

Abstract
Extensive research has been carried out in the past two decades to study the pathobiology of nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), which is an oncogenic fusion protein found exclusively in a specific type of T-cell lymphoid malignancy, namely ALK-positive anaplastic large cell lymphoma. Results from these studies have provided highly useful insights into the mechanisms by which a constitutively tyrosine kinase, such as NPM-ALK, promotes tumorigenesis. Several previous publications have comprehensively summarized the advances in this field. In this review, we provide readers with a brief update on specific areas of NPM-ALK pathobiology. In the first part, the NPM-ALK/signal transducer and activator of transcription 3 (STAT3) signaling axis is discussed, with an emphasis on the existence of multiple biochemical defects that have been shown to amplify the oncogenic effects of this signaling axis. Specifically, findings regarding JAK3, SHP1 and the stimulatory effects of several cytokines including interleukin (IL)-9, IL-21 and IL-22 are summarized. New concepts stemming from recent observations regarding the functional interactions among the NPM-ALK/STAT3 axis, β catenin and glycogen synthase kinase 3β will be postulated. Lastly, new mechanisms by which the NPM-ALK/STAT3 axis promotes tumorigenesis, such as its modulations of Twist1, hypoxia-induced factor 1α, CD274, will be described. In the second part, we summarize recent data generated by mass spectrometry studies of NPM-ALK, and use MSH2 and heat shock proteins as examples to illustrate the use of mass spectrometry data in stimulating new research in this field. In the third part, the evolving field of microRNA in the context of NPM-ALK biology is discussed.
Keywords: NPM-ALK, STAT3, anaplastic large cell lymphoma, oncogenic tyrosine kinase, signalling
NPM-ALK and ALK-positive anaplastic large cell lymphoma
It has been 18 years since the identification and initial characterization of the fusion protein NPM-ALK, which is the result of a specific chromosomal translocation that brings the nucleophosmin (NPM) gene on chromosome 5q35 to the anaplastic large cell lymphoma kinase (ALK) gene on 2p23 [Morris et al. 1994; Shiota et al. 1994]. Since its discovery, the oncogenic role of NPM-ALK has been established using a variety of experimental models, and the mechanisms by which NPM-ALK exerts its oncogenic effects have been extensively investigated and summarized [Amin and Lai, 2007; Chiarle et al. 2008; Palmer et al. 2009; Inghirami and Pileri, 2011]. The role of ALK in several types of ALK-expressing solid tumors as well as the therapeutic use of ALK inhibitors in treating these cancers also have been reviewed in a number of recent papers [Azarova et al. 2011; Crystal and Shaw, 2011; Ogawa et al. 2011; Sasaki and Janne, 2011]. In this article, we intend to provide a brief update on the pathobiology of NPM-ALK in the context of ALK-positive anaplastic large cell lymphoma (ALK+ALCL). Our focus will be on a number of specific areas of NPM-ALK pathobiology our laboratories have recently been studying, as well as specific aspects of NPM-ALK signaling that have not been reviewed in detail elsewhere.
The discovery and the initial characterization of NPM-ALK have been detailed elsewhere [Amin and Lai, 2007; Chiarle et al. 2008; Palmer et al. 2009; Inghirami and Pileri, 2011], and only the key points relevant to this review will be highlighted here. The t(2;5)(p23;q35) chromosomal translocation and the resulting fusion protein NPM-ALK are found in the majority of ALK+ALCL, which represents a small subset of T-cell non-Hodgkin lymphoma recognized as a distinct pathologic entity in the World Health Organization classification scheme [Delsol et al. 2008]. Structurally, NPM-ALK is composed of a short portion of the N-terminus of NPM which contains the oligomerization domain; this segment is fused with the C-terminal tyrosine kinase domain and intracellular tail of the ALK protein [Morris et al. 1994] (Figure 1). The oligomerization domain of NPM provides the biochemical basis for the NPM-ALK proteins to undergo oligomerization, which is a requirement for the tyrosine auto-phosphorylation and constitutive activation of NPM-ALK [Fujimoto et al. 1996; Iwahara et al. 1997].
Figure 1.
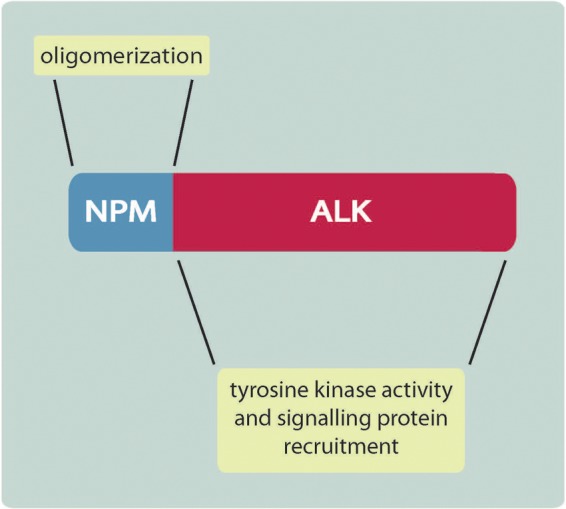
ALK, anaplastic lymphoma kinase; NPM, nucleophosmin.
The expression of the full-length ALK, a cell- surface receptor carrying a tyrosine kinase domain in its cytoplasmic portion, is normally restricted to the brain and nervous system [Iwahara et al. 1997; Pulford et al. 1997]. Normal lymphocytes do not express ALK. In ALK+ALCL cells, the expression of NPM-ALK is driven by the promoter of NPM which is ubiquitously expressed [Wang et al. 1993]. As a consequence of this abnormal gene fusion, the tyrosine kinase activity of ALK is constitutively active, and exerting its biological effects in a ‘foreign’ cell type.
Studies investigating how NPM-ALK promotes oncogenesis have turned out to be highly rewarding in the field of cancer biology, and NPM-ALK has proven to be an excellent study model for understanding how oncogenic tyrosine kinases work. As summarized in the previous review papers, NPM-ALK is known to promote tumorigenecity by binding to and constitutively activating a host of cellular signaling proteins, including those in the signaling pathways of signal transducer and activator of transcription 3 (STAT3), MEK/ERK, mammalian target of rapamycin (mTOR) and phosphoinositide 3 kinase (PI3K)/Akt [Amin and Lai, 2007; Chiarle et al. 2008; Palmer et al. 2009; Inghirami and Pileri, 2011]. The activation of these pathways results in increased cell proliferation and resistance to apoptosis.
The NPM-ALK/STAT3 signaling axis
One of the better characterized pathways deregulated by NPM-ALK is that of STAT3 (Figure 2). It has been shown that NPM-ALK promotes the tyrosine phosphorylation of STAT3 on one of its tyrosine residues, Y705, which is critical for the dimerization and activation of STAT3 [Zamo et al. 2002; Zhang et al. 2002]. Generally, activated STAT3 proteins migrate to the nucleus, where they function as transcription factors in the regulation of a large number of genes [Levy and Darnell, 2002]. STAT3 activation, as evidenced by the expression of phospho-STAT3 and the nuclear localization of the STAT3 protein detectable by immunohistochemistry, is indeed a marker consistently expressed by ALK+ALCL tumors [Zamo et al. 2002; Zhang et al. 2002; Khoury et al. 2003]. Inhibition of STAT3 using a dominant negative construct was shown to induce effective apoptosis and cell-cycle arrest in ALK+ALCL cell lines [Amin et al. 2004]. The importance of STAT3 in ALK+ALCL is further highlighted by the study published by Chiarle and colleagues, in which STAT3 was shown to be required in the setting of NPM-ALK-induced lymphomagensis [Chiarle et al. 2005]. Constitutive activation of STAT3 is associated with the upregulation of many proteins associated with cell proliferation and survival [Zamo et al. 2002; Amin et al. 2004; Chiarle et al. 2005; Piva et al. 2006, 2010; Anastasov et al. 2010; Zhang et al. 2011a], and this appears to be true for other cancer cell types in which STAT3 is constitutively active.
Figure 2.
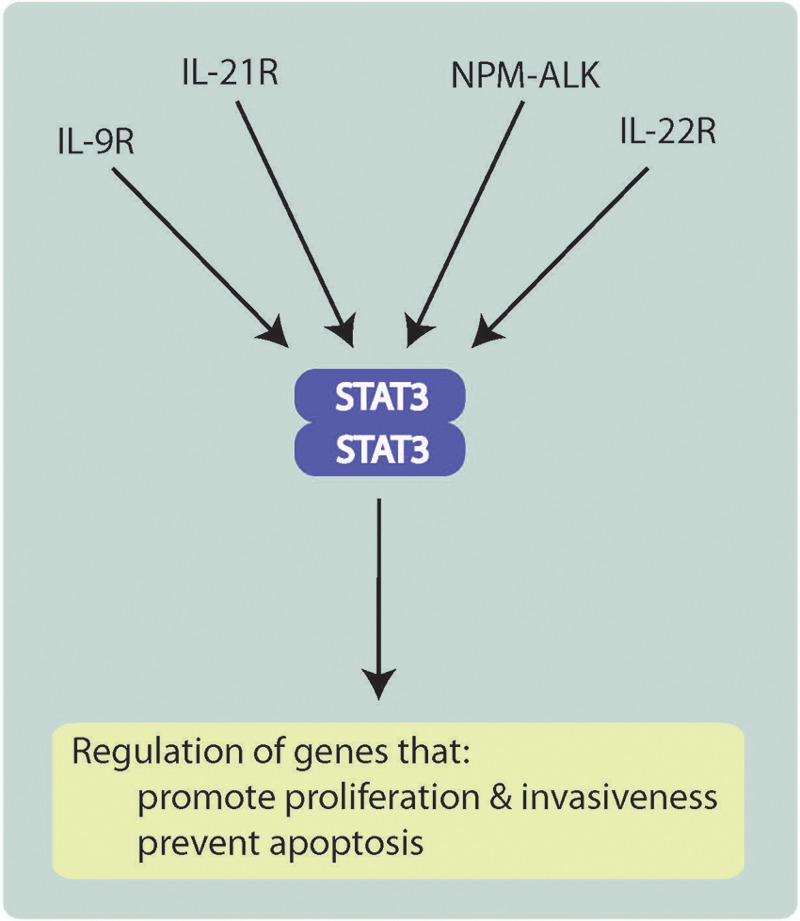
ALK, anaplastic lymphoma kinase; IL, interleukin; NPM, nucleophosmin; STAT3, signal transducer and activator of transcription 3.
Additional studies of how STAT3 promotes the tumorigenesis of ALK+ALCL have revealed novel insights. One example is the observation that STAT3 functionally interacts with DNA methyltransferases, and thereby promotes promoter hypermethylation and gene silencing [Zhang et al. 2005, 2011b]. One gene is SHP1, which encodes a tyrosine phosphatase that negatively regulates the STAT3 signaling pathway [Zhang et al. 2005; Han et al. 2006a, 2006b; Honorat et al. 2006]. Previous studies have shown that SHP1 promoter methylation and loss of SHP1 expression are indeed frequently observed in ALK+ALCL cell lines and tumors [Khoury et al. 2004]. Functional studies have supported that loss of SHP1 is pathogenetically important in ALK+ALCL, as restoration of SHP1 by gene transfection inhibits the growth of these cells [Han et al. 2006a; Hegazy et al. 2010]. Interestingly, restoration of SHP1 not only dampens the level of STAT3 activation, it also promotes proteosomal degradation of NPM-ALK [Han et al. 2006a]. Thus, through silencing SHP1 expression, the NPM-ALK/STAT3 signaling axis is sustained and enhanced.
Accumulating evidence supports that constitutive activation of STAT3 in ALK+ALCL is multifactorial, a concept that was first brought forward by Zhang and colleagues [Zhang et al. 2002]. Further support for this concept came when it was found that JAK3, a tyrosine kinase that normally relays cytokine stimulation to STAT3 activation, is constitutively activated in ALK+ALCL cell lines and tumors [Zamo et al. 2002; Amin et al. 2003; Lai et al. 2005]. In ALK+ALCL cells, JAK3 coimmunoprecipitates with NPM-ALK and STAT3, and it appears to directly enhance the tyrosine phosphorylation/activation status of both proteins [Amin et al. 2003]. In this same study, small interfering RNA (siRNA) knockdown or pharmacologic inhibition of JAK3 resulted in significant apoptosis in ALK+ALCL cells, supporting the notion that JAK3 is biologically important in this lymphoma.
Since the JAK proteins are normally activated via cytokine stimulation, the finding of JAK3 activation in ALK+ALCL cells raises the possibility that cytokine stimulation may contribute to the activation status of NPM-ALK and STAT3, and thus, the tumorigenecity of ALK+ALCL. JAK3 is activated by interleukins of the interleukin (IL)-2 common γ chain family (IL-2Rγ), which includes IL-2, IL4, IL-7, IL-9, IL15 and IL-21 [Witthuhn et al. 1994; Yin et al. 1995; Asao et al. 2001; Habib et al. 2002]. Our research team demonstrated that ALK+ALCL cell lines produce both IL-9 and IL-21 [Qiu et al. 2006; Dien Bard et al. 2009]. Moreover, we provided evidence that these two cytokines promote tumorigenesis by activating JAK3/STAT3 signaling via an autocrine stimulatory loop. In keeping with our conclusion that IL-9 is important in the context of NPM-ALK–driven lymphomagenesis, overexpression of NPM-ALK in IL-9 transgenic mice was found to induce lymphoma formation [Lange et al. 2003].
Intriguingly, in a recently published study, it was reported that STAT3 promotes the binding of DNA methyltransferases to the IL-2Rγ promoter so as to repress the expression of IL-2Rγ [Zhang et al. 2011b]. These findings certainly challenge the notion that IL-9 and IL-21 autocrine stimulatory loops contribute to the pathogenesis of ALK+ALCL, as the signaling of these two cytokines is IL-2Rγ dependent [Kimura et al. 1995; Asao et al. 2001; Habib et al. 2002]. While this discrepancy cannot be fully explained without further experiments, one possibility may be related to the use of different cell clones of ALK+ALCL cell lines by different laboratories. The finding by Zhang and colleagues that stimulation with IL-2, IL-9 and IL-15 did not stimulate STAT3 even in ALK+ALCL cells transfected with the IL-2Rγ gene suggests that these cell clones are intrinsically insensitive to these cytokines [Zhang et al. 2011b].
In addition to IL-9 and IL-21, our group found that IL-22 also contributes to STAT3 activation in ALK+ALCL cells [Dien Bard et al. 2008]. In contrast to the signaling of IL-9 and IL-21, IL-22 signaling is independent of IL-2Rγ. Unlike the receptors for IL-9 and IL-21, IL-22R1 (which is one of the subunits of the IL-22 receptor) is absent in normal lymphocytes [Wolk et al. 2010]. Thus, the expression of IL-22R1 on the cytoplasmic membrane of ALK+ALCL cells is an aberrant event. Results from in vitro studies support that IL-22 signaling contributes to the pathogenesis of ALK+ALCL, as inhibition of the signaling of this pathway using either IL-22 neutralizing antibodies or naturally occurring IL-22 decoy receptors (e.g. IL-22BP) decreased STAT3 activation and significantly inhibited cell growth and colony formation in soft agar; opposite biological effects were observed when recombinant IL-22 was added to ALK+ALCL cells [Dien Bard et al. 2008]. Interestingly, we also found that the aberrant expression of IL-22R1 is directly attributed to NPM-ALK, as gene transfection of NPM-ALK cDNA into Jurkat cells induced IL-22R1 expression. Thus, NPM-ALK can convert an ‘IL-22–unresponsive’ phenotype to an ‘IL-22-responsive’ phenotype. The functional relationship among NPM-ALK, STAT3 and IL-22 signaling also illustrates an excellent example by which NPM-ALK contributes to STAT3 activation via an alternative pathway.
Most recently, our laboratories described yet another mechanism by which STAT3 activation can be enhanced in ALK+ALCL. We found that β catenin, a transcriptional factor and the downstream mediator of the Wnt canonical pathway, is constitutively active in ALK+ALCL cells [Anand et al. 2011]. Downregulation of β catenin using siRNA resulted in a marked reduction in the STAT3 levels and activated/phosphorylated STAT3. While NPM-ALK did not regulate the protein level, nuclear localization or tyrosine phosphorylation of β catenin in ALK+ALCL cells, it upregulated β-catenin transcriptional activity via a yet-to-be defined mechanism. These observations regarding the relationship between β catenin and NPM-ALK are in contrast with that between β catenin and BCR-ABL, an oncogenic tyrosine kinase expressed in chronic myeloid leukemia. In these cells, BCR-ABL contributes to the stabilization of β catenin by increasing its tyrosine phosphorylation, nuclear translocation and transcriptional activity [Coluccia et al. 2007].
In normal cells, β catenin is inhibited by glycogen synthase kinase 3β (GSK3β), which phosphorylates β catenin at multiple serine/threonine residues and thereby promotes its proteosomal degradation [Jope and Johnson, 2004]. GSK3β itself can be inhibited by the PI3K/Akt and the Wnt canonical pathways [Manoukian and Woodgett, 2002]. Specifically, inactivation of GSK3β is mediated via the phosphorylation of its serine 9 residue. Upon inactivation, its inhibitory effect on β catenin is released, and β catenin is allowed to accumulate and translocate to the nucleus, where it upregulates the transcription of multiple genes that promote cell growth. In a recent report, it has been shown that NPM-ALK promotes the phosphorylation of GSK3β on serine 9, likely through PI3K/Akt signaling [McDonnell et al. 2011]. This study further demonstrated that inhibition of GSK3β allowed its substrates, Mcl1 and CDC25A, to accumulate in ALK+ALCL cells. Considering the role of active GSK3β in targeting β catenin for degradation, we believe that these results correlate well with our findings regarding the constitutive activation of β catenin in ALK+ALCL cells.
It has been shown that STAT3 activation contributes to the growth of ALK+ALCL cells by upregulating a large number of genes promoting cell-cycle progression and inhibiting apoptosis, and these details have been summarized previously [Amin and Lai, 2007; Chiarle et al. 2008; Palmer et al. 2009; Inghirami and Pileri, 2011]. More recent studies have revealed that the mechanisms by which STAT3 mediates the oncogenic effects of NPM-ALK are more diverse than originally thought. In this regard, the interaction between STAT3 and DNA methytransferases leads to gene silencing of SHP1 as was discussed earlier. STAT3-mediated epigenetic silencing is also responsible for the loss of T-cell signaling molecules, including CD3ϵ, ZAP70, LAT and SLP76, all of which are important in determining T-cell identity and T-cell receptor signaling [Ambrogio et al. 2009]. A recent paper has revealed that STAT3 binds to the promoter of hypoxia-induced factor 1α (HIF1α) and upregulates HIF1α expression in ALK+ALCL cells [Marzec et al. 2011]. This finding suggests that HIF1α could represent an important mechanism by which STAT3 exerts its oncogenic effects in ALK+ALCL, as HIF1α is known to be involved in tumor growth, angiogenesis, invasiveness, metastasis and drug resistance in other types of malignancy [Majmundar et al. 2010; Rohwer and Cramer, 2011]. The same research group also reported that STAT3 in ALK+ALCL upregulates the mRNA and protein expression of CD274 (PD-L1, B7-H1), which is a cell-surface protein implicated in immunosuppression [Marzec et al. 2008]. To be discussed below, STAT3 has been shown to modulate the expression of specific microRNAs (miRNAs) in this type of lymphoma.
Our laboratories have recently published that STAT3 substantially upregulates Twist1, a protein well known for its role in promoting epithelial mesenchymal transition and the metastatic potential of solid tumors [Yang et al. 2004]. We found that ALK+ALCL cell lines and tumors consistently express a high level of Twist1; in contrast, normal T cells do not have detectable Twist1. Results from our in vitro invasion assays support that Twist1 contributes to the invasiveness of ALK+ALCL cells, and this biological response correlated with the activation of Akt and downregulation of p66Shc [Zhang et al. 2012]. Importantly, the knockdown of Twist1 expression in ALK+ALCL results in increased sensitivity to PF-2341066 (Crizotinib; Pfizer, New York, NY)), which is a US Food and Drug Administration approved ALK inhibitor.
Most recently, our group has found that the NPM-ALK/STAT3 signaling axis strongly upregulates the Sox-2 (sex-determining region Y-box-2) transcription factor (Gelebart et al. 2012). The significance of this finding is underlined by the fact that the major function of Sox-2 in normal cells is to promote self-renewal and pluripotency of embryonic stem cells [Niwa et al. 2000; Chew et al. 2005]. Furthermore, Sox-2 represents one of the few transcription factors involved in the reprogramming and conversion of terminally differentiated fibroblasts into inducible pluripotent stem cells [Park et al. 2008; Okita and Yamanaka, 2010]. Using confocal microscopy, the expression of the Sox-2 protein was detectable in virtually all of the cells in the two ALK+ALCL cell lines tested. However, the transcriptional activity of Sox-2 appears to be restricted to a relatively small subset of cells. Importantly, the subset with active Sox-2 activity was more tumorigenic and invasive than cells without Sox2 activity. While the biological role of Sox-2 in ALK+ALCL still needs to be further examined, Sox-2 represents the latest member of downstream targets of the NPM-ALK/STAT3 axis.
Mass spectrometry studies: identifying novel pathways that mediate NPM-ALK-mediated tumorigenesis
Thus far, we have discussed the role of STAT3 as a key mediator for NPM-ALK-induced tumorigenesis. However, to fully understand the pathobiology of NPM-ALK, several laboratories including ours have performed extensive mass spectrometry studies to profile proteins that are physically bound to NPM-ALK [Crockett et al. 2004; Galietta et al. 2007; Wu et al. 2009]. Results from these studies have proven to be highly useful in unveiling novel oncogenic signaling functions of NPM-ALK. One example is the identification of MSH2 as an NPM-ALK-binding protein [Wu et al. 2009]. MSH2 is a protein known to be pivotal for DNA mismatch repair (MMR). Specifically, MSH2 heterodimerizes with MSH6 (known as MutSα) or MSH3 (as known as MutSβ), to detect single base mismatches and small insertion–deletion loops [Li, 2008]. Once the MSH2 heterodimers bind to the site of DNA error, a second MMR heterodimer consisting of MutL orthologs is recruited (e.g. MLH1•PMS2), followed by the recruitment of additional proteins that mediate the removal of the erroneous DNA bases, using the unaffected strand as a template to resynthesize the DNA [Li, 2008]. Cells lacking MSH2 can form neither MutSα nor MutSβ, and are completely deficient in MMR function [Reitmair et al. 1995; Andrew et al. 1998; Edelmann and Edelmann, 2004]. Failure of MMR function, such as in the loss of MSH2, results in a mutator phenotype and subsequent tumorigenesis [Reitmair et al. 1995]. The importance of MMR function to tumor suppression has been characterized most extensively in Lynch syndrome, an early onset cancer syndrome affecting a heterogeneous group of tissues (e.g. colon, endometrium, urothelium). In addition, acquired inactivation of MMR contributes to a proportion of sporadic cancers [Coleman and Tsongalis, 2001]. Pertinent to this review, the congenital loss of MMR function through the inheritance of two mutated copies of a gene encoding a MMR protein, including MSH2, is associated with the development of lymphoid cancer presenting in childhood [Felton et al. 2007].
Based on this background, the finding that MSH2, but not its normal binding partners, MSH3 and MSH6, binds to NPM-ALK led us to hypothesize that NPM-ALK may interfere with MMR, by virtue of sequestrating MSH2 away from MSH3 and MSH6. In support of this hypothesis, we found that gene transfection of NPM-ALK cDNA into GP293 cells indeed significantly impaired MMR, as measured by 6-thioguanine sensitivity [Young et al. 2011]. Furthermore, site-directed mutagenesis of tyrosine 191 on NPM-ALK, significantly disrupted NPM-ALK•MSH2 binding and partially restored 6-thioguanine sensitivity. Lastly, we also found that NPM-ALK promoted tyrosine phosphorylation of MSH2, which might be a critical step for its functional interference of MSH2 and MMR function. While further experiments are needed to understand this phenomenon more completely, MSH2 represents a novel target of NPM-ALK, and interference of the MMR represents a novel oncogenic function of this fusion protein. Interestingly, it has been reported that another oncogenic tyrosine kinase, BCR-ABL, can interfere with MMR in vitro [Stoklosa et al. 2008]. It is tempting to speculate that BCR-ABL may also interact with MSH2 in a similar manner as NPM-ALK.
Mass spectrometry data have also revealed an interaction between NPM-ALK and various heat shock proteins (Hsps) [Crockett et al. 2004; Boccalatte et al. 2009; Wu et al. 2009]. Most studies regarding Hsps in the context of NPM-ALK pathobiology have been focused on Hsp90, which is a molecular chaperone that plays a critical role in helping proteins, termed clients, fold correctly. There are a multitude of Hsp90 clients in cells and they include protein kinases, transcription factors and chromatin remodeling factors (reviewed by Taipale and colleagues and Trepel and colleagues) [Taipale et al. 2010; Trepel et al. 2010]. A number of tyrosine kinases critical in oncogenesis including Bcr-Abl [An et al. 2000], Her2/Neu [Citri et al. 2002], Flt3 [Minami et al. 2002] and Raf-1 [Stancato et al. 1993] are known Hsp90 clients. Moreover, the inhibition of Hsp90 with benzoquinone ansamycin drugs, such as herbimycin A, geldanamycin and related compounds such as 17-allylamino,17-demethoxygeldanamycin (17-AAG) impairs the activity of these kinases [Okabe et al. 1992; Stancato et al. 1993; Miller et al. 1994; Schulte et al. 1995; An et al. 2000; Naoe et al. 2001; Citri et al. 2002; Minami et al. 2002]. These drugs bind the adenosine triphosphate binding amino-terminus of Hsp90 and interfere with client protein interaction, which ultimately results in client protein degradation [Holzbeierlein et al. 2010]. Several studies have implicated NPM-ALK as an important Hsp90 client.
Treatment of ALK+ALCL cell lines with Hsp90 inhibitors resulted in decreased NPM-ALK expression, and this is most likely due to the targeting of this protein for proteasomal degradation [Bonvini et al. 2002; Georgakis et al. 2006]. Previous work by Bonvini and colleagues demonstrated that disruption of the interaction between NPM-ALK and Hsp90, as a result of 17-AAG treatment, correlated with an increase in the association of the Hsp70 chaperone protein with NPM-ALK [Bonvini et al. 2004]. Hsp70 complexes with the CHIP E3 ubiquitin ligase, and Hsp70/CHIP complexes targeted NPM-ALK for proteasomal degradation in 17-AAG-treated cells [Bonvini et al. 2004]. Furthermore, the treatment of ALK+ALCL cell lines with 17-AAG results in cell cycle arrest in G0/G1 and an increase in apoptosis [Georgakis et al. 2006]. It should be noted that the biological effect of Hsp90 inhibition in ALK+ALCL is likely not exclusively due to inhibition of NPM-ALK, as Hsp90 has many client proteins. For example, Hsp90 has also been shown to be important for regulating the activity of matrix metalloproteinase 9 in ALK+ALCL and allowing these cells to invade through matri-gel [Lagarrigue et al. 2010]. Our group has also recently shown that the Hsp90 co-chaperone, cyclophilin 40, is regulated by NPM-ALK signaling and promotes the viability of ALK+ ALCL cell lines [Pearson et al. 2012].
In view of the fact that NPM-ALK is a constitutively active tyrosine kinase, identifying proteins whose phosphorylation status is influenced by NPM-ALK would be expected to provide insights into the pathobiology of this oncoprotein. To that end, mass spectrometry studies were performed by several laboratories, including ours, focusing on the phosphoproteomic changes induced by NPM-ALK [Rush et al. 2005; Boccalatte et al. 2009; Wu et al. 2010]. In order to optimize the sensitivity and specificity of our assay, we employed an approach combining sequential affinity purification of phosphopeptides and liquid chromatography–tandem mass spectrometry. We identified a large number of phosphoproteins that were differentially expressed in lysates of GP293 cells transfected with NPM-ALK compared with GP293 cells transfected with a kinase-dead NPM-ALK [Wu et al. 2010]. Specifically, 506 phosphoproteins (617 phosphopeptides and 767 phosphorylation sites) were identified. Our method was designed to purify all phosphopeptides, and thus, we were able to detect a large number of serine/threonine phosphorylated proteins [Wu et al. 2010].
Our phosphoproteomic studies have revealed several other pathways that were not previously known to be regulated by NPM-ALK. For instance, we found evidence of tyrosine phosphorylation of multiple proteins in the tumor necrosis factor (TNF) signaling pathway. Using western blotting, we validated that the induction of the tyrosine phosphorylation of several TNF pathway signaling molecules by NPM-ALK, including RIP, TRAP1 and FAF1 [Wu et al. 2010]. In support of their functional importance in this type of lymphoma, we knocked down the expression of TRAP1 using siRNA, and found that this treatment significantly sensitized ALK+ALCL cell lines to TRAIL- and doxorubicin-induced apoptosis. We also found that NPM-ALK induced the phosphorylation of multiple proteins in the ubiquitin–proteasome degradation pathway, including proteasome activator 28 subunit γ (PSME3) [Wu et al. 2010]. These findings echo results from a previous mass spectrometry study we performed, in which we found that NPM-ALK associates with at least six E3 ubiquitin ligases [Wu et al. 2009].
Interestingly, NPM-ALK itself showed evidence of serine phosphorylation [Wang et al. 2011]. Using tandem affinity purification mass spectrometry, we found evidence of phosphorylation of three serine residues of NPM-ALK (serine 135, 164 and 497) when ectopically expressed in GP293 cells. Phosphorylation of these residues is functionally important, as site-directed mutagenesis of these sites led to a significant reduction in the tumorigenecity of NPM-ALK in this experimental model. Thus, although the oncogenic activity of NPM-ALK is dependent on its tyrosine autophosphorylation, phosphorylation of its serine residues appears to enhance tumorigenic potential. While we have confirmed that NPM-ALK in ALK+ALCL tumors is serine phosphorylated, it remains to be determined whether NPM-ALK in tumors is actually phosphorylated on the same serine residues (i.e. serine 135, 164 and 497) as in ALK+ALCL cell lines.
The regulation of miRNAs in ALK+ALCL
miRNAs are small (~20 nt) nonprotein-coding RNAs that regulate diverse cellular functions. These molecules bind the 3’ untranslated region of mRNAs, usually resulting in either translational repression or degradation of the targeted mRNA [Fabian et al. 2010]. miRNAs are appreciated to play an important role in many cancers [Kasinski and Slack, 2011], and this includes ALK+ALCL. A recent study has even demonstrated that NPM-ALK itself is targeted by miRNA 96 [Vishwamitra et al. 2012]. In addition, several studies have demonstrated that NPM-ALK signaling represses or promotes the expression of miRNAs that influence the cellular phenotype and pathogenesis of ALK+ ALCL.
In a study by Desjobert and colleagues, the repression of miR-29a was found to be dependent on NPM-ALK activity and STAT3, and this repression is likely mediated by the epigenetic silencing of miR-29a [Desjobert et al. 2011]. The importance of miR-29a repression in ALK+ALCL was demonstrated by experiments showing that ectopic expression of this miRNA in ALK+ ALCL cell lines resulted in decreased protein levels of the antiapoptotic protein, Mcl-1, a miR-29a target. Decreased Mcl-1 expression correlated with an increased sensitivity of cells to doxorubicin. This study also showed that immunocompromised mice injected with miR-29a-expressing Karpas 299 cells formed smaller tumors that were more apoptotic than tumors formed in mice injected with Karpas 299 cells expressing a control miRNA.
Several other miRNAs are repressed by NPM-ALK and STAT3 signaling in ALK+ALCL. This includes miR-21 and miR-219, which in this lymphoma target DNA methyltransferase 1 [Zhang et al. 2011b] and the proliferation promoting receptor ICOS [Zhang et al. 2011a], respectively. Dejean and colleagues demonstrated that miR-16 was significantly downregulated in cells isolated from the lymph nodes of mice expressing an inducible NPM-ALK or TPM3-ALK transgene compared with cells isolated from mice in which the transgene was not induced [Dejean et al. 2011]. Surprisingly, however, this group found that treatment of TPM3-ALK-expressing mouse embryo fibroblasts with Crizotinib did not increase miR-16 expression and even slightly decreased miR-16 levels. This raises the question as to whether this miRNA is indeed regulated by NPM-ALK. Regardless, the overexpression of miR-16 was found to decrease expression of the VEGF growth factor in ALK+ALCL cell lines and an inverse correlation between miR-16 and VEGF expression was observed in patient samples [Dejean et al. 2011]. Moreover, miR-16 injection in the tumors of nude mice was also found to decrease tumor growth in vivo [Dejean et al. 2011]. miR-101 was also found to be downregulated in ALK+ALCL cell lines, but whether this miRNA is regulated by NPM-ALK signaling was not examined [Merkel et al. 2010]. The forced expression of miR-101 in ALK+ALCL cell lines was found to result in an increased number of cells arrested in G1 as well as an increase in apoptosis. These phenotypes are likely a result of the downregulation of the miR-101 targets, the serine/threonine kinase mTOR and the prosurvival protein Mcl-1 [Merkel et al. 2010].
NPM-ALK signaling also leads to the upregulation of several important miRNAs in ALK+ALCL. For example, miR-135b was found to be highly expressed in ALK+ALCL cell lines and patient samples [Lawrie et al. 2008; Matsuyama et al. 2011], and its expression was NPM-ALK and STAT3 dependent [Matsuyama et al. 2011]. Matsuyama and colleagues further showed that miR-135b targets the FOXO1 transcription factor in ALK+ALCL cell lines, which is critical as FOXO1 can promote the expression of the cell cycle inhibitors p21 and p27. Moreover, this group showed that miR-135b is responsible for IL-17 production in this lymphoma, conferring a ‘Th17-like’ immunophenotype to tumor cells.
Future directions
Since the discovery of the NPM-ALK fusion protein in 1994, significant progress has been made with respect to our understanding of the oncogenic impact and properties of NPM-ALK. Nevertheless, a number of questions remain to be addressed. For instance, the functional importance of serine/threonine phosphorylation of NPM-ALK needs to be further clarified. It is also unclear as to whether the pathogenesis of pediatric ALK+ALCL cases is similar to that involved in adult cases. Lastly, as more cancer types are found to possess other ALK translocations as well as ALK mutations, it has yet to be fully established whether we can extrapolate what we have learned from the studies of NPM-ALK in ALK+ALCL to these malignancies.
Footnotes
Funding: Research in the Lai Laboratory is supported by operating research grants from the Canadian Institute of Health Research, the Canadian Cancer Society and the Alberta Cancer Foundation. Research in the Ingham laboratory is supported by the Alberta Cancer Foundation.
Conflict of interest statement: The authors have no conflicting financial interests.
Contributor Information
Raymond Lai, Department of Laboratory Medicine and Pathology, Cross Cancer Institute and University of Alberta, Rm 2338, Cross Cancer Institute, 11560 University Avenue, Edmonton, Alberta, Canada T6G 1Z2.
Robert J. Ingham, Department of Medical Microbiology and Immunology, Li Ka Shing Institute of Virology, University of Alberta, Edmonton, Alberta, Canada
References
- Ambrogio C., Martinengo C., Voena C., Tondat F., Riera L., Di Celle P., et al. (2009) Npm-Alk oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res 69: 8611–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin H., Lai R. (2007) Pathobiology of Alk+ anaplastic large-cell lymphoma. Blood 110: 2259–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin H., McDonnell T., Ma Y., Lin Q., Fujio Y., Kunisada K., et al. (2004) Selective inhibition of STAT3 induces apoptosis and G(1) cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 23: 5426–5434 [DOI] [PubMed] [Google Scholar]
- Amin H., Medeiros L., Ma Y., Feretzaki M., Das P., Leventaki V., et al. (2003) Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene 22: 5399–407 [DOI] [PubMed] [Google Scholar]
- An W., Schulte T., Neckers L. (2000) The heat shock protein 90 antagonist geldanamycin alters chaperone association with P210BCR-ABL and V-Src proteins before their degradation by the proteasome. Cell Growth Differ 11: 355–360 [PubMed] [Google Scholar]
- Anand M., Lai R., Gelebart P. (2011) Beta-catenin is constitutively active and increases STAT3 expression/activation in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 96: 253–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasov N., Bonzheim I., Rudelius M., Klier M., Dau T., Angermeier D., et al. (2010) C/EBPβ expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica 95: 760–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew S., Mckinnon M., Cheng B., Francis A., Penney J., Reitmair A., et al. (1998) Tissues of MSH2-deficient mice demonstrate hypermutability on exposure to a DNA methylating agent. Proc Natl Acad Sci U S A 95: 1126–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asao H., Okuyama C., Kumaki S., Ishii N., Tsuchiya S., Foster D., et al. (2001) Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol 167: 1–5 [DOI] [PubMed] [Google Scholar]
- Azarova A., Gautam G., George R. (2011) Emerging importance of ALK in neuroblastoma. Semin Cancer Biol 21: 267–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccalatte F., Voena C., Riganti C., Bosia A., D’Amico L., Riera L., et al. (2009) The enzymatic activity of 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/ IMP cyclohydrolase is enhanced by NPM-ALK: new insights in ALK-mediated pathogenesis and the treatment of ALCL. Blood 113: 2776–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvini P., Dalla Rosa H., Vignes N., Rosolen A. (2004) Ubiquitination and proteasomal degradation of nucleophosmin-anaplastic lymphoma kinase induced by 17-allylamino-demethoxygeldanamycin: role of the co-chaperone carboxyl heat shock protein 70-interacting protein. Cancer Res 64: 3256–3264 [DOI] [PubMed] [Google Scholar]
- Bonvini P., Gastaldi T., Falini B., Rosolen A. (2002) Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel HSP90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the HSP90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res 62: 1559–1566 [PubMed] [Google Scholar]
- Chew J., Loh Y., Zhang W., Chen X., Tam W., Yeap L., et al. (2005) Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol 25: 6031–6046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R., Simmons W., Cai H., Dhall G., Zamo A., Raz R., et al. (2005) STAT3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med 11: 623–629 [DOI] [PubMed] [Google Scholar]
- Chiarle R., Voena C., Ambrogio C., Piva R., Inghirami G. (2008) The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 8: 11–23 [DOI] [PubMed] [Google Scholar]
- Citri A., Alroy I., Lavi S., Rubin C., Xu W., Grammatikakis N., et al. (2002) Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 21: 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman W., Tsongalis G. (2001) The role of genomic instability in the development of human cancer. In: Coleman W., Tsongalis G. (eds), The Molecular Basis of Human Cancer, 1st ed. Humana Press, Totowa, NJ: pp. 115–142 [Google Scholar]
- Coluccia A., Vacca A., Dunach M., Mologni L., Redaelli S., Bustos V., et al. (2007) BCR-ABL stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. EMBO J 26: 1456–1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockett D., Lin Z., Elenitoba-Johnson K., Lim M. (2004) Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene 23: 2617–2629 [DOI] [PubMed] [Google Scholar]
- Crystal A., Shaw A. (2011) New targets in advanced NSCLC: EML4-ALK. Clin Adv Hematol Oncol 9: 207–214 [PubMed] [Google Scholar]
- Dejean E., Renalier M., Foisseau M., Agirre X., Joseph N., De Paiva G., et al. (2011) Hypoxia-microrna-16 downregulation induces VEGF expression in anaplastic lymphoma kinase (Alk)-positive anaplastic large-cell lymphomas. Leukemia 25: 1882–1890 [DOI] [PubMed] [Google Scholar]
- Delsol G., Falini B., Muller-Hermelink H., Campo E., Jaffe E., Gascoyne R., et al. (2008) Anaplastic Large Cell Lymphoma (ALCL), ALK-positive. 4th ed. Lyon: International Agency for Research on Cancer (IARC) [Google Scholar]
- Desjobert C., Renalier M., Bergalet J., Dejean E., Joseph N., Kruczynski A., et al. (2011) MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood 117: 6627–6637 [DOI] [PubMed] [Google Scholar]
- Dien Bard J., Gelebart P., Anand M., Amin H., Lai R. (2008) Aberrant expression of IL-22 receptor 1 and autocrine IL-22 stimulation contribute to tumorigenicity in ALK+ anaplastic large cell lymphoma. Leukemia 22: 1595–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dien Bard J., Gelebart P., Anand M., Zak Z., Hegazy S., Amin H., et al. (2009) IL-21 contributes to JAK3/STAT3 activation and promotes cell growth in ALK-positive anaplastic large cell lymphoma. Am J Pathol 175: 825–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann L., Edelmann W. (2004) Loss of DNA mismatch repair function and cancer predisposition in the mouse: animal models for human hereditary nonpolyposis colorectal cancer. Am J Med Genet C Semin Med Genet 129C: 91–99 [DOI] [PubMed] [Google Scholar]
- Fabian M., Sonenberg N., Filipowicz W. (2010) Regulation of MRNA translation and stability by micrornas. Annu Rev Biochem 79: 351–379 [DOI] [PubMed] [Google Scholar]
- Felton K., Gilchrist D., Andrew S. (2007) Constitutive deficiency in DNA mismatch repair. Clin Genet 71: 483–498 [DOI] [PubMed] [Google Scholar]
- Fujimoto J., Shiota M., Iwahara T., Seki N., Satoh H., Mori S., et al. (1996) Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5). Proc Natl Acad Sci U S A 93: 4181–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galietta A., Gunby R., Redaelli S., Stano P., Carniti C., Bachi A., et al. (2007) NPM/ALK binds and phosphorylates the RNA/DNA-binding protein PSF in anaplastic large-cell lymphoma. Blood 110: 2600–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelebart P., Hegazy S.A., Wang P., Bone K.M., Anand M., Sharon D., et al. (2012) Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer Journal 2: e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakis G., Li Y., Rassidakis G., Medeiros L., Younes A. (2006) The HSP90 inhibitor 17-AAG synergizes with doxorubicin and U0126 in anaplastic large cell lymphoma irrespective of ALK expression. Exp Hematol 34: 1670–1679 [DOI] [PubMed] [Google Scholar]
- Habib T., Senadheera S., Weinberg K., Kaushansky K. (2002) The common gamma chain (gamma C) is a required signaling component of the IL-21 receptor and supports IL-21-induced cell proliferation via JAK3. Biochemistry 41: 8725–8731 [DOI] [PubMed] [Google Scholar]
- Han Y., Amin H., Franko B., Frantz C., Shi X., Lai R. (2006a) Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 108: 2796–2803 [DOI] [PubMed] [Google Scholar]
- Han Y., Amin H., Frantz C., Franko B., Lee J., Lin Q., et al. (2006b) Restoration of SHP1 expression by 5-AZA-2’-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia 20: 1602–1609 [DOI] [PubMed] [Google Scholar]
- Hegazy S., Wang P., Anand M., Ingham R., Gelebart P., Lai R. (2010) The tyrosine 343 residue of nucleophosmin (NPM)-anaplastic lymphoma kinase (ALK) is important for its interaction with SHP1, a cytoplasmic tyrosine phosphatase with tumor suppressor functions. J Biol Chem 285: 19813–19820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbeierlein J., Windsperger A., Vielhauer G. (2010) HSP90: a drug target? Curr Oncol Rep 12: 95–101 [DOI] [PubMed] [Google Scholar]
- Honorat J., Ragab A., Lamant L., Delsol G., Ragab-Thomas J. (2006) SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood 107: 4130–4138 [DOI] [PubMed] [Google Scholar]
- Inghirami G., Pileri S. (2011) Anaplastic large-cell lymphoma. Semin Diagn Pathol 28: 190–201 [DOI] [PubMed] [Google Scholar]
- Iwahara T., Fujimoto J., Wen D., Cupples R., Bucay N., Arakawa T., et al. (1997) Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous System. Oncogene 14: 439–449 [DOI] [PubMed] [Google Scholar]
- Jope R., Johnson G. (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102 [DOI] [PubMed] [Google Scholar]
- Kasinski A., Slack F. (2011) Epigenetics and genetics. Micrornas en route to the clinic: progress in validating and targeting micrornas for cancer therapy. Nat Rev Cancer 11: 849–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury J., Medeiros L., Rassidakis G., Yared M., Tsioli P., Leventaki V., et al. (2003) Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK+ and ALK-anaplastic large cell lymphoma. Clin Cancer Res 9: 3692–3699 [PubMed] [Google Scholar]
- Khoury J., Rassidakis G., Medeiros L., Amin H., Lai R. (2004) Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood 104: 1580–1581 [DOI] [PubMed] [Google Scholar]
- Kimura Y., Takeshita T., Kondo M., Ishii N., Nakamura M., Van Snick J., et al. (1995) Sharing of the IL-2 receptor gamma chain with the functional IL-9 receptor complex. Int Immunol 7: 115–120 [DOI] [PubMed] [Google Scholar]
- Lagarrigue F., Dupuis-Coronas S., Ramel D., Delsol G., Tronchere H., Payrastre B., et al. (2010) Matrix metalloproteinase-9 is upregulated in nucleophosmin-anaplastic lymphoma kinase-positive anaplastic lymphomas and activated at the cell surface by the chaperone heat shock protein 90 to promote cell invasion. Cancer Res 70: 6978–6987 [DOI] [PubMed] [Google Scholar]
- Lai R., Rassidakis G., Lin Q., Atwell C., Medeiros L., Amin H. (2005) JAK3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol 36: 939–944 [DOI] [PubMed] [Google Scholar]
- Lange K., Uckert W., Blankenstein T., Nadrowitz R., Bittner C., Renauld J., et al. (2003) Overexpression of NPM-ALK induces different types of malignant lymphomas in IL-9 transgenic mice. Oncogene 22: 517–527 [DOI] [PubMed] [Google Scholar]
- Lawrie C., Saunders N., Soneji S., Palazzo S., Dunlop H., Cooper C., et al. (2008) MicroRNA expression in lymphocyte development and malignancy. Leukemia 22: 1440–1446 [DOI] [PubMed] [Google Scholar]
- Levy D., Darnell J., Jr (2002) Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3: 651–662 [DOI] [PubMed] [Google Scholar]
- Li G. (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18: 85–98 [DOI] [PubMed] [Google Scholar]
- Majmundar A., Wong W., Simon M. (2010) Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40: 294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoukian A., Woodgett J. (2002) Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv Cancer Res 84: 203–229 [DOI] [PubMed] [Google Scholar]
- Marzec M., Liu X., Wong W., Yang Y., Pasha T., Kantekure K., et al. (2011) Oncogenic kinase NPM/ALK induces expression of HIF1α mRNA. Oncogene 30: 1372–1378 [DOI] [PubMed] [Google Scholar]
- Marzec M., Zhang Q., Goradia A., Raghunath P., Liu X., Paessler M., et al. (2008) Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 105: 20852–20857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama H., Suzuki H., Nishimori H., Noguchi M., Yao T., Komatsu N., et al. (2011) miR-135b Mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood 118: 6881–6892 [DOI] [PubMed] [Google Scholar]
- McDonnell S., Hwang S., Basrur V., Conlon K., Fermin D., Wey E., et al. (2011) NPM-ALK signals through glycogen synthase kinase 3β to promote oncogenesis. Oncogene 31: 3733-3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel O., Hamacher F., Laimer D., Sifft E., Trajanoski Z., Scheideler M., et al. (2010) Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK-anaplastic large-cell lymphoma. Proc Natl Acad Sci U S A 107: 16228–16233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller P., Diorio C., Moyer M., Schnur R., Bruskin A., Cullen W., et al. (1994) Depletion of the erbB-2 gene product p185 by benzoquinoid ansamycins. Cancer Res 54: 2724–2730 [PubMed] [Google Scholar]
- Minami Y., Kiyoi H., Yamamoto Y., Yamamoto K., Ueda R., Saito H., et al. (2002) Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by HSP90 inhibitors. Leukemia 16: 1535–1540 [DOI] [PubMed] [Google Scholar]
- Morris S., Kirstein M., Valentine M., Dittmer K., Shapiro D., Saltman D., et al. (1994) Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 263: 1281–1284 [DOI] [PubMed] [Google Scholar]
- Naoe T., Kiyoe H., Yamamoto Y., Minami Y., Yamamoto K., Ueda R., et al. (2001) FLT3 tyrosine kinase as a target molecule for selective antileukemia therapy. Cancer Chemother Pharmacol 48(Suppl. 1): S27–S30 [DOI] [PubMed] [Google Scholar]
- Niwa H., Miyazaki J., Smith A. (2000) Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet 24: 372–376 [DOI] [PubMed] [Google Scholar]
- Ogawa S., Takita J., Sanada M., Hayashi Y. (2011) Oncogenic mutations of ALK in neuroblastoma. Cancer Sci 102: 302–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe M., Uehara Y., Miyagishima T., Itaya T., Tanaka M., Kuni-Eda Y., et al. (1992) Effect of herbimycin A, an antagonist of tyrosine kinase, on bcr/abl oncoprotein-associated cell proliferations: abrogative effect on the transformation of murine hematopoietic cells by transfection of a retroviral vector expressing oncoprotein P210bcr/Abl and preferential inhibition on Ph1-positive leukemia cell growth. Blood 80: 1330–1338 [PubMed] [Google Scholar]
- Okita K., Yamanaka S. (2010) Induction of pluripotency by defined factors. Exp Cell Res 316: 2565–2570 [DOI] [PubMed] [Google Scholar]
- Palmer R., Vernersson E., Grabbe C., Hallberg B. (2009) Anaplastic lymphoma kinase: signalling in development and disease. Biochem J 420: 345–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I., Zhao R., West J., Yabuuchi A., Huo H., Ince T., et al. (2008) Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451: 141–146 [DOI] [PubMed] [Google Scholar]
- Pearson J.D., Mohammed Z., Bacani J.T., Lai R., Ingham R.J. (2012) The heat shock protein-90 co-chaperone, Cyclophilin 40, promotes ALK-positive, anaplastic large cell lymphoma viability and its expression is regulated by the NPM-ALK oncoprotein. BMC Cancer 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piva R., Agnelli L., Pellegrino E., Todoerti K., Grosso V., Tamagno I., et al. (2010) Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J Clin Oncol 28: 1583–1590 [DOI] [PubMed] [Google Scholar]
- Piva R., Pellegrino E., Mattioli M., Agnelli L., Lombardi L., Boccalatte F., et al. (2006) Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and Bcl2A1 as critical target genes. J Clin Invest 116: 3171–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulford K., Lamant L., Morris S., Butler L., Wood K., Stroud D., et al. (1997) Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 89: 1394–1404 [PubMed] [Google Scholar]
- Qiu L., Lai R., Lin Q., Lau E., Thomazy D., Calame D., et al. (2006) Autocrine release of interleukin-9 promotes JAK3-dependent survival of ALK+ anaplastic large-cell lymphoma cells. Blood 108: 2407–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitmair A., Schmits R., Ewel A., Bapat B., Redston M., Mitri A., et al. (1995) MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat Genet 11: 64–70 [DOI] [PubMed] [Google Scholar]
- Rohwer N., Cramer T. (2011) Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat 14: 191–201 [DOI] [PubMed] [Google Scholar]
- Rush J., Moritz A., Lee K., Guo A., Goss V., Spek E., et al. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol 23: 94–101 [DOI] [PubMed] [Google Scholar]
- Sasaki T., Janne P. (2011) New strategies for treatment of ALK-rearranged non-small cell lung cancers. Clin Cancer Res 17: 7213–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T., Blagosklonny M., Ingui C., Neckers L. (1995) Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J Biol Chem 270: 24585–24588 [DOI] [PubMed] [Google Scholar]
- Shiota M., Fujimoto J., Semba T., Satoh H., Yamamoto T., Mori S. (1994) Hyperphosphorylation of a novel 80 kDa protein-tyrosine kinase similar to Ltk in a human Ki-1 lymphoma cell line, AMS3. Oncogene 9: 1567–1574 [PubMed] [Google Scholar]
- Stancato L., Chow Y., Hutchison K., Perdew G., Jove R., Pratt W. (1993) Raf exists in a native heterocomplex with Hsp90 and p50 that can be reconstituted in a cell-free system. J Biol Chem 268: 21711–21716 [PubMed] [Google Scholar]
- Stoklosa T., Poplawski T., Koptyra M., Nieborowska-Skorska M., Basak G., Slupianek A., et al. (2008) BCR/ABL inhibits mismatch repair to protect from apoptosis and induce point mutations. Cancer Research 68: 2576–2580 [DOI] [PubMed] [Google Scholar]
- Taipale M., Jarosz D., Lindquist S. (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11: 515–528 [DOI] [PubMed] [Google Scholar]
- Trepel J., Mollapour M., Giaccone G., Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10: 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishwamitra D., Li Y., Wilson D., Manshouri R., Curry C.V., Shi B., et al. (2012) MicroRNA 96 is a post-transcriptional suppressor of anaplastic lymphoma kinase expression. American Journal of Pathology 180: 1772–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Umekawa H., Olson M. (1993) Expression and subcellular locations of two forms of nucleolar protein B23 in rat tissues and cells. Cell Mol Biol Res 39: 33–42 [PubMed] [Google Scholar]
- Wang P., Wu F., Zhang J., McMullen T., Young L., Ingham R., et al. (2011) Serine phosphorylation of NPM-ALK, which is dependent on the auto-activation of the kinase activation loop, contributes to its oncogenic potential. Carcinogenesis 32: 146–153 [DOI] [PubMed] [Google Scholar]
- Witthuhn B., Silvennoinen O., Miura O., Lai K., Cwik C., Liu E., et al. (1994) Involvement of the JAK-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 370: 153–157 [DOI] [PubMed] [Google Scholar]
- Wolk K., Witte E., Witte K., Warszawska K., Sabat R. (2010) Biology of interleukin-22. Semin Immunopathol 32: 17–31 [DOI] [PubMed] [Google Scholar]
- Wu F., Wang P., Young L., Lai R., Li L. (2009) Proteome-wide identification of novel binding partners to the oncogenic fusion gene protein, NPM-ALK, using tandem affinity purification and mass spectrometry. Am J Pathol 174: 361–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Wang P., Zhang J., Young L., Lai R., Li L. (2010) Studies of phosphoproteomic changes induced by nucleophosmin-anaplastic lymphoma kinase (ALK) highlight deregulation of tumor necrosis factor (TNF)/Fas/TNF-related apoptosis-induced ligand signaling pathway in ALK-positive anaplastic large cell lymphoma. Mol Cell Proteomics 9: 1616–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Mani S., Donaher J., Ramaswamy S., Itzykson R., Come C., et al. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927–939 [DOI] [PubMed] [Google Scholar]
- Yin T., Yang L., Yang Y. (1995) Tyrosine phosphorylation and activation of JAK family tyrosine linases by interleukin-9 in MO7E cells. Blood 85: 3101–3106 [PubMed] [Google Scholar]
- Young L., Bone K., Wang P., Wu F., Adam B., Hegazy S., et al. (2011) Fusion tyrosine kinase NPM-ALK deregulates MSH2 and suppresses DNA mismatch repair function novel insights into a potent oncoprotein. Am J Pathol 179: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamo A., Chiarle R., Piva R., Howes J., Fan Y., Chilosi M., et al. (2002) Anaplastic lymphoma linase (ALK) Activates STAT3 and protects hematopoietic cells from cell death. Oncogene 21: 1038–1047 [DOI] [PubMed] [Google Scholar]
- Zhang J., Wang P., Wu F., Li M., Sharon D., Ingham R., et al. (2012) Aberrant expression of the transcriptional factor Twist1 promotes invasiveness in ALK-positive anaplastic large cell lymphoma. Cell Signal 24: 852–858 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Raghunath P., Xue L., Majewski M., Carpentieri D., Odum N., et al. (2002) Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol 168: 466–474 [DOI] [PubMed] [Google Scholar]
- Zhang Q., Wang H., Kantekure K., Paterson J., Liu X., Schaffer A., et al. (2011a) Oncogenic tyrosine kinase NPM-ALK induces expression of the growth-promoting receptor ICOS. Blood 118: 3062–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Wang H., Liu X., Bhutani G., Kantekure K., Wasik M. (2011b) IL-2R common gamma-chain is epigenetically silenced by nucleophosphin-anaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc Natl Acad Sci U S A 108: 11977–11982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Wang H., Marzec M., Raghunath P., Nagasawa T., Wasik M. (2005) STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci U S A 102: 6948–6953 [DOI] [PMC free article] [PubMed] [Google Scholar]