

Abstract
1-Hexadecyl-3-trifluoroethylglycero-sn-2-phosphomethanol (MJ33) is a fluorinated phospholipid analog that inhibits the phospholipase A2 (PLA2) activity of peroxiredoxin 6 (Prdx6). Prdx6 PLA2 activity is required for activation of NADPH oxidase 2 and subsequent generation of reactive oxygen species (ROS). In vitro, MJ33 inhibited agonist-stimulated production of ROS by the isolated perfused mouse lung, lung microvascular endothelial cells, and polymorphonuclear leukocytes. MJ33 (0.02–0.5 µmol MJ33/kg body weight) in mixed unilamellar liposomes was administered to C57BL/6 mice by either intratracheal (i.t.) or i.v. routes. Lung MJ33 content, measured by liquid chromatography/mass spectroscopy, showed uptake of 67–87% of the injected dose for i.t. and 23–42% for i.v. administration at 4 hours postinjection. PLA2 activity of lung homogenates was markedly inhibited (>85%) at 4 hours postadministration. Both MJ33 content and PLA2 activity gradually returned to near control levels over the subsequent 24–72 hours. Mice treated with MJ33 at 12.5–25 µmol/kg did not show changes (compared with control) in clinical symptomatology, body weight, hematocrit, and histology of lung, liver, and kidney during a 30- to 50-day observation period. Thus, the toxic dose of MJ33 was >25 µmol/kg, whereas the PLA2 inhibitory dose was approximately 0.02 µmol/kg, indicating a high margin of safety. MJ33 administered to mice prior to lung isolation markedly reduced ROS production and tissue lipid and protein oxidation during ischemia followed by reperfusion. Thus, MJ33 could be useful as a therapeutic agent to prevent ROS-mediated tissue injury associated with lung inflammation or in harvested lungs prior to transplantation.
Introduction
The NADPH oxidases (NOXs) are a widely distributed family of proteins in mammalian organs that enzymatically generate reactive oxygen species (ROS) (Krause et al., 2012; Takac et al., 2012). NOX2, the original family member to be described, has been the best studied from both biochemical and physiologic standpoints (Babior, 2002). ROS generated by NOX2 activity regulate many important cell and physiologic processes and also have an important role in host defense (Ushio-Fukai and Alexander, 2004; Leto and Geiszt, 2006; van der Vliet, 2008). However, activation of NOX2 can be associated with the deleterious effects of inflammation (Takac et al., 2012).
Inflammation is a complex process that involves cellular recruitment and activation, release of various effector molecules, and additional mechanisms that contribute to the host response to trauma, infection, and other environmental insults. However, an overly exuberant inflammatory response can amplify the injury that occurs with a primary insult. Oxidant stress subsequent to generation of ROS is considered to be one of the mechanisms that may lead to tissue injury during inflammatory states. Molecular targets of oxidant stress include lipids, proteins, and DNA. Among other effects, peroxidation of membrane lipids represents a major threat to cellular integrity, oxidation of proteins can alter metabolic function, and oxidative damage to DNA can impair cell division and repair. Thus, modulation of oxidant stress can have important implications for clinical management of conditions associated with inflammation.
In this study, our focus was the lung, which is especially at risk for inflammatory injury associated with sepsis or inhalation of toxic agents. Previous studies have examined the effect of antioxidants in protecting lungs from ROS-mediated injury with mixed results. This latter approach has an obvious drawback since an ROS scavenger has to compete with tissue components for reaction with ROS, unavoidably resulting in some tissue oxidation. Thus, ROS scavenging is unlikely to be fully protective against cellular injury. On the other hand, targeting the pathways that generate ROS has theoretical appeal since it could be more effective in preventing tissue oxidation. Since NOX2 is a major source of ROS with inflammation, there have been some efforts devoted to investigation of inhibitors of this enzyme. A relatively recent review pointed out that most currently available NOX inhibitors have low selectivity, potency, and bioavailability, precluding their therapeutic use and work in this area is continuing (Krause et al., 2012).
We recently demonstrated that peroxiredoxin 6 (Prdx6) is a key component of the NOX2 activation pathway in pulmonary endothelial cells and alveolar macrophages (Chatterjee et al., 2011) and a subsequent publication showed a similar requirement for a polymorphonuclear leukocyte (PMN) cell line (Ellison et al., 2012). Peroxiredoxins are a highly conserved family of peroxidases that are widely expressed throughout all kingdoms of life (Rhee et al., 2005). Prdx6 is a novel 1-cysteine member of the family that uniquely exhibits both glutathione peroxidase and phospholipase A2 (PLA2) activities (Manevich and Fisher, 2005; Schremmer et al., 2007; Fisher, 2011). These activities of Prdx6 play important roles in lung antioxidant defense and phospholipid metabolism. Activation of NOX2 is associated solely with the PLA2 activity of Prdx6 as demonstrated by rescue experiments in Prdx6 null cells (Chatterjee et al., 2011; Ellison et al., 2012). Phosphorylation of Prdx6 via a mitogen-activated protein kinase (Wu et al., 2009) followed by translocation of phosphorylated Prdx6 to the cell membrane are crucial steps in the NOX2 activation pathway (Chatterjee et al., 2011).
The requirement of Prdx6 PLA2 activity for activation of NOX2 provides a new target for modulation of ROS production. This study proposes the use of 1-hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol (MJ33), a known inhibitor of the PLA2 activity of Prdx6 that does not inhibit its peroxidase activity (Fisher and Dodia, 1996; Kim et al., 1997). MJ33 (Supplemental Fig. 1) is a fluorinated analog of the sn-2 tetrahedral of a phospholipid transition state (Jain et al., 1991b) that shows relatively tight binding to Prdx6 (Manevich and Fisher, 2005) and inhibits the PLA2 activity of phosphorylated recombinant Prdx6 by greater than 98% (Wu et al., 2009). We recently demonstrated that use of a system to target MJ33 to the pulmonary endothelium of mice blocks the increases in vascular cell adhesion molecule expression and alveolar permeability after intratracheal administration of bacterial lipopolysaccharide (Hood et al., 2012).
In this study, we evaluated the effect of MJ33 on the agonist-modulated activation of NOX2 activity in the intact isolated lung, in lung endothelial cells, and in PMNs. Furthermore, we determined the uptake of MJ33 by the intact lung, the in vivo dose-effect relationship for inhibition of PLA2 activity and the potential toxicity of MJ33. Finally, we evaluated the effect of MJ33 on oxidative injury in a model of lung ischemia/reperfusion (I/R). These studies are oriented toward the potential use of MJ33 as a therapeutic agent.
Materials and Methods
Reagents.
MJ33 is a fluorinated lipid analog that was originally synthesized by Dr. Mahendra Jain, one of the co-authors of this study. The compound is lipid soluble and somewhat soluble in aqueous media with a critical micelle concentration of approximately 10 µM. This agent has been shown to have specificity for PLA2, specifically pancreatic (type 1B) PLA2 and Prdx6 PLA2, but does not inhibit cytosolic (type IV) PLA2, phospholipases C/D, or other lipases (Jain et al., 1991a; Fisher et al., 1992; Gelb et al., 1994; Kim et al., 1997). MJ33 is a chemically nonreactive compound that mimics the tetrahedral transition state of PLA2 substrates and inhibits activity through its binding to the PLA2 protein (Gelb et al., 1994). The inhibitor is effective at mol fraction (MJ33/ lipid substrate) of <0.01 indicating a high binding affinity (Fisher et al., 1992). MJ33 inhibits NOX2 by preventing the generation of the lipid products of PLA2 activity that are necessary for NOX2 activation (Chatterjee et al., 2011). At this time, a requirement for Prdx6 in the activation of other NOX isoforms has not been demonstrated. For the present studies, MJ33 (as the lithium salt) was purchased from Sigma-Aldrich (St. Louis, MO).
Authentic lipids, including 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphocholine (17:0 Lyso PC) used as an internal standard for mass spectrometry (MS), were purchased from Avanti Polar Lipids (Alabaster, MA). [3H]dipalmitoylphosphatidylcholine ([3H]DPPC) was purchased from American Radiolabeled Chemicals (St. Louis, MO). Horseradish peroxidase (HRP) and the fluorescent dyes dihydrodichlorofluorescein (H2DCF) diacetate, dihydroethidine (HE), and Amplex Red were obtained from Life Technologies (Carlsbad, CA). Angiotensin II (AngII) was purchased from Bachem Bioscience (Torrance, CA). Ammonium acetate, methanol, chloroform, bovine superoxide dismutase (SOD), and concanavalin A (Con A) were purchased from Sigma-Aldrich. High-performance liquid chromatography–grade water and acetonitrile were obtained from Fisher Scientific (Fair Lawn, NJ). Gases were supplied by BOC Gases (Lebanon, NJ). A Kinetex C8 column was obtained from Phenomenex (Torrace, CA) and WST-1 was from Dojindo (Kumamoto, Japan).
Cell Isolation and Culture.
These studies used murine wild-type and NOX2 null pulmonary microvascular endothelial cells (mPMVECs), human pulmonary artery smooth muscle cells (hPASMCs), murine PMNs, and A549 cells. mPMVECs were primary isolates from mouse lungs as described previously (Chatterjee et al., 2006) and used at passages 7–12. A549, a lung carcinoma cell line, was obtained from the American Type Culture Collection (ATCC, Manassas, VA). mPMVEC and A549 human lung epithelial cells were grown at 37°C in Dulbecco’s modified Eagle’s medium (DMEM)–low glucose medium supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT), 25 mM HEPES, and 5 mM l-glutamine without antibiotics; for endothelial cells, growth supplement from bovine neural tissue was added at 1.5 mg/100 ml (E2759; Sigma-Aldrich). Immortalized hPASMCs, a gift from Dr. Elena Goncharova (Krymskaya et al., 2011), were grown in DMEM–low glucose medium supplemented with 5% FBS, 2 mM l-glutamine with antibiotics, and 1% insulin, transferrin, and selenium.
Bone marrow PMNs were isolated from mouse femurs as described previously (Nick et al., 2000). Briefly, mice were euthanized by intraperitoneal pentobarbital and fibias and femurs were surgically removed and flushed with Hanks balanced salt solution (minus Ca+2 and Mg+2) using a 25-gauge needle. Marrow cords were suspended in isolation buffer and marrow cells were obtained by repeated aspiration (Lieber et al., 2004). The suspension was layered on a three-step Percoll gradient (72, 64, and 52%). Following centrifugation at 1000g for 30 minutes, cells at the interface of the 72 and 64% Percoll layers were removed and washed with isolation buffer prior to use.
Mice.
Ten-week-old C57BL/6 wild-type and NOX2 (gp91phox) null mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Animal care was in compliance with all regulations as set by the University of Pennsylvania Institutional Animal Care and Use Committee and by the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Animals were kept under high-efficiency particulate filtered air in the vivarium maintained at 25°C ± 3°C.
Preparation and Administration of Liposomes.
Mixed unilamellar liposomes were prepared using authentic lipids with the mole fractional composition 0.5 DPPC, 0.25 egg phosphatidylcholine (PC), 0.15 cholesterol, and 0.1 phosphatidylglycerol; this mixture reflects the approximate composition of lung surfactant (Fisher et al., 2005). Liposomes were prepared by evaporating the mixture of lipids under N2 gas overnight to form a film that was re-suspended with vigorous mixing in phosphate-buffered saline (PBS) with MJ33 at 1–10 mol %. The mixture was frozen and thawed three times by exposure to liquid N2, followed by incubation in a 50°C water bath and then extrusion at 50°C for 20 cycles through a 100-nm pore size filter (Avanti Polar Lipids Mini-Extruder). The recovery of lipid after the filtration step, based on [3H] label in DPPC, was 97% ± 2.3% (mean ± S.E., n = 8) of the initial dpm. Liposome size (approximately 100 nm) was confirmed by measurement of dynamic light scattering using a 90 Plus Particle Sizer (Brookhaven Institute, Holtsville, NY). All experiments with in vivo administration of MJ33 used the liposomal formulation as the drug carrier.
For i.v. injection, mice were placed in a restraining apparatus, the two lateral tail veins were dilated by placing the tail in warm water (40°C) for several minutes, and a 1 ml syringe with a 26G3/8 needle was inserted into one of the veins. Liposomes were injected as a 100 µl bolus. For intratracheal (i.t.) injection, a midline neck incision was performed to expose the trachea of anesthetized mice. A small puncture hole between the third and the fourth ring in the trachea was made using fine micro-dissecting eye scissors and liposomes were infused directly using a 1 ml syringe connected to PE-10 tubing. Liposomes were instilled as a 50 µl bolus. A different i.t. method was used for toxicity studies to avoid a surgical incision. With animals in a Plexiglas restraining apparatus, a light-guided laryngoscope (45° angle) was used to visualize the trachea and liposomes were infused through a catheter.
ROS Production by Cells.
mPMVECs or hPASMCs in culture were treated with control liposomes or liposomes containing MJ33 (1 mol %) for 30 minutes and then incubated in the dark with an ROS-sensitive indicator dye, H2DCF diacetate, or HE, for an additional 15 minutes. Cells were washed and treated with agonist to activate NOX2 and then ROS production by cells was measured by fluorescence microscopy. Dichlorofluorescein (DCF) fluorescence was measured at excitation/emission of 490/530 nm and HE fluorescence at 470/610 nm using an epifluorescence microscope fitted with a ×20 objective (Nikon Diaphot TMD; Nikon, Melville, NY). Cells were randomly selected from the phase contrast images, and the intensity of cellular fluorescence was measured using Metamorph software (Molecular Devices, Sunnyvale, CA). For each field, the intensity of 6–10 cells was averaged, and 4–5 fields were analyzed for each treatment condition. Additional experiments were performed by measuring total DCF fluorescence of mPMVECs grown in multiwell plates using a fluorescence microplate reader (FLX 800; Biotek, Winoski, VT). An increase in DCF or HE fluorescence reflects the oxidation of deacetylated H2DCF, whereas increased HE fluorescence reflects the generation of hydroxyethidium or other oxidation products.
Superoxide (O2.−) production by PMNs was measured colorimetrically by the change in absorbance with reduction of the tetrazolium salt, WST-1 (Tan and Berridge, 2000). WST is reduced by O2.− to a soluble blue formazan dye (Marshall et al., 1995). PMNs (5 × 105) were equilibrated for 15 minutes at 37°C without or with MJ33 (1 mol %) in liposomes and then WST (500 µM) and Con A (250 µg/ml) were added to the incubation medium. Where indicated, SOD (100 U) was added to the incubation medium. The reduction of WST-1 was measured at 450 nm using a Cary 50 Bio UV-Visible spectrophotometer (Agilent Technologies, Foster City, CA). SOD-sensitive O2.– production was calculated from results in the presence and absence of SOD using a molar extinction coefficient of 37,000 M−1 cm−1 (Tan and Berridge, 2000)
Isolated Lung Perfusion.
Mice were anesthetized with i.p. sodium pentobarbital at 50 mg/kg body weight. Liposomes with or without MJ33 (1 mol %) were instilled into the lung through an endotracheal catheter placed at the level of the tracheal carina. After 15 minutes, the lungs were cleared of blood, removed from the thorax, and placed in a chamber for continuous perfusion as described previously (Fisher et al., 1992, 2005). Lungs were continuously ventilated through a tracheal cannula with air containing 5% CO2 and perfused with recirculating Krebs-Ringer bicarbonate solution supplemented with 10 mM glucose and 3% bovine serum albumin. ROS generation was measured by the addition of Amplex Red (25 µM) plus HRP (25 µg/ml) to the perfusate (Chatterjee et al., 2011). Amplex Red does not permeate the cell membrane and thus detects extracellular H2O2. AngII (50 µM) was added to the lung perfusate as a NOX2 agonist. Aliquots of the perfusate were removed at 15 minute intervals, and fluorescence intensity of the oxidized Amplex Red product was measured (excitation/emission, 545/610 nm) using a spectrofluorometer (Photon Technology International, Birmingham, NJ). At the end of the perfusion period, the lung was removed from the chamber and dried in an oven to constant weight. Oxidation of Amplex Red was calculated as nanomoles per gram dry weight of lung using an extinction coefficient of 54,000 cm−1 M−1. The slope of the line for Amplex Red oxidation versus time was calculated by the least mean squares method.
Isolated lungs from wild-type and NOX2 null mice also were studied for I/R injury. In some mice, MJ33 (4 nmol) was administered i.v. 15 minutes before harvest of the lung. The isolated perfused and ventilated lung was equilibrated for 30 minutes and then subjected to ischemia by stopping the perfusion pump while ventilation continued; this model results in ischemia without tissue hypoxia (Eckenhoff et al., 1992; Al-Mehdi et al., 1998; Zhang et al., 2005). The perfusion pump was restarted after 60 minutes of ischemia; reperfusion was continued for another 60 minutes at which time the lungs were harvested for measurement of lung oxidant injury as assessed by lipid peroxidation (thiobarbituric acid reactive substances, 8-isoprostanes) and protein oxidation (protein carbonyls). These procedures for analysis of lung oxidative injury were reported previously (Liu et al., 2010). ROS production during reperfusion was measured by addition of Amplex Red (plus HRP) to the perfusion fluid during the initial equilibration period as described above.
Assay for PLA2 Activity.
To measure the effect of MJ33 on PLA2 activity in vivo, MJ33 in liposomes was injected via i.t. or i.v. routes. Both male and female mice were used for these studies. Mice were sacrificed at intervals by exsanguination under pentobarbital anesthesia and lungs were surgically removed, cleared of blood, and then homogenized for analysis of PLA2 activity. The substrate for measurement of PLA2 activity was mixed unilamellar liposomes containing tracer [3H]DPPC (4400 dpm/nmol) incubated with lung homogenate at 37°C for 1 hour under acidic, Ca2+-free conditions (40 mM sodium acetate: 5 mM EDTA buffer, pH 4.0) (Chen et al., 2000). The reaction was stopped by addition of chloroform/methanol (1:2) and lipids were extracted by the Bligh and Dyer method (Bligh and Dyer, 1959). Radiolabeled free fatty acids were separated by a two-step thin layer chromatography procedure using hexane/ether/acetic acid as a solvent system along with authentic palmitic acid as a standard. The free fatty acid spots were identified using I2 vapor, scraped from the plate, and analyzed by scintillation counting using an internal standard for quench correction. Values for recovered dpm were corrected for blank values obtained in the absence of enzyme. PLA2 activity was calculated based on the specific radioactivity of DPPC (Chen et al., 2000).
Detection of MJ33 in Lung Tissue by MS.
MJ33 was measured in lungs from mice that had been injected with varying amounts of MJ33 via an i.t. or i.v. route. Animals were sacrificed at varying times post-MJ33 injection. Lungs were cleared of blood by perfusion through the pulmonary artery and homogenized for analysis of MJ33 levels using a triple quadrupole mass spectrometer (Supplemental Fig. 1A). In some experiments, lungs were lavaged before homogenization by instillation and aspiration (×5) of 800 µl cold PBS. For extraction of lipids, lung homogenate (0.5 ml), bronchoalveolar lavage fluid, or equivalent H2O as a control was transferred to a 10-ml glass test tube containing 1 ml PBS buffer (1 M, pH 6.8) that was spiked with 2 ng 17:0 Lyso PC (Supplemental Fig. 1B). The latter was chosen as an internal standard (STD) since it is not an endogenous phospholipid, as confirmed by the lack of a corresponding peak in the MS transition. After adding 2 ml methanol to each tube, the samples were vortexed for 30 seconds, and 4 ml chloroform was added to the samples and shaken for 30 minutes. After centrifugation at 2500g for 5 minutes, the organic layer was transferred to a new clean glass tube, and then evaporated to dryness under nitrogen, reconstituted in 100 µl methanol, and maintained at 4°C in the auto-sampler tray before analysis.
Reverse-phase chromatography for liquid chromatography (LC)/MS measurements was performed using a Waters Alliance 2690 high-performance liquid chromatography system (Waters Corp., Milford, MA) with gradient elution at 30°C in the linear mode with a Kinetex C8 column (100 × 2.6 mm i.d., 2.6 µm) using a flow rate of 0.2 ml/min. Solvent A was water and solvent B was 95% acetonitrile, each with 10 mM ammonium acetate. The linear gradient was as follows: 30% B at 0 minutes, 30% B at 2 minutes, 100% B at 8 minutes, 100% B at 16 minutes, 30% B at 16 minutes, and 30% B at 25 minutes.
MS was conducted on a Thermo Finnigan TSQ Quantum Ultra AM mass spectrometer (Thermo Fisher, San Jose, CA) equipped with an electrospray ionization source operated in the positive ion mode. Unit resolution was maintained for both parent and product ions for multiple reaction monitoring analyses. Operating conditions were as follows: spray voltage, 3500 V; vaporizer temperature, 300°C; and heated capillary temperature, 280°C. Nitrogen was used for the sheath gas and auxiliary gas set at 25 and 10 (in arbitrary units), respectively. Collision induced dissociation was performed using argon as the collision gas at 1.5 mTorr in the second (rf only) quadrupole. An additional DC offset voltage was applied to the region of the second multipole ion guide (Q0) at 5 V. The following Selected Reaction Monitoring transitions were monitored; MJ33, mass-to-charge ratio (m/z) 493 → 113 (collision energy 18 eV) as a qualifier, m/z 493 → 119 (collision energy 15 eV) as a quantifier, 17:0 Lyso PC, m/z 510 → 104 (collision energy 18 eV) as a qualifier, and m/z 510 → 184 (collision energy 15 eV) as a quantifier. The mass spectrum of MJ33 showed the fragments 381, 139, and 113. A linear calibration curve was constructed with authentic MJ33 in the range from 0.1 ng/ml to 10 ng/ml (y = 0.004x + 0.00007; r2 0.999). No peak corresponding to MJ33 was detected in the control samples. All data analysis was performed using Xcalibur software, version 2.0 SR2 (Thermo Fisher) from raw mass spectral data.
Proliferation, Cell Viability, and Clonogenic Survival in Response to MJ33.
For proliferation studies, T25 cell culture flasks were each plated with 1 × 105 cells; MJ33 was added to the plates on the next day. The rate of proliferation was determined on a daily basis for up to 10 days. Attached cells were made into a single suspension by incubation with 0.25% trypsin/EDTA for 3–5 minutes, and counted to obtain the total cell number on each plate. Cell viability was assessed by a trypan blue exclusion assay (Lee et al., 2000). Clonogenic survival studies were performed as described previously (Lee and Shogen, 2008). Briefly, 7 ml 15% FBS-DMEM (low glucose at 1 g/l) was added to 6-well plates with 102–104 cells that were incubated at 37oC for 5 h to allow for cell attachment and then incubated for various durations with MJ33 at 0–25 µM. Cultures were fixed with 99.5% isopropyl alcohol, stained with 1% crystal violet and counted. Colonies with more than 50 cells were scored as positive. The surviving fraction was calculated as follows: number of colonies counted/number of cells plated × fractional plating efficiency. To evaluate a potential role of cellular contact inhibition on MJ33 cytotoxicity for mPMVEC, feeder cells (heavily x-irradiated at 30 Gy with a 137Cs source, rendering them unable to generate clones) were added at 103, 104, or 105 cells per well to generate confluent or plateau-phase growth patterns.
Toxicity Studies in Mice.
Toxicity of MJ33 after its administration by i.v. or i.t. routes was evaluated by daily measurement of animal weight and observation of the level of spontaneous activity during the subsequent 60 days, measurement of hematocrit at 30 days after dosing, and histologic examination of lungs, liver, and kidneys at 10 days after MJ33 administration. Death was not used as an end point in accordance with policies of the University of Pennsylvania Institutional Animal Care and Use Committee. Only female mice were used for these studies because of the difficulties of housing male mice together for prolonged periods.
Measurement of Hematocrit.
To measure the hematocrit, a capillary pipette containing anticoagulant was inserted in the lateral canthus of the eye and 40 µl blood was collected from the retro-orbital sinus. The capillary tubes were centrifuged for 10 minutes at 12,500g (Clay Adams Autocrit Ultra3; Becton Dickinson, Franklin Lakes, NJ), and the separate bands of hematocrit were read on a hematocrit scale.
Histologic Evaluation.
Histologic evaluation of lungs, liver, and kidneys of mice was performed after administration of MJ33 (500 nmol) via i.v. or i.t. routes. Anesthetized mice were exsanguinated 10 days post-treatment by cutting the caudal vena cava. A cannula was inserted into the trachea and fixed with a ligature. Lungs and heart were removed en bloc, and lungs were inflated via the cannula by gentle infusion of fixative (10% phosphate-buffered formalin, pH 7.0) over 5 minutes to reach a constant fluid pressure of 25 cm H2O. The trachea was tied with a ligature, and the lungs were placed in a glass vial containing 10% formalin and were kept on ice for 24 hours. Livers and left kidneys also were collected and fixed in formalin. All tissue samples were processed by the Pathology Core at the Children’s Hospital of Philadelphia (Abramson Research Center, Philadelphia, PA). After fixation, tissues were embedded in paraffin and blocks were sectioned and stained with hematoxylin and eosin. The tissue sections were examined independently by two different investigators and a result was determined by consensus.
Statistical Analysis.
All measured values are presented as the mean ± S.E. for each group. Analysis of variance with the post hoc Tukey test (SysStat, San Jose, CA) was used for multiple group comparisons. Differences within a group before and after treatment were evaluated using a paired t test. Statistical significance was set at 95% (P < 0.05).
Results
Effect of MJ33 on Agonist-Induced ROS Production.
The effect of MJ33 on agonist-induced ROS generation was evaluated in isolated lungs of mice with Amplex Red (plus HRP) added to the perfusate to detect H2O2. Unilamellar liposomes containing MJ33 at 0, 0.2, or 4 nmol were injected i.v. into anesthetized mice and the lungs were isolated 15 minutes later for in vitro perfusion. Amplex Red oxidation was plotted versus time of perfusion (Fig. 1A). The addition of AngII to the perfusate resulted in an approximately 7-fold increase in the rate of Amplex Red oxidation (Fig. 1A). The effect of AngII was inhibited by approximately 25% by 0.2 nmol MJ33 and was essentially abolished in lungs that had been pretreated with 4 nmol MJ33. This result indicates ROS production in the presence of AngII and a dose-dependent inhibition by treatment with MJ33. Similar inhibition of Amplex Red oxidation was observed after pretreatment with 4 nmol MJ33 by the i.t. route (not shown). ROS production after AngII treatment of lungs from NOX2 null mice was decreased by >90% compared with wild-type lungs, indicating that NOX2 is the predominant source of ROS in this model (Fig. 1A). However, ROS production by NOX2 null lungs after AngII was greater (P < 0.05) than control (evaluated at 60 minutes of perfusion), suggesting that another (non-NOX2) albeit minor (approximately 9%) source of AngII-stimulated ROS production. There was no significant difference in AngII-stimulated ROS production between the wild type treated with 4 nmol MJ33 and the NOX2 null lungs, suggesting that this dose of MJ33 totally inhibited NOX2-dependent ROS production but had no effect on the NOX2-independent ROS production (Fig. 1A).
Fig. 1.

Inhibition of agonist-induced ROS production by MJ33. (A) Effect of MJ33 on AngII- induced ROS generation by isolated perfused wild-type (WT) and NOX2 null mouse lungs as measured by Amplex Red oxidation. Liposomes containing 0, 0.2, or 4 nmol MJ33 were administered intravenously over 1 min to the intact mouse. The lung was isolated 15 minutes later, placed in the temperature controlled (37°) chamber, and perfused in the absence or presence of AngII (50 µM). Perfusate samples were taken at 15-minute intervals and analyzed for oxidized Amplex Red; results are plotted versus time of perfusion. The numbers in parentheses indicate the slope (in nmol/g dry weight per minute) of the lines calculated by the least mean squares method. Results are mean ± S.E. for n = 3 (in some cases, the S.E. bars are within the plotted points). The slope of the line for NOX2 null mice minus AngII (not shown) was essentially identical to the control. (B) ROS production by isolated mPMVECs as indicated by epifluorescence microscopy for DCF. Cells were incubated for 30 minutes with liposomes with or without MJ33 (1 mol %), then loaded with H2DCF diacetate (5 µM) and stimulated with 10 µM AngII. Integrated fluorescence of 10–15 cells per field was quantitated using Metamorph software. Basal conditions represent no AngII. DCF fluorescence intensities are expressed in arbitrary units. Values are mean ± S.E. for n = 3. *P < 0.05 versus the corresponding basal. (C) Effect of MJ33 in liposomes (1 mol %) on the respiratory burst of mouse bone marrow PMNs stimulated with Con A (250 µg/ml). ROS production was evaluated at 30-second intervals by the change in absorbance due to reduction of WST-1. The control was Con A stimulation in the presence of SOD. This figure is representative for n = 3.
The effect of MJ33 on ROS production was evaluated further with mPMVECs in monolayer culture. ROS production was determined by oxidation of DCF or HE as assessed by fluorescence microscopy. mPMVECs that were stimulated by AngII showed increased DCF fluorescence (Fig. 1B) or HE fluorescence (not shown) that was abolished by pretreatment of cells with MJ33. Similar results were obtained for the effect of MJ33 on ROS production by mPMVEC by measuring cellular fluorescence with a multiplate reader (not shown). Significant ROS production was not seen after AngII treatment of NOX2 null mPMVEC (Fig. 1B).
PMNs isolated from mouse bone marrow also showed a robust respiratory burst, as measured by WST reduction in response to Con A (250 µg/ml) (Fig. 1C). Reduction of WST reached a plateau value in approximately 4 minutes. This stimulated O2.− generation was inhibited by pretreatment of PMNs with MJ33 (4 µM). The rate of Con A–stimulated O2.− generation, calculated from the change of WST-1 absorbance during the linearly increasing portion of the curve and corrected for the residual after SOD treatment was 484 ± 9 pmol/min per 106 cells for control cells and 104 ± 74 for cells pretreated with MJ33 (mean ± S.E., n = 3), a 79% reduction in the presence of MJ33. There was essentially no SOD-insensitive O2.− production by these cells (Fig. 1C). PMNs from NOX2 null mice showed minimal WST-1 reduction after Con A (41±18 pmol/min per 106 cells), indicating NOX2 as the predominant (>90%) source of O2.− under these experimental conditions (Fig. 1C). MJ33 had no effect on this residual O2.− production by NOX2 null PMNs (not shown).
The results for Amplex Red oxidation by the isolated lung stimulated with AngII indicated that about 10% of ROS production was through non-NOX2 sources. Although there are several possible enzymatic sources for non-NOX2 ROS production, NOX1 would be a likely possibility because this is the major source of ROS induced by AngII in vascular smooth muscle (Lassègue et al., 2001). We thus evaluated the possible effect of MJ33 on AngII-stimulated ROS production by hPASMCs. These cells showed an increase in ROS production in the presence of AngII that was less robust than the response of mPMVECs (Supplemental Fig. 2). ROS production by these cells was unaffected by the presence of MJ33, indicating that NOX1 does not require the PLA2 activity of Prdx6 for activation.
Uptake and Retention of MJ33 in the Lung.
As the initial step to evaluate potential use of MJ33 in vivo, the uptake and retention of MJ33 by the lung were determined after i.t. or i.v. administration of the agent. MJ33 accumulation in the lung homogenate and bronchoalveolar lavage fluid was evaluated by MS under positive electron spray ionization conditions. The mass spectrum of authentic MJ33 exhibited an intense parent ion at m/z 493 corresponding to the parent MH+ (not shown); the tandem mass spectrometry (MS/MS) spectrum indicated the product ions m/z 139 and m/z 113 (Supplemental Fig. 1). Since there is no isotopically labeled MJ33 commercially available, we chose 17:0 Lyso PC as an internal standard (STD). MS/MS analysis of the Lyso PC standard showed the characteristic PC fragment, m/z 184 (not shown). The calibration curve used to quantify MJ33 in lung tissue was constructed using the area ratio of the MJ33 to the area ratio of the STD. To confirm the presence of MJ33 in the lung homogenate, we used LC-MS/MS. The MJ33 detected in the lung homogenate had the same MH+ ion as authentic MJ33 and the same product ions, m/z 139 and m/z 113. The internal STD was used to confirm the relative retention time of the MJ33 detected in lung homogenate versus the relative retention time of authentic MJ33.
In control lung (liposomes only, no MJ33), a peak corresponding to MJ33 was not seen (Fig. 2A, top) but the presence of a peak corresponding to the STD (Fig. 2A, bottom) indicated the effective extraction of lipids from the sample. There were significant peaks corresponding to MJ33 and the STD after i.v. (Fig. 2B) or i.t. (Fig. 2C) injection of 0.4 nmol MJ33. At 4 hours after i.v. injection of 0.4, 4, or 10 nmol MJ33, 23–42% of the administered dose was found in the lung (Table 1). The retention in the lung at 4 hours after i.t. injection of MJ33 was 67–87% of the injected dose (0.4–10 nmol). The percentage of injected MJ33 present in the lung declined slightly at 24 and 48 hours after dosing but MJ33 was undetectable in the lung after either i.v. or i.t. administration at 72 hours post-treatment (Table 1). MJ33 was not detected in the bronchoalveolar lavage fluid at 4 hours after i.t. administration of 0.4 nmol (not shown). These results indicate that the MJ33 was retained in the lung for a considerable period and was either intracellular or tightly bound to the cell surface.
Fig. 2.
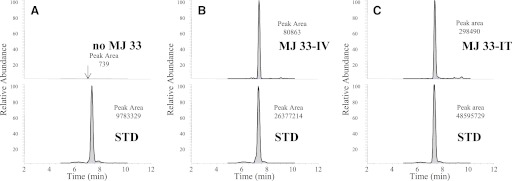
Measurement of MJ33 content in the lung using LC/MS. MJ33 (0.4 nmol in liposomes) was injected i.v. (B) or i.t. (C) in the intact mouse. The lungs were isolated 4 h later, cleared of blood, and homogenized. An internal standard (STD), 17:0 lysophosphatidylcholine, was added to the lung homogenate and the lipids were extracted. The chromatograms are shown in the upper panels for MJ33 and in the lower panels for the STD. The elution time for all MJ33 and STD peaks was 7.3 minutes. MJ33 was detected from m/z 493 → 139 and the STD from m/z 510 → 184. Liposomes without MJ33 were injected i.t. as a baseline control (A). The integrated peak area for each panel is indicated.
TABLE 1.
Lung uptake of MJ33 at increasing time after i.v. or i.t. injection as determined by MS
Data are presented as mean ± range for n = 2; all other values are single measurements. The amount of MJ33 was calculated based on the area ratios (Fig. 2) using a calibration curve constructed with the same amount of internal standard and variable amount of MJ33.
| Time Postinjection | Amount Injected | Uptake %, of Injected Dose |
|
|---|---|---|---|
| i.t. | i.v. | ||
| h | nmol | % | |
| 4 | 0.4 | 68 ± 4 | 23 ± 2 |
| 4 | 87 | 42 | |
| 10 | 67 | 33 | |
| 24 | 0.4 | 35 | 20 |
| 4 | 51 | 32 | |
| 10 | 55 | 31 | |
| 48 | 10 | 46 | 40 |
| 72 | 0.4 | nd | nd |
| 4 | nd | nd | |
| 10 | nd | nd | |
nd, none detected.
Inhibition of Lung PLA2 Activity.
The effect of MJ33 on lung PLA2 activity after i.t. or i.v. administration was evaluated using dosing protocols similar to those used for MS analysis. The enzymatic assay was performed at pH 4 in the absence of Ca2+, conditions that are relatively specific for the PLA2 activity of lung Prdx6 (Fisher et al., 1992; Fisher and Dodia, 1996). The patterns for inhibition of lung PLA2 activity were similar for MJ33 administered by either the i.v. (Fig. 3A) or i.t. (Fig. 3B) route. There was a marked (approximately 85%) decrease in lung PLA2 activity at the 4-hour time point that was similar for all three doses (0.4, 4, 10 nmol) of MJ33. However, the persistence of the inhibitory effect of MJ33 beyond 4 hours and the degree of recovery were dose dependent. For doses of 4 and 10 nmol, there was only slight recovery at 24 hours and partial recovery at 48 hours post-treatment but activity had returned close to the control values at 72 hours post-treatment. This pattern of effect on PLA2 activity agreed reasonably well with the pharmacokinetics of MJ33 in lung tissue as evaluated by MS/MS (Table 1). There were no differences between male and female mice in the time-related effect of MJ33 on lung PLA2 activity.
Fig. 3.

PLA2 activity of lung homogenate from male and female mice measured at varying times after i.v. (A) or i.t. (B) injection of MJ33 (0–10 nmol) in liposomes. PLA2 activity was measured by assay in Ca2+-free medium, pH 4. Results are mean ± S.E. for n = 3–5. *P < 0.05 versus control (no MJ33) for each time point.
In Vitro Cytotoxicity of MJ33.
Cytotoxicity of MJ33 was evaluated with A549 human lung epithelial cells and mPMVEC during the exponential growth phase and with confluence. Plating efficiency, calculated as the percentage of seeded cells that grow into macroscopic colonies in the absence of MJ33, was approximately 45% for mPMVECs and approximately 55% for A549 cells. There was no effect of 10–25 µM MJ33 on A549 cell division during a 10-day observation period (Fig. 4A). Survival of A549 cells exposed continuously during the exponential growth phase to 5–25 µM MJ33 was unaffected as determined by colony formation (clonogenic assay) at 2 or 7 days of exposure (Fig. 4B). We previously showed that acute exposure (<24 hours) to MJ33 at 10–25 µM did not reduce the viability of mPMVECs, as measured by a (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT) (Lien et al., 2012), and this study showed no decrease in survival for confluent cells exposed for 24 hours to 10 or 20 µM MJ33 (Fig. 4C). A clonogenic survival assay with exposures of mPMVEC for up to 7 days also was used to assess cytotoxicity. Continuous exposure to 5–10 µM MJ33 for 24 hours had no effect on survival of exponentially growing mPMVEC, although survival was decreased with longer exposure to MJ33 at concentrations >5 µM (Fig. 4C). The presence of a feeder cell layer resulted in a statistically significant (P < 0.05) but relatively modest (approximately 10%) increase of mPMVEC survival on exposure to 20 µM MJ33 for 24 hours (not shown). For recombinant Prdx6 assayed in vitro, PLA2 activity was inhibited by 50% at 0.3 µM and essentially 100% at 0.4 µM MJ33 (not shown). Thus, these studies with primary lung endothelial cells and a lung cancer cell line show a lack of toxicity for confluent cells with MJ33 up to 25 µM while MJ33 at concentrations > 5 µM appeared to be cytotoxic to the primary lung endothelial cells, but not the lung cancer cell line, during the exponential growth phase. The toxic concentration of MJ33 significantly exceeds the concentration required for inhibition of PLA2 activity.
Fig. 4.

Effect of MJ33 on the proliferation of cells in culture. (A) Proliferation of A549 cells during exposure to 0, 10, or 25 µM MJ33 during a 10-day incubation. (B) Clonogenic assay of A549 cells after a 2- or 7-day exposure to MJ33 at 0–25 µM. At the end of exposure, the number of colonies were counted and expressed as a fraction of the number of cells that were originally plated. (C) Clonogenic assay of mPMVECs treated with 0–25 µM MJ33 for 1, 2, or 7 days. Cells were studied during the exponential growth phase or at confluence (confl-24h). Mean ± S.E. for n = 3–4.
In Vivo Toxicity.
Toxicity studies of intact mice evaluated the effect of MJ33 on their activity level, body weight, hematocrit, and histology of lungs, liver, and kidneys. Mice (n = 5 per group) received a single dose of 250, 500, or 2500 nmol MJ33 (approximately 12.5–125 µmol/kg body weight) by the i.v. or i.t. route followed by a 7-week observation period (5 weeks for the higher dose). By clinical observation, mice in all three treatment groups exhibited generally normal behavior; some mice appeared to be slightly less active during the initial 1–3 days after MJ33 administration but then recovered during the subsequent days. Clinical manifestations such as hyperactivity, aggressive behavior, alopecia, dehydration, salivation, nasal discharge, constipation, or diarrhea were not observed and none of the mice died or had to be euthanized because of poor clinical appearance. A small weight loss was seen for control mice at the end of 1 week of observation that was possibly related to the experimental procedures; these mice during subsequent weeks progressively gained weight to surpass the initial body weight by week 4 (Fig. 5). The initial weight loss was slightly greater in most groups of mice dosed with MJ33, but the mice subsequently resumed their normal rate of weight gain. By 5 weeks postinjection, the mean weights for all groups except 2500 nmol i.t. were similar and exceeded their initial weight (Fig. 5). None of the changes in weight for the single dose groups was statistically different from control (P > 0.05).
Fig. 5.

Change in body weight for mice that were administered 250, 500, or 2500 nmol MJ33 in liposomes i.v. (A) or i.t. (B). An additional i.v. group received multiple doses (i.v.) at 2500 nmol per day for 4 days. Mouse body weights were measured at weekly intervals. Values are mean ± S.E. for n = 5 for each group. Initial body weight of mice was 21.5 ± 0.15 g (n = 45). No deaths occurred during the observation periods. *P < 0.05 compared with the corresponding control (no MJ33).
MJ33 was also given in repeated doses of 2.5 µmol i.v. daily for 4 days. The mean body weight in these mice decreased by about 12% compared with the initial weight in the first week of treatment and did not show a subsequent recovery as seen with the single injection condition (Fig. 5A). The difference in body weight beyond 3 weeks compared with control was statistically significant (P < 0.05). Like the single dose studies, clinical symptomology with the multiple dose regimen was not observed.
The hematocrit, the volume percentage of erythrocytes in blood, was measured prior to treatment and at 30 days after 0, 250, or 500 nmol MJ33 i.t. or i.v.. There was no significant effect of MJ33 on hematocrit at these doses (Table 2).
TABLE 2.
Hematocrit levels at 30 days after i.v. or i.t. injection of MJ33
Pretreatment hematocrit was 47.4 ± 0.2. Values are mean ± S.E. for n = 5.
| MJ33 Dose | Hematocrit |
|
|---|---|---|
| i.t. | i.v. | |
| Control (nmol)a | 47.7 ± 0.3 | 45.8 ± 0.9 |
| 250 | 47.2 ± 0.4 | 46 ± 0.3 |
| 500 | 46.4 ± 0.7 | 47.0 ± 0.4 |
Blank liposomes with MJ33.
Tissue sections of lungs, liver, and kidneys were examined by two observers with good agreement between their respective analyses. The lungs of MJ33-treated and control animals appeared similar with no evidence of alveolar edema, vascular congestion, inflammation, or destruction or fibrosis of alveolar septae (Fig. 6). Likewise, no histologic differences were found in the liver or kidneys in the MJ33 treated groups compared with control (Supplemental Fig. 3).
Fig. 6.

Effect of MJ33 on lung histology. The mice were administered MJ33, 500 nmol in liposomes or blank liposomes (control) by either i.t. or i.v. routes. The sections were stained with hematoxylin and eosin. Scale bar, 200 µm in (A); 50 µm in (B).
Effect of MJ33 on Lung I/R Injury.
ROS production measured by Amplex Red oxidation was increased significantly during the reperfusion phase of I/R. (ROS production during the ischemic period cannot be measured by this method.) The rate of ROS production with reperfusion (Fig. 7) was approximately 50% greater than the rate observed in the presence of AngII (Fig. 1). The rate of ROS production with reperfusion was decreased by 66% versus I/R by the presence of MJ33. ROS production was decreased further (approximately 80% versus I/R) with NOX2 null lungs indicating that the major fraction but not all of NOX2-dependent ROS production was inhibited by MJ33. Based on the result with NOX2 null lungs, approximately 20% of ROS production appeared to be from sources other than NOX2 in this model of lung injury.
Fig. 7.

ROS production during reperfusion in mouse lungs after 60 min ischemia. ROS production was measured by the Amplex Red method as described in Fig. 1. The dye was added to the perfusate during the 15-minute equilibration period prior to ischemia. A zero reperfusion time perfusate sample was used for background subtraction. Values represent mean ± S.E. for n = 3. The numbers in parentheses indicate the slope of the line (nmol/g dry weight per minute) calculated by least mean squares.
Oxidative stress associated with I/R was evaluated by measuring indices of lung lipid peroxidation (thiobarbituric acid reactive substance, 8-isoprostanes) and protein oxidation (protein carbonyls). Altered lung permeability was evaluated by the wet/dry weight ratio. Control was the isolated wild-type lung that was continuously perfused for 2 hours (Table 3). Values for continuously perfused NOX2 null lungs (not shown) were essentially identical to the control wild type. In wild-type mice, the I/R protocol resulted in a significant (P < 0.05) increase of 81–110% in the biochemical indices of oxidative stress. The wet/dry ratio of the lung also was significantly increased after I/R. These changes were largely abolished by pretreatment of the mice with MJ33. I/R in the NOX2 null lungs indicated that the bulk of the oxidative damage was due to activation of the NOX2 system. However, the values obtained for I/R with NOX2 null lungs were significantly greater than control, indicating that approximately 20% of the lung injury with I/R could be attributed to non-NOX2 sources of ROS (Table 3).
TABLE 3.
Effect of MJ33 on indices of oxidative stress associated with lung I/R
Studies were done with isolated lungs from wild-type or NOX2 null mice. Results are mean ± S.E. for n = 3.
| TBARS | 8-Isoprostanes | Protein Carbonyls | Lung Weight Wet/Dry | |
|---|---|---|---|---|
| nmol/mg prot | ||||
| Control | 74.6 ± 2.5*† | 210 ± 7*† | 5.07 ± 0.19*† | 5.85 ± 0.04*† |
| I/R, wild type | 136 ± 3.0† | 440 ± 23† | 9.25 ± 0.12† | 7.35 ± 0.07† |
| I/R + MJ33, wild type | 95.5 ± 2.1* | 310 ± 5*† | 7.00 ± 0.01*† | 6.35 ± 0.02* |
| I/R, NOX2 null | 82.3 ± 1.8* | 250 ± 5* | 6.09 ± 0.07* | 6.16 ± 0.007* |
Control, continuous perfusion; I/R, 1 hour ischemia followed by 1 hour reperfusion; TBARS, thiobarbituric and reactive substance.
P < 0.05 versus I/R, wild type; †P < 0.05 versus I/R, NOX2 null.
Discussion
Previous studies with mPMVECs, lung alveolar macrophages, and PMNs have shown that the PLA2 activity of Prdx6 is required for activation of NOX2 and the subsequent production of ROS (Chatterjee et al., 2011; Ellison et al., 2012). We proposed that inhibiting PLA2 activity could be useful therapeutically as a means to limit the activation of NOX2 and ROS production that are in part responsible for cell injury associated with lung inflammation (Kratzer et al., 2012; Min et al., 2012). In this study, we evaluated MJ33, a known inhibitor of the PLA2 activity of Prdx6 (Fisher and Dodia, 1996; Kim et al., 1997), as an inhibitor of NOX2 activation. As shown in this study, MJ33 does prevent the activation of NOX2 and generation of ROS. An important advantage of MJ33 is that this agent does not inhibit the peroxidase activity of Prdx6 (Fisher and Dodia, 1996) because this activity has been shown to be important for antioxidant defense (Wang et al., 2004, 2006a,b, 2008; Hood et al., 2012; Lien et al., 2012). This dichotomy is consistent with the clearly distinct active sites for the two enzymatic activities (Chen et al., 2000; Fisher, 2011). Thus, the important role of Prdx6 as a peroxide scavenger and its function in antioxidant defense is preserved in the presence of MJ33.
We first tested the effect of MJ33 on ROS generation by model systems including the isolated perfused mouse lung and mPMVECs in culture that were stimulated by AngII, and isolated bone marrow PMNs that were stimulated with Con A. Agonist-treated PMNs from NOX2 null mice showed scant ROS production in the absence of MJ33, indicating NOX2 as the predominant source of ROS; we have previously shown NOX2 as the predominant source of ROS in AngII-treated mPMVECs (Chatterjee et al., 2011). Study of NOX2 null lungs indicated that approximately 90% of ROS production by AngII-stimulated lungs was via NOX2. The addition of MJ33 to the lung perfusate or the cell incubation medium markedly inhibited the agonist-induced ROS production in each of these systems. MJ33 inhibits the activation of NOX2 by preventing the generation of lipid products of PLA2 activity (Chatterjee et al., 2011). Studies with pulmonary artery smooth muscle cells where NOX1 is the predominant source of ROS after AngII treatment (Lassègue et al., 2001) indicate that MJ33 does not inhibit this NOX isoform (Supplemental Fig. 2). These results suggest that MJ33 could be useful as a biologically active agent to inhibit NOX2-mediated ROS generation in vivo.
To better understand possible therapeutic applications of MJ33 in an intact animal, we measured the inhibitory effect of MJ33 on lung PLA2 activity with increasing concentration of the agent delivered by either i.t. or i.v. routes. PLA2 activity in the lung homogenate was significantly reduced to approximately 15% of the control level by MJ33 delivered by either route when measured at 4 hours after exposure, and remained significantly reduced at 4–24 hours after treatment. This 85% reduction in PLA2 activity (measured at pH 4 in Ca2+ free buffer) with MJ33 is similar to the effect of Prdx6 “knock-out,” indicating that the residual activity reflects some PLA2 source other than Prdx6 (Fisher et al., 2005). Varying doses of Prdx6 from 0.4 nmol to 10 nmol (i.t. and i.v.) were equally effective in inhibiting enzyme activity, but the recovery time of PLA2 activity was dose dependent. Based on these observations, a daily treatment with 4 nmol MJ33 (0.2 µmol/kg body weight) in the mouse could effectively maintain near total inhibition of lung Prdx6 PLA2 activity.
We overcame the inability to label MJ33 with fluorophores or other tags by developing a method using LC-MS/MS to assay the pharmacodynamics of MJ33. The method used three parameters to positively identify MJ33 in lung homogenate: first, demonstrating the same parent ion as authentic MJ33 (m/z 493); second, demonstrating an identical product ion as authentic MJ33 (m/z 139); and third, demonstrating the same relative retention time to the internal standard as authentic MJ33 (7.3 minutes). The LC/MS method was linear in the range from 0.2 to 10 ng/ml and the limit of detection was 0.1 ng/ml.
After an i.t. injection, the percentage of injected MJ33 remaining in the lung at 4 h was 67–87% for doses of 0.4–10 nmol. Uptake by the lung at 4 hours after i.v. injection of MJ33 was 23–42% of the injected dose; this presumably reflects a first pass effect of the injected lipophilic agent. MJ33 after either i.t. or i.v. injection gradually disappeared from the lung during the subsequent 72 hours. The fate of the MJ33 after leaving the lung is not known but the agent could be stored in the fat or excreted since it is unlikely to be metabolized.
The measurement of lung MJ33 showed generally good agreement with our data for MJ33-induced inhibition of PLA2 activity. Thus, either the i.t. or i.v. routes of administration appear to be satisfactory to inhibit lung Prdx6 PLA2 activity. The optimal route for clinical use could depend on the etiology of the inflammatory lung disease. This study has not determined the effect of route of administration on the distribution of MJ33 among the various cells that comprise the lung. Conceivably, the distribution could favor endothelium after i.v. administration and epithelium after i.t. dosing but that remains to be determined.
After showing effectiveness as an enzyme inhibitor in vivo, the next step was to evaluate potential toxicity. The only biologic effects thus far described for MJ33 is its ability to inhibit several forms of PLA2, mainly pancreatic (secreted) PLA2 and Prdx6 PLA2 (Jain et al., 1991b; Kim et al., 1997; Chen et al., 2000). Inhibition of the pancreatic enzyme is unlikely to be toxic over the short term and mice null for this enzyme seem to do quite well (Huggins et al., 2002). Mice that are null for Prdx6 have normal growth rates and life spans (Mo et al., 2003; Wang et al., 2003) although they show defective lung lipid metabolism with age-dependent accumulation of surfactant-related lipids (Fisher et al., 2005). Prdx6 null mice also showed increased oxidant sensitivity related to the loss of the Prdx6 peroxidase activity (Wang et al., 2003, 2004, 2006a), but this function of Prdx6 is unaffected by MJ33 (Fisher et al., 1999). Thus, a toxic effect of MJ33, if found, would likely be due to the chemical itself and unrelated to its inhibition of PLA2 activity.
Assay of cellular proliferation and clonogenic survival of a lung cancer epithelial cell line and primary lung endothelial cells were used to evaluate potential cytotoxicity of MJ33. Toxicity of MJ33 at concentrations up to 20–25 µM was not demonstrated for A549 cells in the rapid growth phase, for confluent A549 cells, or for confluent mPMVECs. mPMVECs that were treated with MJ33 for increasing time of exposure (up to 7 days) showed inhibition of clonogenic potential at concentrations >5 µM. Based on a dose of 4 nmol i.t. to inhibit lung PLA2 activity for 24 hours, one can estimate an average peak MJ33 concentration in the lung of <20 µM, a dose that is nontoxic for confluent cells.
To assess the potential toxicity of MJ33 in intact mice, MJ33 was administered far above the 0.4–4 nmol dosages that fully inhibit lung Prdx6 PLA2 activity. Mice treated with a single dose of MJ33 at up to 2500 nmol showed neither overt clinical signs of toxicity nor weight changes that were significantly different than control. There was no change in hematocrit or lung histology after treatment. Based on these results, MJ33 appears to be a reliable nontoxic inhibitor of NOX2 activation. Considering that 0.4 nmol to the mouse fully inhibited PLA2 activity and 2500 nmol in a single dose was not toxic, MJ33 appears to have a safety margin of at least 6000, although multiple doses of MJ33 (10 µmol total dose) did result in nonlethal toxicity.
The potential utility of MJ33 as an agent to prevent oxidative stress was evaluated in a model of lung I/R injury. The lung injury in I/R is recognized in part as a manifestation of oxidative stress associated with NOX2 activation (Eckenhoff et al., 1992; Dodd-O and Pearse, 2000; Zhang et al., 2005; Brandes et al., 2010). The role of oxidative stress with I/R in this study was demonstrated by increased ROS production, increased tissue lipid peroxidation, increased protein oxidation, and increased lung permeability. These manifestations were markedly reduced by pretreatment of mice with MJ33. These results suggest potential clinical utility for MJ33 in conditions in which pretreatment in anticipation of I/R is a possibility. A clear example would be pretreatment of donor lungs in anticipation of lung transportation.
Our study based on NOX2 null mice indicates that approximately 80% of the ROS production and oxidative injury with lung I/R is due to NOX2 activation and that there was no significant difference between NOX2 null mice and wild-type mice pretreated with MJ33. The ROS production that was not from NOX2 could derive from NOX1 activation, the mitochondrial respiratory chain, or xanthine oxidase among other sources and does not appear to be inhibited by MJ33. NOX3 and NOX5 are not present in rodent lung and NOX4 is constitutively active so they should not contribute to the increase in ROS production with 2 h I/R. Of note, Prdx6 can also function as an ROS scavenger through its peroxidase activity, and this activity is not inhibited by MJ33.
Based on its low toxicity, MJ33 would appear to offer a major advantage over currently available NOX2 inhibitors (Jaquet et al., 2009; Krause et al., 2012). Among these agents, diphenyleneiodonium, a commonly used inhibitor of NOX2 in vitro, is not specific and its toxicity related to general inhibition of flavoproteins would preclude its use as a therapeutic agent (Gatley and Martin, 1979; Jaquet et al., 2009). Apocynin is perhaps the most specific inhibitor described to date. Importantly, this agent can ameliorate lung injury associated with IR, hemorrhagic shock, and Gram-negative sepsis (Wang et al., 1994; Dodd-O and Pearse, 2000; Zhou et al., 2002; Abdelrahman et al., 2005; Zhu et al., 2008; Yang et al., 2009). However, the mechanism for action of this inhibitor is not clear and the margin between its therapeutic and toxic doses after systemic administration is relatively narrow (Tang et al., 2008). Several other drawbacks to the use of apocynin have been noted, including observations that it initially stimulates ROS production and that its activation requires ROS plus a peroxidase (Brandes et al., 2010). Recently, a TAT-peptide inhibitor of protein kinase Cδ has been shown to block NOX2 activation and protect against sepsis-induced lung injury (Kilpatrick et al., 2010), but studies of toxicity and specificity for the agent have not yet been reported and clinical use of the TAT-peptide may be problematic. Although these other inhibitors of NOX2 have limited clinical utility, their effectiveness supports the concept that inhibition of this enzymatic pathway can have a beneficial effect on lung inflammation.
Supplementary Material
Acknowledgments
The authors thank Dr. Scott Worthen for assistance with PMN isolation, Dr. Elena Goncharova for gift of the pulmonary artery smooth muscle cells, Kris Debolt for assistance with cell culture, and Kevin Yu for assistance with histology.
Abbreviations
- AngII
angiotensin II
- Con A
Concanavalin A
- DCF
dichlorofluorescein
- DMEM
Dulbecco’s modified Eagle’s medium
- DPPC
dipalmitoylphosphatidylcholine
- FBS
fetal bovine serum
- H2DCF
dihydrodichlorofluorescein
- HE
hydroethidine
- hPASMC
human pulmonary artery smooth muscle cell
- HRP
horseradish peroxidase
- I/R
ischemia/reperfusion
- i.t.
intratracheal
- LC
liquid chromatography
- MJ33
1-hexadecyl-3-trifluorethyglycero-sn-2-phosphomethanol
- mPMVEC
mouse pulmonary microvascular endothelial cell
- MS
mass spectroscopy
- MS/MS
tandem mass spectrometry
- m/z
mass-to-charge ratio
- NOX
NADPH oxidase
- PBS
phosphate-buffered saline
- PC
phosphatidylcholine
- PLA2
phospholipase A2
- PMN
polymorphonuclear leukocyte
- Prdx6
peroxiredoxin 6
- ROS
reactive oxygen species
- SOD
bovine superoxide dismutase
- STD
internal standard
Authorship Contributions
Participated in research design: Lee, Chatterjee, Zagorski, Feinstein, Fisher.
Conducted experiments: Lee, Dodia, Chatterjee, Zagorski.
Contributed new reagents or analytic tools: Mesaros, Blair, Jain.
Performed data analysis: Lee, Dodia, Chatterjee, Mesaros.
Wrote or contributed to the writing of the manuscript: Lee, Mesaros, Fisher.
Footnotes
This research was supported by the National Institutes of Health [Grants R01-HL105509 (to A.B.F.) and P30-ESO13508 (to I.A.B.)].
This research was presented in part at the following meetings: Lee I, Zagorski J, Dodia C, Feinstein S, Fisher AB (2012) Safety evaluation of MJ33 as a potential in vivo inhibitor of NADPH oxidase (NOX2) activation. Experimental Biology 2012; 2012 Apr 21-25; San Diego, CA; and Fisher AB, Lee I, Dodia C, Chatterjee S, Zagorski J, Mesaros C, Blair IA, and Feinstein SI (2012) A novel and non-toxic inhibitor of the activation of NADPH oxidase (NOX2). Society for Free Radicals in Biology and Medicine 19th Annual Meeting; 2012 Nov 14-18, 2012; San Diego, CA.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Abdelrahman M, Mazzon E, Bauer M, Bauer I, Delbosc S, Cristol JP, Patel NS, Cuzzocrea S, Thiemermann C. (2005) Inhibitors of NADPH oxidase reduce the organ injury in hemorrhagic shock. Shock 23:107–114 [DOI] [PubMed] [Google Scholar]
- Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. (1998) Endothelial NADPH oxidase as the source of oxidants in lungs exposed to ischemia or high K+. Circ Res 83:730–737 [DOI] [PubMed] [Google Scholar]
- Babior BM. (2002) The leukocyte NADPH oxidase. Isr Med Assoc J 4:1023–1024 [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917 [DOI] [PubMed] [Google Scholar]
- Brandes RP, Weissmann N, Schröder K. (2010) NADPH oxidases in cardiovascular disease. Free Radic Biol Med 49:687–706 [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Feinstein SI, Dodia C, Sorokina E, Lien YC, Nguyen S, Debolt K, Speicher D, Fisher AB. (2011) Peroxiredoxin 6 phosphorylation and subsequent phospholipase A2 activity are required for agonist-mediated activation of NADPH oxidase in mouse pulmonary microvascular endothelium and alveolar macrophages. J Biol Chem 286:11696–11706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Levitan I, Wei Z, Fisher AB. (2006) KATP channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation 13:633–644 [DOI] [PubMed] [Google Scholar]
- Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. (2000) 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem 275:28421–28427 [DOI] [PubMed] [Google Scholar]
- Dodd-O JM, Pearse DB. (2000) Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion lung injury. Am J Physiol Heart Circ Physiol 279:H303–H312 [DOI] [PubMed] [Google Scholar]
- Eckenhoff RG, Dodia C, Tan Z, Fisher AB. (1992) Oxygen-dependent reperfusion injury in the isolated rat lung. J Appl Physiol 72:1454–1460 [DOI] [PubMed] [Google Scholar]
- Ellison MA, Thurman GW, Ambruso DR. (2012) Phox activity of differentiated PLB-985 cells is enhanced, in an agonist specific manner, by the PLA2 activity of Prdx6-PLA2. Eur J Immunol 42:1609–1617 [DOI] [PubMed] [Google Scholar]
- Fisher AB. (2011) Peroxiredoxin 6: a bifunctional enzyme with glutathione peroxidase and phospholipase A₂ activities. Antioxid Redox Signal 15:831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher AB, Dodia C. (1996) Role of phospholipase A2 enzymes in degradation of dipalmitoylphosphatidylcholine by granular pneumocytes. J Lipid Res 37:1057–1064 [PubMed] [Google Scholar]
- Fisher AB, Dodia C, Chander A, Jain M. (1992) A competitive inhibitor of phospholipase A2 decreases surfactant phosphatidylcholine degradation by the rat lung. Biochem J 288:407–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher AB, Dodia C, Feinstein SI, Ho YS. (2005) Altered lung phospholipid metabolism in mice with targeted deletion of lysosomal-type phospholipase A2. J Lipid Res 46:1248–1256 [DOI] [PubMed] [Google Scholar]
- Fisher AB, Dodia C, Manevich Y, Chen JW, Feinstein SI. (1999) Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J Biol Chem 274:21326–21334 [DOI] [PubMed] [Google Scholar]
- Gatley SJ, Martin JL. (1979) Some aspects of the pharmacology of diphenyleneiodonium, a bivalent iodine compound. Xenobiotica 9:539–546 [DOI] [PubMed] [Google Scholar]
- Gelb MH, Jain MK, Berg OG. (1994) Inhibition of phospholipase A2. FASEB J 8:916–924 [DOI] [PubMed] [Google Scholar]
- Hood ED, Greineder CF, Dodia C, Han J, Mesaros C, Shuvaev VV, Blair IA, Fisher AB, Muzykantov VR. (2012) Antioxidant protection by PECAM-targeted delivery of a novel NADPH-oxidase inhibitor to the endothelium in vitro and in vivo. J Control Release 163:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins KW, Boileau AC, Hui DY. (2002) Protection against diet-induced obesity and obesity- related insulin resistance in Group 1B PLA2-deficient mice. Am J Physiol Endocrinol Metab 283:E994–E1001 [DOI] [PubMed] [Google Scholar]
- Jain MK, Tao WJ, Rogers J, Arenson C, Eibl H, Yu BZ. (1991a) Active-site-directed specific competitive inhibitors of phospholipase A2: novel transition-state analogues. Biochemistry 30:10256–10268 [DOI] [PubMed] [Google Scholar]
- Jain MK, Yu BZ, Rogers J, Ranadive GN, Berg OG. (1991b) Interfacial catalysis by phospholipase A2: dissociation constants for calcium, substrate, products, and competitive inhibitors. Biochemistry 30:7306–7317 [DOI] [PubMed] [Google Scholar]
- Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. (2009) Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal 11:2535–2552 [DOI] [PubMed] [Google Scholar]
- Kilpatrick LE, Sun S, Li H, Vary TC, Korchak HM. (2010) Regulation of TNF-induced oxygen radical production in human neutrophils: role of delta-PKC. J Leukoc Biol 87:153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Sundaresh CS, Feinstein SI, Dodia C, Skach WR, Jain MK, Nagase T, Seki N, Ishikawa K, Nomura N, et al. (1997) Identification of a human cDNA clone for lysosomal type Ca2+-independent phospholipase A2 and properties of the expressed protein. J Biol Chem 272:2542–2550 [DOI] [PubMed] [Google Scholar]
- Kratzer E, Tian Y, Sarich N, Wu T, Meliton A, Leff A, Birukova AA. (2012) Oxidative stress contributes to lung injury and barrier dysfunction via microtubule destabilization. Am J Respir Cell Mol Biol 47:688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause KH, Lambeth D, Krönke M. (2012) NOX enzymes as drug targets. Cell Mol Life Sci 69:2279–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krymskaya VP, Snow J, Cesarone G, Khavin I, Goncharov DA, Lim PN, Veasey SC, Ihida-Stansbury K, Jones PL, Goncharova EA. (2011) mTOR is required for pulmonary arterial vascular smooth muscle cell proliferation under chronic hypoxia. FASEB J 25:1922–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. (2001) Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res 88:888–894 [DOI] [PubMed] [Google Scholar]
- Lee I, Lee YH, Mikulski SM, Lee J, Covone K, Shogen K. (2000) Tumoricidal effects of onconase on various tumors. J Surg Oncol 73:164–171 [DOI] [PubMed] [Google Scholar]
- Lee I, Shogen K. (2008) Mechanisms of enhanced tumoricidal efficacy of multiple small dosages of ranpirnase, the novel cytotoxic ribonuclease, on lung cancer. Cancer Chemother Pharmacol 62:337–346 [DOI] [PubMed] [Google Scholar]
- Leto TL, Geiszt M. (2006) Role of Nox family NADPH oxidases in host defense. Antioxid Redox Signal 8:1549–1561 [DOI] [PubMed] [Google Scholar]
- Lieber JG, Webb S, Suratt BT, Young SK, Johnson GL, Keller GM, Worthen GS. (2004) The in vitro production and characterization of neutrophils from embryonic stem cells. Blood 103:852–859 [DOI] [PubMed] [Google Scholar]
- Lien YC, Feinstein SI, Dodia C, Fisher AB. (2012) The roles of peroxidase and phospholipase A2 activities of peroxiredoxin 6 in protecting pulmonary microvascular endothelial cells against peroxidative stress. Antioxid Redox Signal 16:440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Feinstein SI, Wang Y, Dodia C, Fisher D, Yu K, Ho YS, Fisher AB. (2010) Comparison of glutathione peroxidase 1 and peroxiredoxin 6 in protection against oxidative stress in the mouse lung. Free Radic Biol Med 49:1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevich Y, Fisher AB. (2005) Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic Biol Med 38:1422–1432 [DOI] [PubMed] [Google Scholar]
- Marshall NJ, Goodwin CJ, Holt SJ. (1995) A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regul 5:69–84 [PubMed] [Google Scholar]
- Min JH, Codipilly CN, Nasim S, Miller EJ, Ahmed MN. (2012) Synergistic protection against hyperoxia-induced lung injury by neutrophils blockade and EC-SOD overexpression. Respir Res 13:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Feinstein SI, Manevich Y, Zhang Q, Lu L, Ho YS, Fisher AB. (2003) 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett 555:192–198 [DOI] [PubMed] [Google Scholar]
- Nick JA, Young SK, Arndt PG, Brown NJ, Suratt BT, Janes M, Avdi NJ, Henson PM, Worthen SG. (2000) Role of p38 MAP Kinase in a murine model of pulmonary inflammation. J Immunol 164:2151–2159 [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chae HZ, Kim K. (2005) Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med 38:1543–1552 [DOI] [PubMed] [Google Scholar]
- Schremmer B, Manevich Y, Feinstein SI, Fisher AB. (2007) Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Subcell Biochem 44:317–344 [DOI] [PubMed] [Google Scholar]
- Takac I, Schröder K, Brandes RP. (2012) The Nox family of NADPH oxidases: friend or foe of the vascular system? Curr Hypertens Rep 14:70–78 [DOI] [PubMed] [Google Scholar]
- Tan AS, Berridge MV. (2000) Superoxide produced by activated neutrophils efficiently reduces the tetrazolium salt, WST-1 to produce a soluble formazan: a simple colorimetric assay for measuring respiratory burst activation and for screening anti-inflammatory agents. J Immunol Methods 238:59–68 [DOI] [PubMed] [Google Scholar]
- Tang XN, Cairns B, Cairns N, Yenari MA. (2008) Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience 154:556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M, Alexander RW. (2004) Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem 264:85–97 [DOI] [PubMed] [Google Scholar]
- van der Vliet A. (2008) NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radic Biol Med 44:938–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Suzuki Y, Tanigaki T, Rank DR, Raffin TA. (1994) Effect of the NADPH oxidase inhibitor apocynin on septic lung injury in guinea pigs. Am J Respir Crit Care Med 150:1449–1452 [DOI] [PubMed] [Google Scholar]
- Wang X, Phelan SA, Forsman-Semb K, Taylor EF, Petros C, Brown A, Lerner CP, Paigen B. (2003) Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J Biol Chem 278:25179–25190 [DOI] [PubMed] [Google Scholar]
- Wang Y, Feinstein SI, Fisher AB. (2008) Peroxiredoxin 6 as an antioxidant enzyme: protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. J Cell Biochem 104:1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. (2004) Lung injury and mortality with hyperoxia are increased in peroxiredoxin 6 gene-targeted mice. Free Radic Biol Med 37:1736–1743 [DOI] [PubMed] [Google Scholar]
- Wang Y, Feinstein SI, Manevich Y, Ho YS, Fisher AB. (2006a) Peroxiredoxin 6 gene-targeted mice show increased lung injury with paraquat-induced oxidative stress. Antioxid Redox Signal 8:229–237 [DOI] [PubMed] [Google Scholar]
- Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. (2006b) Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am J Respir Cell Mol Biol 34:481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Feinstein SI, Manevich Y, Chowdhury I, Pak JH, Kazi A, Dodia C, Speicher DW, Fisher AB. (2009) Mitogen-activated protein kinase-mediated phosphorylation of peroxiredoxin 6 regulates its phospholipase A(2) activity. Biochem J 419:669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Sharma AK, Marshall M, Kron IL, Laubach VE. (2009) NADPH oxidase in bone marrow-derived cells mediates pulmonary ischemia-reperfusion injury. Am J Respir Cell Mol Biol 40:375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. (2005) Activation of endothelial NADPH oxidase during normoxic lung ischemia is KATP channel dependent. Am J Physiol Lung Cell Mol Physiol 289:L954–L961 [DOI] [PubMed] [Google Scholar]
- Zhou R, Hu DY, Liu LM, Zhou XW. (2002) Protective effects of apocynin on “two-hit” injury induced by hemorrhagic shock and lipopolysaccharide. Acta Pharmacol Sin 23:1023–1028 [PubMed] [Google Scholar]
- Zhu C, Bilali A, Georgieva GS, Kurata S, Mitaka C, Imai T. (2008) Salvage of nonischemic control lung from injury by unilateral ischemic lung with apocynin, a nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor, in isolated perfused rat lung. Transl Res 152:273–282 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.