPV1) | 12.2 ± 4.0 (rTRPV1) | 6.50 | 120.75 |
The I-RTX potencies are taken from Lim et al.27. rTRPV1, rat TRPV1; hTRPV1, human TRPV1; ant., antagonist activity; cinnamic acid derivatives, SB366791, DVV24, and compounds 1–3; urea derivatives, compounds 6 and 7; aminoquinazolines, compound 16 and DVV54.
Kd and Ki values are shown as means ± the standard error of the mean of three independent experiments.
None of the tested compounds displayed agonist activity. The observed Kd and Ki values of the cinnamic acid derivatives depend on the nature of the substituents on the aromatic rings. Substitution of the chlorine at position 4 in SB366791 with a trifluoromethyl (DVV24) resulted in a 3-fold increase in the binding affinity for rTRPV1 and a 1.7-fold increase of the binding affinity for hTRPV1. The phenolic precursor 1 shows a modest 1.2-fold lower affinity (higher Kd) than the corresponding methoxy derivative (SB366791), whereas the affinity of precursor 2 is comparable with that of DVV24. The introduction of a more hydrophobic fluoroethyl substituent at position 3 (compound 3) on the phenol in 1 drastically decreased the binding affinity for rTRPV1. For the ureas, methylation (6) of the secondary amine of 7 resulted in a 16-fold higher binding affinity for rTRPV1. The aminoquinazolines 16 and DVV54 showed Kd values in the low nanomolar range and thus have the highest affinity for rTRPV1 among the tested compounds. Replacement of the methyl ether in 16 with fluorine (DVV54) resulted in a 3-fold decrease in its binding affinity for rTRPV1 and a 4-fold decrease for hTRPV1.
These results demonstrate that small structural changes can lead to enormous shifts in binding affinity as well as functional potency (3 vs DVV24 and 7 vs 6). There were modest species differences in both Kd and Ki values, which are, among the tested compounds, most pronounced for the aminoquinazoline derivatives. Compounds had binding affinities for hTRPV1 3-fold higher to 5-fold lower than that of rTRPV1. Values for antagonistic potency ranged from 2-fold higher to 3-fold lower, respectively. The required affinity of a PET radioligand depends on the density of the target protein (Bmax) and should be at least 4-fold higher than the Bmax.28 Using enzyme-linked immunosorbent assays, TRPV1 protein concentrations in the rat spinal cord range from 0.42 to ∼1.05 pmol/mg of protein, whereas brain TRPV1 protein levels are at least 10–20-fold lower (0.016–0.042 pmol/mg of protein).29 The density of central TRPV1 channels is much lower than that of other brain receptors such as CB1 (highest-density regions, 0.084–0.209 pmol/mg of tissue)30 and the dopamine D2 receptor (striatum, 0.267 pmol/mg of tissue).31
Biodistribution Studies
The kinetics and tissue distribution of [11C]DVV24, [18F]DVV54, and 123I-RTX were studied in normal male Naval Medical Research Institute (NMRI) mice 2, 10, and 60 min post-tracer injection. The results of the biodistribution studies are presented in Figure 6, expressed as the percentage of injected dose (% ID) and standardized uptake value (SUV). [11C]DVV24 and 123I-RTX were efficiently cleared from blood (2 min/60 min ratios of 4.0 and 8.8, respectively), while [18F]DVV54 showed slower kinetics (2 min/60 min ratio of 1.9). All three tracers were cleared from plasma mainly through the hepatobiliary pathway as the decrease in liver uptake parallels the increase of radioactivity in the intestines. However, in the case of [11C]DVV24, 35% of the ID was eliminated via urinary excretion, which was also observed for [18F]DVV54 and 123I-RTX, albeit to a much lesser extent (≤10% ID). The total level of initial brain uptake of [11C]DVV24 was high, with 1.3% ID at 2 min post-injection (p.i.), and wash-out from brain was rapid (0.2% ID at 60 min p.i.). In the case of [18F]DVV54, only 0.4% ID was found in the brain at 2 min p.i., and the level of brain uptake of 123I-RTX was negligible, in accordance with their high clogD and PSA (123I-RTX) values. This limits their potential for in vivo mapping of the central TRPV1 receptors, yet from a pharmacological perspective, administration of the nonradioactive TRPV1 antagonists DVV54 and I-RTX is likely devoid of central effects.
Figure 6.

Tissue distribution of [11C]DVV24 (A), [18F]DVV54 (B), and 123I-RTX (C) in control NMRI mice (n = 3 or 4 per time point). Data are presented as the percentage of injected dose (% ID) ± the standard deviation (SD) and standardized uptake value (SUV) ± SD.
The retention of [11C]DVV24 in the trigeminal nerve [organ-to-blood radioactivity ratio (AR) of 2.7 at 60 min p.i.], a TRPV1-positive tissue, is prominent (Figure 7). Persistent binding of [11C]DVV24 was also observed in spleen, pancreas, lungs, skin, and bone tissue. Despite the high affinity of DVV54 and I-RTX for TRPV1, no retention of their radiolabeled analogues was observed in the trigeminal nerve (for [18F]DVV54, SUV = 0.21; for 123I-RTX, SUV = 0.08), or in the spleen, pancreas, or lungs. Given the high degree of homology of mouse and rat TRPV1, these results are unlikely to be explained in terms of interspecies differences. A more likely hypothesis is that [18F]DVV54 and 123I-RTX do not reach their target in sufficient concentration because of their lipophilic nature and extensive plasma protein binding. With regard to [18F]DVV54, an increase in radioactivity concentration in bone tissue was observed over time (AR = 3.3 at 60 min p.i.), probably caused by accumulation of [18F]F–, which indicates that [18F]DVV54 is susceptible to defluorination.
Figure 7.
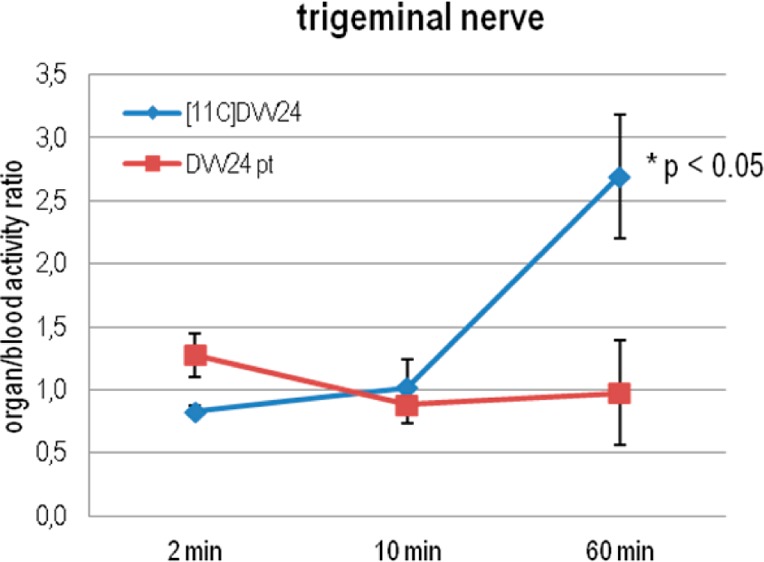
Trigeminal nerve-to-blood radioactivity ratios of [11C]DVV24 in control mice (blue) and mice pretreated with the nonradioactive reference DVV24 (10 mg/kg, 1 h before tracer injection, subcutaneous) at 2, 10, and 60 min p.i. of the tracer (n = 4 per time point). Data are expressed as means ± the standard deviation.
To determine whether retention of [11C]DVV24 in the different tissues is due to TRPV1 binding, biodistribution studies with [11C]DVV24 were performed in NMRI mice pretreated with DVV24 (Figure A.3 of the Supporting Information). Pretreatment of mice with DVV24 resulted in a significant decrease in the ARs (60 min p.i.) of the trigeminal nerve (Figure 7), pancreas, spleen, lungs, and bone tissue (Figure A.4 of the Supporting Information). These results are in accordance with those obtained with our previously developed cinnamic acid derivative [11C]SB36679118 and suggest specific binding of [11C]DVV24 to the TRPV1 receptor in the trigeminal nerve. As demonstrated for [11C]SB366791 using TRPV1–/– mice, the persistent binding of [11C]DVV24 observed in the pancreas, spleen, lungs, and bone tissue is presumably caused by non-TRPV1 binding or accumulation of radiometabolite(s). Nevertheless, it appears that [11C]DVV24 cannot be used for in vivo imaging of TRPV1 because of its relatively low TRPV1 binding affinity. Therefore, no further studies were performed to investigate its selectivity for TRPV1.
Radiometabolites
To evaluate the in vivo stability of [11C]DVV24, [18F]DVV54, and 123I-RTX, the fraction of radiometabolites in the plasma of NMRI mice was determined at 2, 10, or 60 min p.i. of the desired tracer. An overview of the results is provided in Table 3. Both [11C]DVV24 and 123I-RTX showed a rapid in vivo metabolism. At 10 min p.i. of [11C]DVV24, <10% of the recovered radioactivity was in the form of intact tracer, while in the case of 123I-RTX, approximately 25% of intact tracer was present in plasma after 60 min. [18F]DVV54 displayed high in vivo stability, with approximately 70% of the intact tracer at 60 min after injection of the tracer. Consequently, this may be translated into unfavorable target-to-background ratios. All detected radiometabolites were more polar than the intact tracers. In the case of [18F]DVV54, the observed accumulation of radioactivity in bone tissue in the biodistribution studies suggests that one of the detected radiometabolites is [18F]F–.
Table 3. Overview of the Fraction of the Plasma and Brain Radiometabolites Determined 2, 10, or 60 min after the Injection of [11C]DVV24, [18F]DVV54, or 123I-RTX in NMRI Mice (n = 2 per time point)a.
| % intact tracer in plasma |
% intact tracer
in brain |
||||
|---|---|---|---|---|---|
| 2 min p.i. | 10 min p.i. | 60 min p.i. | 2 min p.i. | 10 min p.i. | |
| [11C]DVV24 | 34.3/43.4 | 7.5/8.8 | – | 86.6/87.5 | 52.2/58.3 |
| [18F]DVV54 | 93.9/95.1 | – | 64.7/71.7 | – | – |
| 123I-RTX | 84.2/86.3 | – | 24.3/26.4 | – | – |
Data are presented as the percentage of intact tracer relative to total recovered activity.
Because [11C]DVV24 efficiently crossed the blood–brain barrier, radiometabolites in brain of NMRI mice were quantified at 2 and 10 min p.i. of this tracer (Table 3). The percentage of intact tracer in brain tissue decreased from approximately 90% at 2 min p.i. to approximately 55% at 10 min p.i. Also in this case, the detected radiometabolites were more polar than the parent tracer.
Conclusions
We have efficiently synthesized two new TRPV1 PET radioligands, [11C]DVV24 and [18F]DVV54, in good yields, with high radiochemical purity and high specific activity. Additionally, the 123I-radiolabeled analogue of the potent TRPV1 antagonist I-RTX was efficiently synthesized. Preliminary biological data of [11C]DVV24 were promising, and the tracer showed TRPV1-specific retention in the trigeminal nerve. However, its application for in vivo mapping of TRPV1 in brain is less likely because of its experimentally observed relatively low binding affinity for TRPV1 and the presence of a large fraction of radiometabolites in brain. However, these data do suggest that this class of cinamic acid derivatives may be used in vivo as TRPV1 antagonists, although they are rapidly metabolized. Despite their high binding affinity for TRPV1, ex vivo, [18F]DVV54 and 123I-RTX showed no retention in the trigeminal nerve and negligible brain uptake probably because of their lipophilic nature. These experimentally observed biological characteristics limit their potential to visualize TRPV1 receptors in vivo. In the future, less lipophilic TRPV1 PET radioligands with higher affinity will need to be developed to allow in vivo imaging of TRPV1. Nevertheless, the explicit species differences and the low TRPV1 protein density under normal physiological conditions are significant hurdles in the evaluation of PET radioligands that target TRPV1.
Methods
Chemicals and General Instrumentation
All reagents and solvents were purchased from TCI Europe (Zwijndrecht, Belgium), Acros Organics (Geel, Belgium), or Fisher Scientific (Erembodegem, Belgium) and used without further purification. [123I]NaI was purchased from GE Healthcare (Diegem, Belgium), and Pierce iodination tubes were obtained from Thermo Scientific (Erembodegem, Belgium). Capsaicin was obtained from Calbiochem (now EMD Chemicals, San Diego, CA). Resiniferatoxin (RTX) was purchased from LC Laboratories (Woburn, MA) or Sigma-Aldrich (Bornem, Belgium). [3H]RTX (1 GBq/mmol) was obtained from Perkin-Elmer (Boston, MA) through a licensing agreement. Dulbecco’s modified Eagle’s medium (DMEM, 1×), Dulbecco’s phosphate-buffered saline (DPBS, 1×), Ham’s F-12 medium, and Geneticin were purchased from Invitrogen (Grand Island, NY). Tetracycline was purchased from EMD Chemicals. Fetal bovine serum was obtained from Gemini Bio-Products (West Sacramento, CA), and bovine serum albumin (Cohn fraction V) was obtained from Sigma-Aldrich (St. Louis, MO). Bovine glycoprotein (fraction VI) and [45Ca]CaCl2 were purchased from MP Biomedicals (Solon, OH).
For ascending thin layer chromatography (TLC), precoated aluminum-backed plates [silica gel 60 with fluorescent indicator UV 254 nm, 0.2 mm thickness (Macherey-Nagel, Düren, Germany)] were used. Column chromatography was performed using silica gel [silica 60–200, 60 Å (Acros, Geel, Belgium)]. Mass measurements were perfomed on an ultra-high resolution time-of-flight mass spectrometer [maXis impact LC/MS (Bruker, Bremen, Germany)] equipped with an orthogonal electrospray ionization (ESI) interface. Acquisition and processing of data were conducted using HyStar and Compass DataAnalysis (version 3.2, Bruker), respectively. Calculated monoisotopic mass values were obtained using ChemCalc (http://www.chemcalc.org). 1H nuclear magnetic resonance (NMR) and 13C NMR spectra were acquired with 400 and 500 MHz Bruker (Fällanden, Switzerland) Avance II spectrometers, respectively (5 mm probe). Chemical shifts are reported in parts per million relative to tetramethylsilane (δ = 0). Coupling constants are reported in hertz. Splitting patterns are defined by s (singlet), d (doublet), t (triplet), or m (multiplet). Calculated logD and PSA values were obtained using Marvinsketch version 5.7.0 and the calculator from Daylight (http://www.daylight.com/meetings/emug00/Ertl/tpsa.html), respectively. HPLC analysis was performed on a Merck Hitachi L2130 LaChrom Elite pump (Hitachi, Tokyo, Japan) connected to an L2400 LaChrom Elite UV spectrometer (Hitachi). Radioactivity in the HPLC effluent was monitored by a 3 in. NaI(Tl) scintillation detector connected to a single-channel analyzer [GABI box (Raytest, Straubenhardt, Germany)]. Data were acquired and analyzed using a RaChel (Lablogic, Sheffield, U.K.) or GINA Star (Raytest) data acquisition system. Quantification of radioactivity during biodistribution and metabolite studies was performed using an automated gamma counter equipped with a 3 in. NaI(Tl) well crystal coupled to a multichannel analyzer, mounted in a sample changer [Wallac 1480 Wizard 3q (Wallac, Turku, Finland)]. The values are corrected for background radiation, physical decay, and counter dead time.
All animal experiments were performed in compliance with the principles set by the Belgian law relating to the conduct of animal experimentation, after approval from the university animal ethics committee. Animals were housed in individually ventilated cages in a thermoregulated (22 °C), humidity-controlled facility under a 12 h light/12 h dark cycle, with free access to food and water.
Synthesis
N-(3-Hydroxyphenyl)-4-(trifluoromethyl)cinnamide (2)
The title compound was synthesized as described previously:18 290 mg, 0.94 mmol, 64% yield; TLC (1:1 heptane/EtOAc) Rf = 0.47; HPLC (0.05 M CH3CN/NH4OAc, pH 5.5, 65:35 v/v; 1 mL/min) tR = 5.3 min (>99%); HR-LCMS (ESI) calcd for C16H13F3NO2 (MH+) 308.0898, measured 308.0904; 1H NMR (400 MHz, MeOD-d4) δ 6.56–6.61 (1H, m, H4Ar–OH), 6.90 (1H, d, J = 15.7, -CHCHCO-), 7.04–7.08 (1H, m, H6Ar–OH), 7.12–7.18 (1H, m, H5Ar–OH), 7.30 (1H, br s, -CONH-), 7.67–7.80 [4H, m (overlapping peaks), H2Ar–CF3, H6Ar–CF3, H3Ar–CF3, H5Ar–CF3, and -CHCHCO-]; 13C NMR (500 MHz, MeOD-d4) δ 108.4, 112.4, 112.5, 125.3, 126.7, 126.8, 126.9, 129.4, 130.6, 132.1, 132.4, 140.1, 140.7, 140.9, 159.0, 165.8
N-[3-(2-Fluoroethoxy)phenyl]-4-chlorocinnamide (3)
2-Fluoroethyl tosylate (87 mg, 0.4 mmol) and Cs2CO3 (195 mg, 0.6 mmol) were added to a solution of 1 (109 mg, 0.4 mmol) in DMF (2 mL). After being stirred for 5 h at 120 °C, the reaction mixture was allowed to cool to room temperature and diluted with water (2 mL). The mixture was extracted with CH2Cl2. The organic phase was washed with 1 M NaOH, water, and brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a mixture of heptane and EtOAc (2:1 v/v) as eluent to afford 3 as a beige solid (95 mg, 0.3 mmol): 74% yield; TLC (1:1 heptane/EtOAc) Rf = 0.56; HPLC (XTerra RP-C18, 5 μm, 4.6 mm × 250 mm; 0.05 M CH3CN/NH4OAc, pH 5.5, 65:35 v/v; 1 mL/min) tR = 7.6 min (>98%); HR-LCMS (ESI) Calcd for C17H16ClFNO2 (MH+) 320.0854, measured 320.0854; 1H NMR (400 MHz, MeOD-d4) δ 4.60–4.84 (2H, m, -CH2CH2F), 5.25–5.37 (2H, m, -CH2CH2F), 7.31 (1H, m, H4Ar–O), 7.79–8.17 [9H, m (overlapping peaks), -CHCHCO-, H2Ar–Cl, H6Ar–Cl, H3Ar–Cl, H5Ar–Cl, H2Ar–O, H5Ar–O, and H6Ar–O].
N-(3-Methoxyphenyl)-4-(trifluoromethyl)cinnamide (DVV24)
EDCI (1.49 g, 7.76 mmol) and HOBt (1.05 g, 7.76 mmol) were added to a solution of 4-(trifluoromethyl)cinnamic acid (1.5 g; 6.9 mmol) in DMF (50 mL). After the mixture had been stirred for 20 min at room temperature, 3-methoxyaniline (0.96 g, 7.76 mmol) was added to the reaction mixture and stirring was continued for 16 h at room temperature. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in EtOAc, washed with 1 M HCl and water, dried over MgSO4, filtered, and again concentrated under reduced pressure. The residue was purified by column chromatography on basic alumina eluted with EtOAc. The solvent was removed under reduced pressure, and the desired product was crystallized from diethyl ether (yellowish crystals, 199 mg, 0.62 mmol): 9% yield; TLC (1:1 heptane/EtOAc) Rf = 0.65; HPLC (XTerra RP-C18, 5 μm, 4.6 mm × 250 mm; 0.05 M CH3CN/NH4OAc, pH 5.5, 65:35 v/v; 1 mL/min) tR = 8.1 min (>99%); HR-LCMS (ESI) calcd for C17H15F3NO2 (MH+) 322.1055, measured 322.1059; 1H NMR (400 MHz, MeOD-d4) δ 4.62 (3H, s, -OCH3), 6.58–6.62 (1H, m, H4Ar-OCH3), 6.78 (1H, d, J = 15.7, -CHCHCO-), 7.06 (1H, d, J = 8.0, H6Ar-OCH3), 7.10–7.16 (1H, m, H5Ar-OCH3), 7.30 (1H, br s, -CONH-), 7.57–7.69 [4H, m (overlapping peaks), H2Ar–CF3, H6Ar–CF3, H3Ar–CF3, H5Ar–CF3, and -CHCHCO-]; 13C NMR (500 MHz, MeOD-d4) δ 55.7, 107.1, 111.0, 113.4, 125.2, 126.7, 126.8, 126.9, 129.4, 130.6, 132.2, 140.0, 140.8, 141.0, 161.6, 165.9 (one Ar peak obscured).
N-Methyl-N-(4-methylphenyl)ethylenediamine (4) and N-(4-Methylphenyl)ethylenediamine (5)
A solution of N-methyl-p-toluidine (4.00 g, 33 mmol; for 4) or p-toluidine (3.54 g, 33 mmol; for 5) and 2-bromoethylamine hydrobromide (3.38 g, 16.5 mmol) in toluene (50 mL) was refluxed for 24 h. After the mixture had cooled to room temperature, the solvent was removed under reduced pressure. The residue was suspended in diethyl ether (50 mL) and washed with an aqueous solution of 20% potassium carbonate. The organic layer was dried over MgSO4 and filtered, and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using a mixture of MeOH and CH2Cl2 (1:9, v/v) as the eluent, yielding 4. After purification with silica gel column chromatography, compound 5 was precipitated using 1.25 M HCl in MeOH and obtained as white crystals. 4: 406 mg, 2.47 mmol, 15% yield. 5: 446 mg, 2.00 mmol, 12% yield.
N-(2-Bromophenyl)-N′-{2-[N″-methyl-N″-(4-methylphenyl)amino]ethyl}urea (6)
To a solution of 4 (406 mg, 2.47 mmol) in CH2Cl2 (3 mL) was added a solution of 2-bromophenyl isocyanate (509 mg, 2.57 mmol) in CH2Cl2 (2 mL), and the mixture was stirred for 1 h at room temperature. The precipitate was filtered off, washed with diethyl ether, and dried in a vacuum oven to yield 430 mg (1.19 mmol) of 6 as a white powder: 48% yield; TLC (95:5 CH2Cl2/MeOH) Rf = 0.83; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 40:60 v/v; 0.8 mL/min) tR = 11.7 min (>99%); HR-LCMS (ESI) calcd for C17H21BrN3O (MH+) 362.0868, measured 362.0869; 1H NMR (400 MHz, MeOD-d4) δ 2.19 (3H, s, -Ar-CH3), 2.92 (3H, s, -N-CH3), 3.33–3.39 (2H, m, -CH2-), 3.39–3.45 (2H, m, -CH2-), 6.70 (2H, d, J = 8.6, H2Ar–CH3 and H6Ar–CH3), 6.92 (1H, td, J = 1.5 and 7.4, H4Ar–Br), 6.97 (2H, d, J = 8.2, H3Ar–CH3 and H5Ar–CH3), 7.26 (1H, td, J = 1.5 and 7.4, H5Ar–Br), 7.51 (1H, dd, J = 1.4 and 8.0, H3Ar–Br), 7.87 (1H, dd, J = 1.5 and 8.2, H6Ar–Br); 13C NMR (500 MHz, DMSO-d6) δ 20.0, 36.6, 38.3, 52.0, 112.3, 112.6, 116.1, 122.0, 123.4, 124.4, 128.0, 129.6, 132.4, 137.9, 147.2, 155.1 (one Ar peak obscured).
N-(2-Bromophenyl)-N′-{2-[N″-(4-methylphenyl)amino]ethyl}urea (7)
To a solution of 5 (4.46 mg, 2.00 mmol) in CH2Cl2 (3 mL) were added NEt3 (700 mg, 5.00 mmol) and a solution of 2-bromophenyl isocyanate (407 mg, 2.06 mmol) in CH2Cl2 (2 mL). After being stirred for 1 h at room temperature, the reaction mixture was concentrated by vacuum evaporation, and the residue was partitioned between water and CH2Cl2. The organic layer was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. Finally, the desired product (310 mg, 0.89 mmol) was crystallized from diethyl ether: 44% yield; TLC (95:5 CH2Cl2/MeOH) Rf = 0.76; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 40:60 v/v; 0.8 mL/min) tR = 6.7 min (>99%); HR-LCMS (ESI) calcd for C16H19BrN3O (MH+) 348.0712, measured 348.0714; 1H NMR (400 MHz, MeOD-d4) δ 2.18 (3H, s, Ar-CH3), 3.23 (2H, t, J = 6.2, -CH2-), 3.31 (1H, br s, -NH-), 3.40 (t, 2H, J = 6.3, -CH2-), 6.59 (2H, d, J = 8.4, H2Ar–CH3 and H6Ar–CH3), 6.92 (2H, d, J = 8.3, H3Ar–CH3 and H5Ar–CH3), 7.27 (1H, td, J = 1.4 and 8.5, H5Ar–Br), 7.52 (1H, dd, J = 1.3 and 8.0, H3Ar–Br), 7.91 (1H, dd, J = 1.4 and 8.2, H6Ar–Br); 13C NMR (500 MHz, MeOD-d4) δ 9.2, 20.5, 40.5, 45.4, 47.9, 114.3, 124.1, 125.3, 127.4, 129.0, 130.6, 130.7, 133.6, 138.6, 147.6, 158.1.
2-p-Tolyl-3-(trifluoromethyl)pyridine (8)
Palladium tetrakis(triphenylphosphine) [Pd(PPh3)4, 3.24 g, 2.8 mmol] was added to a degassed mixture of 2-chloro-3-(trifluoromethyl)pyridine (12.72 g, 70.1 mmol), 4-methylphenylboronic acid (9.60 g, 70.6 mmol), and 2 M aqueous Na2CO3 (87 mL, 174 mmol) in dimethyl ether (DME) (200 mL). The mixture was kept under nitrogen and stirred for 16 h at 80 °C. After the mixture had cooled to room temperature, DME was removed by vacuum evaporation. The residue was extracted with EtOAc (2 × 250 mL). The organic layer was dried over MgSO4 and filtered, and the solution was concentrated under reduced pressure. The residue was applied on a silica gel column eluted with a heptane/EtOAc mixture (3:1 v/v), yielding 8 as a yellow oil (11 g, 46.4 mmol, 66% yield).
2-(4-Methyl-3-nitrophenyl)-3-(trifluoromethyl)pyridine (9)
Concentrated nitric acid (HNO3, 10 mL) was added dropwise to a solution of 8 (10.48 g, 44.2 mmol) in concentrated H2SO4 (32 mL). After being stirred for 1 h at room temperature, the mixture was poured carefully into an ice/water mixture (200 mL) and extracted with EtOAc (2 × 300 mL). The acidic organic layers were collected and neutralized with 1 M NaOH. Next, the EtOAc layer was dried over MgSO4 and filtered, and the solvent was evaporated under reduced pressure. The residue was dried in a vacuum oven overnight to yield 9 as an ochre powder. The crude product was used as such in the next step without further purification.
2-Nitro-4-[3-(trifluoromethyl)pyridin-2-yl]benzoic Acid (10)
KMnO4 (24.8 g, 157 mmol) was added to a solution of 9 (12.74 g, 44.2 mmol) in a pyridine/water mixture (2:1 v/v, 90 mL), and the mixture was stirred for 4 h at 110 °C. Next, a second amount of KMnO4 (24.8 g, 157 mmol) and 60 mL of water were added to the mixture, and stirring was continued overnight at 110 °C. After cooling to room temperature, the solution was poured over Celite, and the pyridine was removed from the mixture under reduced pressure. The residue was diluted with water, filtered, and washed with EtOAc. Next, the aqueous solution was acidified with 2 M HCl to pH 2. The formed precipitate was collected as a white solid and dried in a vacuum oven overnight to afford 10 (6.9 g, 22.1 mmol): 50% yield (relative to 8).
2-Nitro-4-[3-(trifluoromethyl)pyridin-2-yl]benzamide (11)
A solution of 10 (6.24 g, 20.0 mmol) in SOCl2 (12.5 mL) was refluxed for 4 h at 100 °C. After the solution had cooled to room temperature, toluene (50 mL) was added, and the solvent was removed by vacuum evaporation. This step was repeated twice. The residue was dissolved in CH2Cl2 (100 mL), the solution cooled in an ice bath, and NH3 gas passed through the solution for 30 min. Thereafter, the solution was stirred for 15 min at room temperature and concentrated under reduced pressure. The solid was washed with water and dried in a vacuum oven overnight to yield 11 as a beige solid (6.22 g). The product was used as such in the next step without further purification.
2-Amino-4-[3-(trifluoromethyl)pyridin-2-yl]benzamide (12)
The crude product 11 (6.22 g, 20.0 mmol) was added to a suspension of tin chloride dihydrate (SnCl2 2aq, 13.5 g, 60.0 mmol) in EtOH (200 mL), and the reaction mixture was refluxed for 2 h at 80 °C. The solvent was evaporated and the residue dissolved in EtOAc. Next, the organic phase was washed with 1 M NaOH, water, and brine and dried over MgSO4. After filtration, the solvent was evaporated under reduced pressure to afford the desired compound 12 (3.71 g, 13.2 mmol) as a light yellow powder: 66% yield (relative to 10).
2-Chloromethyl-7-[3-(trifluoromethyl)pyridin-2-yl]-3H-quinazolin-4-one (13)
Compound 12 (3.70 g, 13.2 mmol) was dissolved in 2-chloro-1,1,1-trimethoxyethane (10 mL, 74.2 mmol), and the solution was stirred for 4 h at 130 °C. After cooling to room temperature, the solution was concentrated under reduced pressure to yield 13 (2.58 g), which was used as such in the next step without further purification.
4-Chloro-2-(chloromethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazoline (14)
Phosphorus oxychloride (POCl3, 3.79 mL, 40.7 mmol) was added dropwise to a solution of 13 (2.58 g, 7.58 mmol) in CH2Cl2 (75 mL). After the mixture had refluxed for 1 h at 80 °C and cooled to room temperature, a second amount of POCl3 (3.79 mL, 40.7 mmol) was added to the reaction mixture. This handling was repeated once more, and the reaction mixture was refluxed for 16 h at 80 °C. The reaction mixture was allowed to cool to room temperature and the solvent removed under reduced pressure. The residue was partitioned between EtOAc and a saturated NaHCO3 solution. The organic layer was collected and the aqueous layer extracted with EtOAc. The organic layers were combined, washed with brine, dried over MgSO4, and filtered. The solvent was evaporated under a vacuum and the residue purified by silica gel column chromatography eluted with an EtOAc/heptane mixture (1:1 v/v) to afford 14 (1.82 g, 5.08 mmol) as a light yellow solid: 38% yield (relative to 12).
{2-Chloromethyl-7-[3-(trifluoromethyl)pyridin-2-yl]quinazolin-4-yl}[4-(trifluoromethyl)phenyl]amine (15) and {2-(Benzyloxymethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazolin-4-yl}[4-(trifluoromethyl)phenyl]amine (19)
Compound 14 (1.82 g, 5.08 mmol) or 18 (260 mg, 0.61 mmol) was added to a solution of 4-(trifluoromethyl)aniline (825 mg, 5.12 mmol for 15; 107 mg, 0.67 mmol for 19) in 2-propanol (45 mL) or CH3CN (6 mL), respectively. The reaction mixture was stirred for 4 h at 75 °C (for 15) or 2 h at 80 °C (for 19). The reaction mixture was cooled to room temperature and the precipitate filtered off and washed with 2-propanol (only for 15) and diethyl ether. The residue was dried in a vacuum oven overnight, yielding 15 (1.8 g, 3.46 mmol, 68% yield) or 19 (90 mg, 0.16 mmol, 26% yield).
15: TLC (1:2 heptane/EtOAc and 1% NEt3) Rf = 0.49; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 50:50 v/v; 0.8 mL/min) tR = 11.6 min (>98%); HR-LCMS (ESI) calcd for C22H14ClF6N4 (MH+) 483.0811, measured 483.0820; 1H NMR (400 MHz, DMSO-d6) δ 4.82 (2H, s, -CH2Cl), 7.73–7.85 [4H, m (overlapping peaks), H4′, H5′, H2″, and H6″], 7.95 (1H, s, H8), 8.20 (2H, d, J = 8.6, H3″ and H5″), 8.39 (1H, d, J = 8.1, H5), 8.90 (1H, d, J = 8.6, H6), 8.97 (1H, d, J = 4.3, H6′); 13C NMR (500 MHz, DMSO-d6) δ 44.7, 113.4, 122.6, 122.8, 123.3, 123.6, 123.7, 123.9, 124.0, 124.6, 125.8, 125.9, 128.5, 135.6, 135.7, 141.5, 143.4, 145.3, 152.9, 155.2, 159.2, 160.3.
{2-(Methoxymethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazolin-4-yl}[4-(trifluoromethyl)phenyl]amine (16)
Sodium methanolate (NaOCH3, 432 mg, 8.0 mmol) was added to a solution of 15 (190 mg, 0.4 mmol) in MeOH (15 mL). After the mixture had been stirred for 1 h at 60 °C, metallic sodium (220 mg, 9.57 mmol) was added carefully to the cooled reaction mixture, and stirring was continued for 16 h at 60 °C. The solvent was removed under reduced pressure and the residue dissolved in EtOAc. The organic phase was washed with water and brine, dried over MgSO4, and filtered, and the EtOAc was removed by vacuum evaporation. Finally, the residue was dissolved in diethyl ether, and after a solution of 4 M HCl in dioxane (1 mL) had been added, 16 was obtained as a brownish precipitate (66 mg, 0.13 mmol): 35% yield; TLC (1:2 heptane/EtOAc and 1% NEt3) Rf = 0.32; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 45:55 v/v; 0.8 mL/min) tR = 9.7 min (>96%); HR-LCMS (ESI) calcd for C23H17F6N4O (MH+) 479.1307, measured 479.1311; 1H NMR (400 MHz, MeOD-d4) δ 3.53 (3H, s, -OCH3), 4.65 (2H, s, -CH2OCH3), 7.65–7.78 (3H, m, H4′, H2″, and H6″), 7.86–7.93 (3H, m, H5′, H3″, and H5″), 8.11 (1H, s, H8), 8.29 (1H, d, J = 7.8, H5), 8.63 (1H, d, J = 8.4, H6), 8.85 (1H, d, J = 4.4, H6′); 13C NMR (500 MHz, MeOD-d4) δ 60.1, 72.5, 114.6, 120.8, 125.2, 125.4, 126.0, 126.2, 126.5, 127.1, 127.2, 130.8, 136.9, 137.0, 139.9, 141.2, 148.2, 148.3, 153.7, 156.4, 161.5, 164.7 (one Ar peak obscured).
{2-(Fluoromethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazolin-4-yl}[4-(trifluoromethyl)phenyl]amine (DVV54)
In a pressure vial, compound 15 (100 mg, 0.19 mmol) was dissolved in a solution of 1 M TBAF in THF (207 μL). The reaction vial was sealed and the solution heated for 5 min at 150 °C. A fraction of DVV54 was purified from the mixture by RP-HPLC on a preparative XTerra column (Waters, 10 mm × 250 mm, 10 μm) eluted with a mixture of CH3CN/H2O (60:40 v/v); TLC (1:2 heptane/EtOAc and 1% NEt3) Rf = 0.42; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 45:55 v/v; 0.8 mL/min) tR = 11.0 min (>98%); HR-LCMS (ESI) calcd for C22H14F7N4 (MH+) 467.1107, measured 467.1116; 1H NMR (400 MHz, CDCl3) δ 5.53 (2H, d, J = 46.8, -CH2F), 7.24 (1H, s, H8), 7.50–7.73 [4H, m (overlapping peaks), H4′, H5′, H2″, and H6″], 7.99–8.15 [4H, m (overlapping peaks), H5, H6, H3″, and H5″], 8.89 (1H, d, J = 4.6, H6′).
2-(Benzyloxymethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]-3H-quinazolin-4-one (17)
A solution of benzyloxy acetic acid (0.856 g, 5.16 mmol) in heptane (20 mL) was cooled to 0 °C. Oxalyl chloride (1.80 g, 14.2 mmol) and DMF (1 drop) were added to the cooled solution, and the mixture was stirred for 1 h at 0 °C. The solvent was removed under reduced pressure and the crude acid chloride dissolved in dry THF (10 mL). In a separate bowl, 12 (1.32 g, 4.69 mmol) was dissolved in a mixture of dry THF (25 mL) and pyridine (416 μL, 5.16 mmol) and the solution cooled to 0 °C. The solution of the crude acid chloride was added dropwise to the second solution and the mixture allowed to warm to room temperature. After the mixture had been stirred for 1 h at room temperature, an aqueous solution of 10% NaOH was added, and stirring was continued for 1 h. Next, the mixture was concentrated under reduced pressure (to approximately 10 mL), diluted with an equal volume of water, and acidified to pH 2 with concentrated HCl. The resulting solution was extracted with EtOAc (3 × 30 mL); the EtOAc layers were collected and washed with brine. After the mixture had been dried over MgSO4, the solvent was removed under reduced pressure to yield 17 as a brownish oil (510 mg, 1.19 mmol): 24% yield.
4-Chloro-2-(benzyloxymethyl)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazoline (18)
POCl3 (279 μL, 2.99 mmol) was added dropwise to a solution of 17 (490 mg, 1.19 mmol) in a mixture of CHCl3 (7 mL) and 2,6-lutidine (386 mg, 3.60 mmol). The reaction mixture was refluxed for 18 h at 70 °C. After the mixture had cooled to room temperature, the solvent was removed under reduced pressure and the residue partitioned between EtOAc (50 mL) and saturated NaHCO3 (50 mL). The organic layer was collected and the aqueous layer extracted once more with EtOAc (2 × 50 mL). The EtOAc layers were combined, washed with brine, dried over MgSO4, and filtered, and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography eluted with an EtOAc/heptane (mixture 1:1 v/v) to afford 18 (260 mg, 0.61 mmol) in 51% yield.
2-{4-(4-Trifluoromethylphenylamino)-7-[3-(trifluoromethyl)pyridin-2-yl]quinazolin-2-yl}methanol (20)
Compound 19 (50 mg, 0.11 mmol) was dissolved in EtOH (10 mL), and 10 wt % Pd/C (25 mg) was added. The mixture was hydrogenated at room temperature for 62 h using a balloon filled with H2 gas. The mixture was filtered, the filter washed with EtOH, and the solvent removed under reduced pressure. The residue was applied to a silica gel column and eluted with a heptane/EtOAc mixture (1:5 v/v) and 0.5% NEt3, yielding 20 (11 mg, 0.024 mmol): 22% yield; TLC (1:2 heptane/EtOAc and 1% NEt3) Rf = 0.26; HPLC (XBridge RP-C18, 3.5 μm, 3.0 mm × 100 mm; 0.05 M CH3CN/NaOAc, pH 5.5, 45:55 v/v; 0.8 mL/min) tR = 5.5 min (>95%); 1H NMR (400 MHz, MeOD-d4) δ 4.85 (2H, s, -CH2OH); 7.67–7.71 (4H, m, H4′, H5′, H2″, and H6″), 7.96 (1H, s, H8), 8.18 (2H, d, J = 8.9, H3″ and H5″), 8.34 (1H, d, J = 8.0, H5), 8.53 (1H, d, J = 8.4, H6), 8.89 (1H, d, J = 4.0, H6′).
Radiosynthesis
Carbon-11, in the form of [11C]CH4, and fluorine-18 (as [18F]F–) were produced by 14N(p,α)11C and 18O(p,n)18F nuclear reactions, respectively, in a Cyclone 18/9 cyclotron (IBA, Louvain-la-Neuve, Belgium). [11C]CH4 was converted to [11C]CH3I, in a home-built gas phase recirculation module, and trapped on a Porapak N column (divinylbenzene/vinylpyrrolidone polymer). Upon warming, the [11C]CH3I was transferred to the reaction vial in a stream of helium for nucleophilic substitution with the desired precursor compounds. For radiofluorination, [18F]F– was concentrated on an anion-exchange QMA column and eluted with a K222/K2CO3 mixture in a CH3CN/H2O mixture (95:5 v/v) into the reaction vial. After evaporation of the solvent at 110 °C with a stream of helium, anhydrous CH3CN was added and the mixture heated at 110 °C under a stream of helium until the reaction vial was dry. Next, a solution of 15 (1 mg/0.5 mL) in DMF was added to the reaction vial and the reaction mixture heated for 15 min at 150 °C for nucleophilic substitution. For radiosynthesis of 123I-RTX, 200 μL of a solution of RTX in DMSO (1 mg/mL) was added to a Pierce iodination tube containing 300 μL of a 0.1 M phosphate buffer (pH 6.5). Next, Na123I (∼185 MBq) was added and the reaction mixture held for 15 min at room temperature. Purification and quality control of 123I-RTX were performed on an XTerra column [RP-C18, 5 μm, 7.8 mm × 150 mm (Waters)] eluted with a CH3CN/H2O mixture and 0.1% TFA (70:30 v/v) at a flow rate of 1 mL/min (tR = 8 min for RTX; tR = 11 min for 123I-RTX). [11C]DVV24 was purified on an XTerra column [RP-C18, 5 μm, 7.8 mm × 150 mm (Waters)] eluted with an EtOH/0.05 M NH4OAc mixture (pH 5.5) (65:35 v/v) at a flow rate of 1.5 mL/min (tR = 10 min). [18F]DVV54 was isolated after injection on an XBridge column (RP-C18, 5 μm, 4.6 mm × 150 mm) eluted with a CH3CN/0.05 M NaOAc mixture (pH 5.5) (50:50 v/v) at a flow rate of 1 mL/min (tR = 18 min). UV detection of the HPLC eluates was performed at 254 nm. The isolated 123I-RTX and [18F]DVV54 solutions were loaded on a Sep-Pak cartridge [C18 light (Waters)]; the cartridge was washed with saline, and the tracers were eluted from the cartridge with absolute ethanol (0.5 mL). The radioactivity of the isolated radiochemical solutions was determined with a dose calibrator, and samples were diluted with saline ([11C]DVV24 and 123I-RTX) or with a solution of 0.5% Tween 80 in saline ([18F]DVV54), yielding an ethanol concentration of <10%, suitable for intravenous injection. Quality control of [11C]DVV24 was performed using an HPLC system with an XTerra column [RP-C18, 5 μm, 4.6 mm × 250 mm (Waters)] eluted with a CH3CN/0.05 M NH4OAc mixture (pH 5.5) (65:35 v/v) at a flow rate of 1 mL/min and UV detection at 273 nm (tR = 8 min). Analysis of [18F]DVV54 was performed on an XBridge column [RP-C18, 3.5 μm, 3.0 mm × 100 mm (Waters)] eluted with a CH3CN/0.05 M NaOAc mixture (pH 5.5) (45:55 v/v) at a flow rate of 0.8 mL/min and UV detection at 228 nm (tR = 11 min).
Biodistribution Studies
Male NMRI mice (body weight of 34–48 g) were anesthetized with pentobarbital [60 mg/kg intraperitoneally (ip)] and injected with [11C]DVV24 (∼9.25 MBq), [18F]DVV54 (∼1.11 MBq), or 123I-RTX (∼0.37 MBq) intravenously (iv) via a lateral tail vein. For the blocking experiment, mice were pretreated with DVV24 (10 mg/kg, subcutaneously) 1 h before the injection of [11C]DVV24. The mice were sacrificed by decapitation at 2, 10, or 60 min p.i. (n = 3 or 4 per time point) and dissected, and blood, organs, and other body parts were collected in tared tubes. The radioactivity in each tube was measured using an automated gamma counter, and the tubes containing selected organs and blood were weighed. For the calculation of total blood radioactivity, the blood mass was assumed to be 7% of the body mass. The SUV values were calculated as (radioactivity in counts per minute in organ/weight of the organ in grams)/(total counts recovered/body weight in grams).
Plasma Radiometabolites
NMRI mice were anesthetized with pentobarbital (60 mg/kg, ip) and injected iv with [11C]DVV24 (∼9.25 MBq), [18F]DVV54 (∼16.65 MBq), or 123I-RTX (∼2.22 MBq) via a lateral tail vain. The mice were decapitated at 2, 10, or 60 min p.i. (n = 2 per time point) of the tracer, and blood was collected into lithium heparin-containing tubes [4.5 mL lithium heparin PST tubes, BD Vacutainer (BD, Franklin Lakes, NJ)]. After centrifugation (3000 rpm for 10 min) of the blood, plasma was isolated and stored on ice. Because extensive binding of I-RTX to plasma proteins has been reported,32 the plasma proteins in the 123I-RTX-containing plasma samples were precipitated by the addition of CH3CN (same volume as the collected plasma). The mixture was vortexed and centrifuged for 10 min and the supernatant collected and stored on ice. The plasma and supernatant were analyzed by RP-HPLC on a Chromolith column [RP-C18, 3 mm × 100 mm (Merck)] eluted with gradient mixtures of CH3CN (A) and 0.05 M NaOAc (pH 5.5) (B). The nonradioactive reference compounds (20 μg) were co-injected on the Chromolith column to assess the retention time of the intact parent tracer. After passing through a UV detector coupled in series with a 3 in. NaI(Tl) scintillation detector, connected to a single-channel analyzer, the HPLC eluate was collected as 0.5 or 1 mL fractions (model 2110 fraction collector, Bio-Rad, Hercules, CA). The radioactivity in each fraction was measured using an automated gamma counter. The recovery from the HPLC- and Chromolith column-injected radioactivity was 87, 111.5, and 95% (n = 4) for [11C]DVV24, [18F]DVV54, and [123I]-RTX, respectively.
[11C]DVV24 (tR = 10 min): 0 to 4 min isocratic 100% B, 0.5 mL/min; 4 to 9 min linear gradient from 100 to 10% B, 1 mL/min; 9 to 12 min isocratic 10% B, 1 mL/min; 12 to 15 min isocratic 100% B, 0.5 mL/min. [18F]DVV54 (tR = 15 min): 0 to 5 min isocratic 100% B, 0.5 mL/min; 5 to 15 min linear gradient from 100 to 10% B, 1 mL/min; 15 to 18 min isocratic 10% B, 1 mL/min; 18 to 20 min isocratic 100% B, 0.5 mL/min. 123I-RTX (tR = 14 min): 0 to 5 min isocratic 100% B, 0.5 mL/min; 5 to 12 min linear gradient from 100 to 10% B, 1 mL/min; 12 to 19 min isocratic 10% B, 1 mL/min; 19 to 20 min linear gradient from 10 to 100% B, 0.5 mL/min; 20 to 25 min isocratic 100% B, 0.5 mL/min.
Brain Radiometabolites
For each studied time point, two NMRI mice were injected with [11C]DVV24 (∼9.25 MBq iv). The mice were sacrificed by an overdose of pentobarbital (Nembutal, CEVA Santé Animale; 150 mg/kg ip) at 2 or 10 min p.i. of the tracer and perfused by injection of saline into the left ventricle until the liver turned pale. Brain was isolated and homogenized in a mixture of CH3CN and water (4 mL, 1:1 v/v). After centrifugation for 10 min at 3000 rpm, the supernatant was collected, diluted with CH3CN (1 mL), and filtered through a 0.22 μm filter (Millipore, Bedford, MA). The filtrate (0.5–1 mL) was spiked with DVV24 (20 μg) and analyzed on an analytical XTerra column [RP-C18, 5 μm, 4.6 mm × 250 mm (Waters)] eluted with a mixture of CH3CN and 0.05 M NaOAc (pH 5.5) (60:40 v/v) at a flow rate of 1 mL/min. UV detection was performed at 254 nm. The HPLC eluate was collected as 1 mL fractions, and radioactivity in each fraction was measured using an automated gamma counter.
Acknowledgments
We are grateful to Julie Cornelis (Laboratory for Radiopharmacy, KU Leuven) for her skillful help with the animal experiments.
Glossary
Abbreviations
- TRPV1
transient receptor potential vanilloid subfamily member 1
- hTRPV1
human TRPV1
- rTRPV1
rat TRPV1
- CNS
central nervous system
- RTX
resiniferatoxin
- PET
positron emission tomography
- RP-HPLC
reversed phase high-performance liquid chromatography
- % ID
percentage of injected dose
- SUV
standardized uptake value
- AR
organ-to-blood radioactivity ratio.
Supporting Information Available
HPLC chromatograms of [11C]DVV24 and [18F]DVV54 after co-injection with the corresponding authentic reference compounds (Figure A.1), HPLC chromatograms of I-RTX and 123I-RTX (Figure A.2), tissue distribution of [11C]DVV24 in mice after pretreatment with the nonradioactive reference DVV24 (Figure A.3), and organ-to-blood radioactivity ratios of [11C]DVV24 in control mice and mice pretreated with DVV24 for the pancreas, spleen, lungs, and bone tissue (Figure A.4). This material is available free of charge via the Internet at http://pubs.acs.org.
This research was funded by a Ph.D. grant of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT), in part by in vivo molecular imaging research (IMIR), and in part by the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research (Project Z1A BC 005270).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Caterina M. J.; Schumacher M. A.; Tominaga M.; Rosen T. A.; Levine J. D.; Julius D. (1997) The capsaicine receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–24. [DOI] [PubMed] [Google Scholar]
- Bodó E.; Kovács I.; Telek A.; Varga A.; Paus R.; Kovács L.; Bíró T. (2004) Vanilloid receptor-1 (VR1) is widely expressed on various epithelial and mesenchymal cell types of human skin. J. Invest. Dermatol. 123, 410–3. [DOI] [PubMed] [Google Scholar]
- Charrua A.; Reguenga C.; Cordeiro J. M.; Correiade-Sá P.; Paule C.; Nagy I.; Cruz F.; Avelino A. (2009) Functional transient receptor potential vanilloid 1 is expressed in human urothelial cells. J. Urol. 182, 2944–50. [DOI] [PubMed] [Google Scholar]
- Akiba Y.; Kato S.; Katsube K.; Nakamura M.; Takeuchi K.; Ishii H.; Hibi T. (2004) Transient receptor potential vanilloid subfamily 1 expressed in pancreatic islet β cells modulates insulin secretion in rats. Biochem. Biophys. Res. Commun. 321, 219–25. [DOI] [PubMed] [Google Scholar]
- Mezey E.; Tóth Z. E.; Cortright D. N.; Arzubi M. K.; Krause J. E.; Elde R.; Guo A.; Blumberg P. M.; Szallasi A. (2000) Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of rat and human. Proc. Natl. Acad. Sci. U.S.A. 97, 3655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo T.; Biro T.; Gonzalez A. F.; Palkovits M.; Blumberg P. M. (2002) Pharmacological characterization of vanilloid receptor located in the brain. Mol. Brain Res. 98, 51–7. [DOI] [PubMed] [Google Scholar]
- Roberts J. C.; Davis J. B.; Benham C. D. (2004) [3H]Resiniferatocin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 995, 176–83. [DOI] [PubMed] [Google Scholar]
- Tóth A.; Boczán J.; Kedei N.; Lizanecz E.; Bagi Z.; Papp Z.; Edes I.; Csiba L.; Blumberg P. M. (2005) Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res. Mol. Brain Res. 135, 162–8. [DOI] [PubMed] [Google Scholar]
- Cavanaugh D. J.; Chesler A. T.; Jackson A. C.; Sigal Y. M.; Yamanaka H.; Grant R.; O’Donnell D.; Nicoll R. A.; Shah N. M.; Julius D.; Basbaum A. I. (2011) TRPV1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J. Neurosci. 31, 5067–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway S. J. (2008) TRPing the switch on pain: An introduction to the chemistry and biology of capsaicin and TRPV1. Chem. Soc. Rev. 37, 1530–45. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R.; Garcìa-Sanz N.; Morenilla-Palao C.; Ferrer-Montiel A. (2005) Functional aspects and mechanisms of TRPV1 involvement in neurogenic inflammation that leads to thermal hyperalgesia. Pfluegers Arch. 451, 151–9. [DOI] [PubMed] [Google Scholar]
- Szallasi A.; Cruz F.; Geppetti P. (2006) TRPV1: A therapeutic target for novel analgesic drugs?. Trends Mol. Med. 12, 545–54. [DOI] [PubMed] [Google Scholar]
- Moran M. M.; McAlexander M. A.; Biro T.; Szallasi A. (2011) Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discovery 10, 601–20. [DOI] [PubMed] [Google Scholar]
- Ho K. W.; Ward N. J.; Calkins D. J. (2012) TRPV1: A stress response protein in the central nervous system. American Journal of Neurodegenerative Disease 1, 1–14. [PMC free article] [PubMed] [Google Scholar]
- Gibson H. E.; Edwards J. G.; Page R. S.; Van Hook M. J.; Kauer J. A. (2008) TRPV1 channels mediate long-term depression at synapses on hippocampal neurons. Neuron 57, 746–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer J. A.; Gibson H. E. (2009) Hot flash: TRPV channels in the brain. Trends Neurosci. 32, 215–24. [DOI] [PubMed] [Google Scholar]
- Morgese M. G.; Cassano T.; Cuomo V.; Giuffrida A. (2007) Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: Role of CB1 and TRPV1 receptors. Exp. Neurol. 208, 110–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veghel D.; Cleynhens J.; Pearce L. V.; Blumberg P. M.; Van Laere K.; Verbruggen A.; Bormans G. (2012) Synthesis and biological evaluation of [11C]SB366791: A new PET-radioligand for in vivo imaging of the TRPV1 receptor. Nucl. Med. Biol. 40, 141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe M. J.; Rami H. K.; Jerman J. C.; Smart D.; Gill C. H.; Soffin E. M.; Luis Hannan S.; Lappin S. C.; Egerton J.; Smith G. D.; Worby A.; Howett L.; Owen D.; Nasir S.; Davies C. H.; Thompson M.; Wyman P. A.; Randall A. D.; Davis J. B. (2004) Identification and characterization of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 46, 133–49. [DOI] [PubMed] [Google Scholar]
- Blum C. A.; Zheng X.; Brielmann H.; Hodgetts K. J.; Bakthavatchalam R.; Chandrasekhar J.; Krause J. E.; Cortright D.; Matson D.; Crandall M.; Ngo C. K.; Fung L.; Day M.; Kershaw M.; De Lombaert S.; Chenard B. L. (2008) Aminoquinazolines as TRPV1 antagonists: Modulation of drug-like properties through the exploration of 2-position substitution. Bioorg. Med. Chem. Lett. 18, 4573–7. [DOI] [PubMed] [Google Scholar]
- Rami H. K.; Thompson M.; Wyman P.; Jerman J. C.; Egerton J.; Brough S.; Stevens A. J.; Randall A. D.; Smart D.; Gunthorpe M. J.; Davis J. B. (2004) Discovery of small molecule antagonists of TRPV1. Bioorg. Med. Chem. Lett. 14, 3631–4. [DOI] [PubMed] [Google Scholar]
- Wahl P.; Foged C.; Tullin S.; Thomsen C. (2001) Iodo-resiniferatoxin, a new potent vanilloid receptor antagonist. Mol. Pharmacol. 59, 9–15. [DOI] [PubMed] [Google Scholar]
- Thompson M., and Wyman P. A. (2002) Urea derivatives having vanilloid receptor (VR1) antagonist activity. U.S. Patent WO 02/072536 A1.
- Bakthavatchalam R., Blum C. A., Brielmann H., Caldwell T., De Lombaert S., Hodgetts K. J., and Zhen X. (2004) 2-Substituted quinazoline-4-ylamine analogues as capsaicin receptor modulators. U.S. Patent WO 2004/055003 A1.
- Zheng X.; Hodgetts K. J.; Brielmann H.; Hutchison A.; Burkamp F.; Brian Jones A.; Blurton P.; Clarkson R.; Chandrasekhar J.; Bakthavatchalam R.; De Lombaert S.; Crandall M.; Cortright D.; Blum C. A. (2006) From arylureas to biarylamides to aminoquinazolines: Discovery of a novel potent TRPV1 antagonist. Bioorg. Med. Chem. Lett. 16, 5217–21. [DOI] [PubMed] [Google Scholar]
- Ünak T.; Ünak P. (1996) Direct radioiodination of metabolic 8-hydroxy-quinolyl-gluconoride, as a potential anticancer drug. Appl. Radiat. Isot. 47, 645–7. [DOI] [PubMed] [Google Scholar]
- Lim K. S.; Kang D. W.; Kim Y. S.; Kim M. S.; Park S. G.; Choi S.; Pearce L. V.; Blumberg P. M.; Lee J. (2011) Receptor activity and conformational analysis of 5′-halogenated resiniferatoxin analogs as TRPV1 ligands. Bioorg. Med. Chem. Lett. 21, 299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passchier J.; Gee A.; Willemsen A.; Vaalburg W.; van Waarde A. (2002) Measuring drug-related receptor occupancy with positron emission tomography. Methods 27, 278–86. [DOI] [PubMed] [Google Scholar]
- Han P.; Korepanova A. V.; Vos M. H.; Moreland R. B.; Chiu M. L.; Faltynek C. R. (2012) Quantification of TRPV1 protein levels in rat tissues to understand its physiological roles. J. Mol. Neurosci. DOI: 10.1007/s12031-012-9849-7. [DOI] [PubMed] [Google Scholar]
- Dalton V. S.; Zavitsanou K. (2010) Cannabinoid effects on CB1 receptor density in the adolescent brain: An autoradiographic study using the synthetic cannabinoid HU210. Synapse 64, 845–54. [DOI] [PubMed] [Google Scholar]
- Minuzzi L.; Cumming P. (2010) Agonist binding fraction of dopamine D2/3 receptors in rat brain: A quantitative autoradiographic study. Neurochem. Int. 56, 747–52. [DOI] [PubMed] [Google Scholar]
- Seabrook G. R.; Sutton K. G.; Jarolimek W.; Hollingworth G. J.; Teague S.; Webb J.; Clark N.; Boyce S.; Kerby J.; Ali Z.; Chou M.; Middleton R.; Kaczorowski G.; Jones A. B. (2002) Functional properties of the high-affinity TRPV1 (VR1) vanilloid receptor antagonist (4-hydroxy-5-iodo-3-methoxyphenylacetate ester) iodo-resiniferatoxin. J. Pharmacol. Exp. Ther. 303, 1052–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.