

Abstract
5-Methoxy-N,N-dimethyltryptamine (5-MeO-DMT or street name “5-MEO”) is a newer designer drug belonging to a group of naturally occurring indolealkylamines. Our recent study has demonstrated that coadministration of monoamine oxidase A (MAO-A) inhibitor harmaline (5 mg/kg) increases systemic exposure to 5-MeO-DMT (2 mg/kg) and active metabolite bufotenine. This study is aimed at delineating harmaline and 5-MeO-DMT pharmacokinetic (PK) interactions at multiple dose levels, as well as the impact of CYP2D6 that affects harmaline PK and determines 5-MeO-DMT O-demethylation to produce bufotenine. Our data revealed that inhibition of MAO-A–mediated metabolic elimination by harmaline (2, 5, and 15 mg/kg) led to a sharp increase in systemic and cerebral exposure to 5-MeO-DMT (2 and 10 mg/kg) at all dose combinations. A more pronounced effect on 5-MeO-DMT PK was associated with greater exposure to harmaline in wild-type mice than CYP2D6-humanized (Tg-CYP2D6) mice. Harmaline (5 mg/kg) also increased blood and brain bufotenine concentrations that were generally higher in Tg-CYP2D6 mice. Surprisingly, greater harmaline dose (15 mg/kg) reduced bufotenine levels. The in vivo inhibitory effect of harmaline on CYP2D6-catalyzed bufotenine formation was confirmed by in vitro study using purified CYP2D6. Given these findings, a unified PK model including the inhibition of MAO-A- and CYP2D6-catalyzed 5-MeO-DMT metabolism by harmaline was developed to describe blood harmaline, 5-MeO-DMT, and bufotenine PK profiles in both wild-type and Tg-CYP2D6 mouse models. This PK model may be further employed to predict harmaline and 5-MeO-DMT PK interactions at various doses, define the impact of CYP2D6 status, and drive harmaline–5-MeO-DMT pharmacodynamics.
Introduction
Indolealkylamine xenobiotics are serotonin (5-HT) analogs that have high impact as substances of abuse (Yu, 2008; Halberstadt and Geyer, 2011), except that a subgroup of indolealkylamines, namely triptans, are used to treat migraine (Kelley and Tepper, 2012). Some newer indolealkylamine club drugs include 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT or by the street name “5-MEO”) and 5-methoxy-N,N-diisopropyltryptamine (“Foxy” and “Foxy methoxy”) that were placed into Schedule I Controlled Substances by the US Drug Enforcement Administration in 2010 and 2004, respectively (Drug Enforcement Administration, 2004, 2010). It is noteworthy that 5-MeO-DMT is an endotoxin because of its presence within the human body (Barker et al., 2012). 5-MeO-DMT is also an active ingredient in a number of hallucinogenic animal and plant preparations such as the venom of Colorado River Bufo alvarius, virola snuffs, and the Ayahuasca beverage for medical, religious, or recreational uses (Ott, 2001; McKenna, 2004). Although the trafficking, distribution, and abuse of 5-MeO-DMT are likely underreported because it was not a controlled drug before 2011, the System to Retrieve Information from Drug Evidence revealed 23 cases involving 35 drug exhibits identified as 5-MeO-DMT from 1999 to 2009, and the National Forensic Laboratory Information System documented 27 state and local drug cases involving 32 drug exhibits identified as 5-MeO-DMT from 2004 to 2009 (Drug Enforcement Administration, 2010). With the epidemic of abuse, indolealkylamine intoxications have been frequently reported in hospitals in recent years, including several deaths associated with the abuse of 5-MeO-DMT and 5-methoxy-N,N-diisopropyltryptamine (Brush et al., 2004; Sklerov et al., 2005; Tanaka et al., 2006; Hill and Thomas, 2011).
5-MeO-DMT is predominantly metabolized through monoamine oxidase A (MAO-A)–catalyzed deamination metabolism (Shen et al., 2010a). Thus, 5-MeO-DMT is often coabused with MAO-A inhibitors (MAOIs), such as harmaline, to enhance its hallucinogenic effects (Ott, 1999, 2001; Brush et al., 2004; Sklerov et al., 2005). Coabuse of harmaline and 5-MeO-DMT is reasoned to cause complex drug-drug interactions (DDIs). First, both drugs act agonistically on the serotonergic system given the fact that harmaline is an MAOI (Kim et al., 1997) and 5-MeO-DMT is a 5-HT receptor agonist (Roth et al., 1997; Winter et al., 2000; Krebs-Thomson et al., 2006). Second, inhibition of MAO-A by harmaline may alter the pharmacokinetics (PK) of 5-MeO-DMT. Indeed, our recent studies (Shen et al., 2010b) support the metabolic DDI between MAOI and 5-MeO-DMT, as manifested by the reduction of 5-MeO-DMT depletion in human liver microsomes and elevation of 5-MeO-DMT exposure in animal models by concurrent harmaline exposure. Furthermore, the blockage of MAO-A–controlled deamination metabolism may result in an enhanced O-demethylation of 5-MeO-DMT (Shen et al., 2010b), which is catalyzed by polymorphic CYP2D6 (Zanger et al., 2004) to generate bufotenine (Yu et al., 2003a), an active metabolite with 10-fold higher affinity to the 5-HT2A receptor than 5-MeO-DMT (Roth et al., 1997). In addition, the PK of harmaline is affected by CYP2D6 status, as demonstrated by a faster systemic clearance in CYP2D6-humanized (Tg-CYP2D6) mice than that in wild-type mice (Yu et al., 2003b; Wu et al., 2009). Therefore, concurrent harmaline may interact agonistically with 5-MeO-DMT at both pharmacodynamic and PK levels (Shen et al., 2010a), and CYP2D6 genetic polymorphism may further complicate their PK interactions.
In this study, we aimed to delineate the PK interactions between harmaline and 5-MeO-DMT at multiple dose levels including nontoxic and toxic dose combinations in wild-type and Tg-CYP2D6 mouse models that mimic human CYP2D6 poor and extensive metabolizers, respectively (Corchero et al., 2001; Yu et al., 2004). Both blood and brain 5-MeO-DMT and bufotenine concentrations were measured to determine the impact of harmaline on PK of 5-MeO-DMT and metabolite bufotenine as well as the influence of CYP2D6 status. In addition, a unified mathematical model was developed to quantitatively define harmaline, 5-MeO-DMT, and bufotenine PK, which will be used to understand the complex DDI between harmaline and 5-MeO-DMT as well as the resultant pharmacological effects.
Materials and Methods
Chemicals and Materials.
5-MeO-DMT oxalate, harmaline hydrochloride dihydrate, 5-methyl-N,N-dimethyltryptamine (5-Me-DMT), perchloric acid, the NADPH and l-α-dilauroylphosphatidylcholine were purchased from Sigma-Aldrich (St. Louis, MO). Bufotenine standard solution was bought from Cambridge Isotope Laboratories (Andover, MA). Saline was purchased from B. Braun Medical Inc. (Irvine, CA). All other reagents or organic solvents were either analytical or high-performance liquid chromatography (HPLC) grade. CYP2D6.1 enzyme was expressed and purified as reported (Yu et al., 2002; Zhang et al., 2009).
Animals.
Age-matched Tg-CYP2D6 (Corchero et al., 2001) and wild-type FVB/N mice weighing 25–30 g were used in the study. Animals were housed in an animal care facility under standard conditions (20°C ± 2.0°C, 50–60% relative humidity, and lights on at 6:00 AM and off at 6:00 PM). Food and water were provided ad libitum. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University at Buffalo, The State University of New York.
Pharmacokinetic Studies.
Pharmacokinetic experiments were conducted as previously reported (Shen et al., 2010b, 2011b). To define the impact of MAOI harmaline on 5-MeO-DMT PK, wild-type and Tg-CYP2D6 mice were treated i.p. with various dose combinations of harmaline plus 5-MeO-DMT. In particular, harmaline (2, 5, or 15 mg/kg) was administered to the animals at 0 min, and 5-MeO-DMT (2 or 10 mg/kg) was given at 15 minutes. Control studies have been described in our recent publications, which involve i.v. and i.p. administration of 5-MeO-DMT (2, 10, or 20mg/kg) alone (Shen et al., 2011b) as well as i.v. and i.p. administration of harmaline (5 or 15 mg/kg) alone (Wu et al., 2009). In this study, 0.2–0.3 ml of blood sample was collected from one mouse at various time points (15–255 minutes, n = 4 per time point), and each mouse was sampled once. Serum was prepared with a serum separator (Becton Dickinson, Franklin Lakes, NJ) and stored at −80°C until the quantification of harmaline, 5-MeO-DMT, and bufotenine concentrations by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
It is noteworthy that higher dose combinations, namely 5 and 15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, resulted in obvious toxicity in mice. Therefore, the PK experiments were terminated at 105 and 45 minutes, respectively, when animals showed signs of dying. The serum profiles of harmaline, 5-MeO-DMT, and bufotenine of these dose combinations determined experimentally were used for validating the final PK DDI model.
Brain Drug Distribution.
Experiments were conducted as previously reported (Shen et al., 2011b). Briefly, wild-type and Tg-CYP2D6 mice (n = 4 per time point) were euthanized at various time points after drug administration. Whole brain was immediately removed from the skull, rinsed, homogenized with ice-cold saline, and stored at −80°C until the quantification of 5-MeO-DMT and bufotenine by LC-MS/MS. The contribution of residual blood in the brain tissue to the brain concentration was corrected by subtracting 3.7% of the serum drug concentration from the corresponding brain concentration (Khor et al., 1991).
Drug Metabolism by Purified CYP2D6.
Enzyme incubations were carried out as reported using purified CYP2D6.1 enzyme (Yu et al., 2002; Jiang et al., 2009; Zhang et al., 2009). In particular, each reaction was incubated in 100 mM potassium phosphate buffer (pH 7.4) in a final volume of 200 μl at 37°C for 20 minutes, which consisted of 0.02 µM CYP2D6.1, 0.2 µM cytochrome P450 reductase, 10 µg L-α-dilauroylphosphatidylcholine, and 2 or 10 µM 5-MeO-DMT in the absence or presence of harmaline (2 or 20 µM). All reactions were initiated by the addition of NADPH (1 mM final concentration) and terminated by 5 µl 70% perchloric acid. All reactions were conducted in duplicate. The mixture was centrifuged at 14,000g for 5 minutes, and the supernatant was directly injected for HPLC analysis.
HPLC and LC-MS/MS Analyses.
Bufotenine concentrations within enzyme incubations with 5-MeO-DMT were determined by HPLC analyses. The Agilent 1100 series HPLC system (Agilent Technologies, Palo Alto, CA) consisted of an online vacuum degasser, quaternary pump, autosampler, thermostat controlled column compartment, fluorescence detector, and diode-array detector, and the instrument was controlled by Agilent ChemStation software. 5-MeO-DMT and bufotenine were separated with a Regis REXCHROM phenyl column (250 mm × 4.6 mm, 5 µm; Morton Grove, IL) under the conditions described previously (Yu et al., 2003a). The calibration linear ranges for 5-MeO-DMT and bufotenine were 2–100 pmol on-column. Interday and intraday coefficients of variation were less than 10% for both analytes.
Blood and brain drug and metabolite concentrations were determined by a validated LC-MS/MS method (Shen et al., 2009, 2011b). A simple protein precipitation and a liquid-liquid extraction method were used for the processing of serum and brain samples, respectively. In particular, 60 µl ice-cold acetonitrile containing 50 nM 5-Me-DMT (internal standard) was added to 20 µl serum sample for protein precipitation. After 10-minute centrifugation at 14,000g, the supernatant was subject to LC-MS/MS analysis. For brain drug concentrations, 50 µl brain homogenate was mixed with 5 µl sodium hydroxide (1 M) and 10 µl 5-Me-DMT (100 nM) and the mixture was extracted with 1 ml ethyl acetate. After the centrifugation at 14,000g for 5 minutes, the supernatant was transferred to a new vial and evaporated to dryness. The residue was reconstituted in 50 µl 50% methanol, centrifuged at 14,000g for 5 minutes, and transferred to a new vial for LC-MS/MS analysis. Separation of analytes was achieved with a Phenomenex phenyl-hexyl column (50 mm × 4.6 mm, 3 µm; Torrance, CA). LC-MS/MS quantification was conducted with an API 3000 turbo ionspray ionization triple-quadrupole mass spectrometer (Applied Biosystems, Foster City, CA) coupled to a Shimadzu prominence HPLC system (Kyoto, Japan).
PK Modeling.
Noncompartmental analyses were performed with Phoenix WinNonlin software (version 6.1; Pharsight, Mountain View, CA). The maximum serum concentrations (Cmax) were the observed values between the first and last measured time points without interpolation. The area under the concentration-time curve from the start to the last measured time point (AUC0→t) was calculated by the linear trapezoidal method. AUC0→∞ was estimated by extrapolation from AUC0→t with the addition of Clast/λ, where Clast is the last measured concentration and λ is the apparent terminal slope of linear regression from the semi-logarithmic concentration-time curve.
To better understand the PK interactions between harmaline and 5-MeO-DMT as well as the pharmacodynamics (unpublished data), a mathematical model was proposed to characterize serum harmaline, 5-MeO-DMT, and bufotenine PK. In our previous single-dose PK studies, two-compartmental models with first-order absorption and linear or capacity-limited elimination from the central compartments have been established to describe the PK properties of harmaline and 5-MeO-DMT, respectively (Wu et al., 2009; Shen et al., 2011b). These models were adopted into this study with modifications to characterize the PK profiles of harmaline and 5-MeO-DMT under the DDI condition. The model was also extended to include the PK of bufotenine, the O-demethylated metabolite of 5-MeO-DMT. Although the PK profile of bufotenine after i.v. administration is not available in our study, literature data indicate that this compound exhibits polyexponential PK in rats after i.v. injection (Sanders and Bush, 1967). Therefore, bufotenine PK profiles after i.p. administration of 5-MeO-DMT were fitted to a two-compartment model with linear elimination from its central compartment, assuming that bufotenine was only generated in the central compartment from parent drug, and bufotenine formed from 5-MeO-DMT all entered into the systemic circulation without first-pass elimination at the site of formation. The final model (Fig. 1) was chosen based on the goodness-of-fit criteria, including Akaike’s information criterion, Schwarz criterion, coefficient variation of the estimates, and visual inspection of the fitted profiles.
Fig. 1.

Proposed PK model for harmaline-5-MeO-DMT DDI and bufotenine production in wild-type and Tg-CYP2D6 mice. Abbreviations are defined in the Materials and Methods.
Harmaline PK was defined by the following differential equations (Eqs. 1–3):
| (1) |
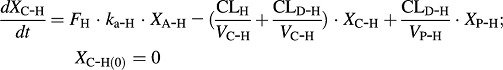 |
(2) |
| (3) |
| (4) |
XA-H, XC-H, and XP-H represent the amount of harmaline in the absorption, central, and peripheral compartments, respectively. Harmaline concentration in the central compartment (CC-H) was determined by Eq. 4.
All PK parameters for harmaline were obtained from our previous analysis (Wu et al., 2009), except that bioavailability (FH) of the 2 mg/kg dose level of harmaline was estimated in the present study. ka-H (0.307 1/min) is the absorption rate constant, and VC-H (2.43 l/kg), and VP-H (2.86 l/kg) are the volume of central and peripheral compartments, respectively. CLD (0.879 l/min per kilogram) is the distribution clearance between the central and peripheral compartments. Values of the FH for the 5 mg/kg dose were 74.2 and 68.8% for wild-type and Tg-CYP2D6 mice, respectively. FH values for the 15 mg/kg dose were 90.3 and 80.1% for wild-type and Tg-CYP2D6 mice, respectively. Note that the total systemic clearance (CLH) of harmaline consists of CYP2D6 dependent (CLCYP2D6-H, 0.0608 l/min per kilogram) and independent (CLother-H, 0.0962 l/min per kilogram) routes that differ in Tg-CYP2D6 and wild-type mice, which are defined by the following formula (Eqs. 5 and 6):
In Tg-CYP2D6 mice:
| (5) |
In wild-type mice:
| (6) |
The PK model for 5-MeO-DMT in wild-type and Tg-CYP2D6 mice is defined according to the following equations (Eqs. 7–9):
| (7) |
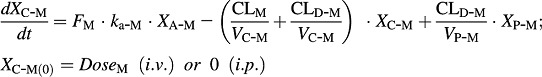 |
(8) |
| (9) |
| (10) |
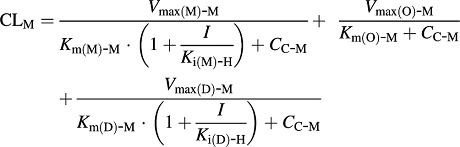
The total systemic clearance (CLM) of 5-MeO-DMT is different in two genotypes of mice (Eqs. 11 and 12).
In Tg-CYP2D6 mice:
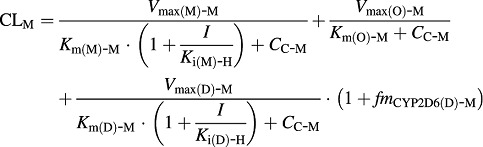 |
(11) |
In wild-type mice:
 |
(12) |
XA-M, XC-M, and XP-M represent the amounts of 5-MeO-DMT in the absorption, central, and peripheral compartments, respectively. 5-MeO-DMT concentration in the central compartment (CC-M) is defined by Eq. 10. FM and ka-M are the bioavailability and first-order absorption rate constant for 5-MeO-DMT, and VC-M and VP-M represent the volumes of distribution of the central and peripheral compartments for 5-MeO-DMT. CLD-M is the distribution clearance between the central and peripheral compartments. Vmax(M)-M represents the maximum metabolic rate of 5-MeO-DMT by MAO-A, and Km(M)-M represents the concentration of 5-MeO-DMT at 50% of Vmax(M)-M. Vmax(O)-M represents the maximum elimination rate of 5-MeO-DMT by other elimination pathways and Km(O)-M represents the concentration of 5-MeO-DMT at 50% of Vmax(O)-M. Vmax(D)-M represents the maximum metabolic rate of 5-MeO-DMT by O-demethylation and Km(D)-M represents the concentration of 5-MeO-DMT at 50% of Vmax(D)-M. The fmCYP2D6(D)-M is the fraction of CYP2D6-mediated 5-MeO-DMT O-demethylation, which only presents in Tg-CYP2D6 mice, in addition to murine 5-MeO-DMT O-demethylase capacity occurring in both wild-type and Tg-CYP2D6 mice. I represents the concentration of harmaline in the central compartment (CC-H) that inhibits the elimination of 5-MeO-DMT. Ki(M)-H is the inhibition constant of harmaline against MAO-A-catalyzed metabolic elimination, which was fixed to 0.048 μM, a value obtained from an in vitro assay (Kim et al., 1997). Ki(D)-H represents the inhibition constant of harmaline against 5-MeO-DMT O-demethylation to form bufotenine, which was estimated simultaneously with other PK parameters. Harmaline acts as a competitive inhibitor against both MAO-A and CYP2D6/O-demethylase activities in the final PK DDI model (Fig. 1), according to the known mechanisms (Kim et al., 1997; Zhao et al., 2011).
The rate of change in bufotenine concentrations in wild-type and Tg-CYP2D6 mice is defined by the following two differential equations (Eqs. 13 and 14):
| (13) |
| (14) |
The input function for Eq. 13 is different in two genotypes of mice, as shown in Eqs. 15 and 16.
In Tg-CYP2D6 mice:
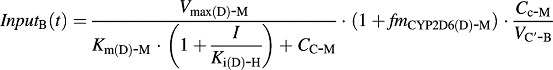 |
(15) |
In wild-type mice:
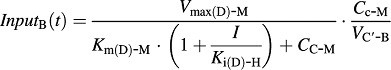 |
(16) |
CC-B is the concentration of bufotenine in the central compartment, XP-B is the amount of bufotenine in the peripheral compartment, VC′-B is the apparent central compartment volume of bufotenine, K10-B is the first-order elimination rate constant of bufotenine from its central compartment, and K12-B and K21-B are the first-order rate constants of distribution between the central and peripheral compartments.
Harmaline, 5-MeO-DMT, and bufotenine data from the present DDI study after i.p. administration of 2, 5, or 15 mg/kg harmaline plus 2 mg/kg 5-MeO-DMT as well as previous studies with 2, 10, and 20 mg/kg 5-MeO-DMT administered i.v. and i.p. alone (Shen et al., 2011b) were fitted simultaneously with ADAPT V (Biomedical Simulations Resource; University of South California, Los Angeles, CA) using a pooled population analysis and the maximum likelihood estimator. The variance model was defined as follows in Eq. 17:
| (17) |
where σ1 and σ2 are the estimated variance model parameters and Y(θ, ti) is the ith predicted value from the PK model.
Statistical Analysis.
All data are presented as mean ± S.D. Statistical analysis was conducted with GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA). Data were compared with the t test, one-way analysis of variance followed by Tukey’s post hoc comparisons, or two-way analysis of variance followed by Bonferroni’s post hoc comparisons. The difference was considered statistically significant when P < 0.05.
Results
CYP2D6 Status Has a Significant Impact on Serum Harmaline PK.
To investigate the PK interactions of harmaline and 5-MeO-DMT, it is necessary to examine harmaline PK. We recently defined harmaline PK profiles in mouse models after i.v. and i.p. administration (Wu et al., 2009). In this study, serum harmaline concentrations were also monitored over time after coadministration of harmaline and 5-MeO-DMT (Figs. 2, A and B, and 3, A and B), although 5-MeO-DMT unlikely alters harmaline PK. Consistent with our previous findings (Wu et al., 2009), elimination of harmaline was faster in Tg-CYP2D6 mice (Figs. 2, A and B, and 3, A and B), given the fact that Tg-CYP2D6 mice exhibit CYP2D6-mediated metabolism besides intrinsic murine elimination capacity. Consequently, systemic exposure to harmaline (AUC) was about 1-fold higher in wild-type mice than that in Tg-CYP2D6 mice treated with the same doses of harmaline and 5-MeO-DMT (Table 1), supporting the importance of CYP2D6 genetic polymorphism in harmaline metabolism and PK (Yu et al., 2003b; Wu et al., 2009).
Fig. 2.

Pharmacokinetics of harmaline (HAR) (A and B), 5-MeO-DMT (C and D), and bufotenine (E and F) in wild-type and Tg-CYP2D6 mice after i.p. coadministration of 2 (●), 5 (▴), or 15 mg/kg (♦) harmaline and 2 mg/kg 5-MeO-DMT. Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively. Serum harmaline, 5-MeO-DMT, and bufotenine concentrations are mean ± S.D. (n = 4 at each time point). The dashed, solid, and dotted lines represent the fitted data of 2, 5, and 15 mg/kg harmaline plus 2 mg/kg 5-MeO-DMT, respectively, which were obtained from simultaneous estimation using the final PK model (Fig. 1).
Fig. 3.

Pharmacokinetics of harmaline (HAR) (A and B), 5-MeO-DMT (C and D), and bufotenine (E and F) in wild-type and Tg-CYP2D6 mice after i.p. coadministration of 5 (▴) or 15 mg/kg (♦) harmaline and 10 mg/kg 5-MeO-DMT. Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively. Serum harmaline, 5-MeO-DMT, and bufotenine concentrations are mean ± S.D. (n = 4 at each time point). The solid and dotted lines represent the simulated data of 5 and 15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, respectively, using the final PK model parameters shown in Table 4. Inserts are enlarged 0–120 minutes PK profiles.
TABLE 1.
Noncompartmental analysis of serum harmaline PK in wild-type and Tg-CYP2D6 mice coadministered with harmaline and 5-MeO-DMT
Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively.
| Mice | Parameters | 5-MeO-DMT 2 mg/kg |
5-MeO-DMT 10 mg/kg |
|||
|---|---|---|---|---|---|---|
| Harmaline 2 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | ||
| Wild-type | Cmax (µM) | 0.450 ± 0.113 | 2.61 ± 0.48* | 11.0 ± 1.5*† | 3.18 ± 0.22 | 10.9 ± 0.6† |
| AUC (µM⋅min)a | 27.9 ± 7.1 | 198 ± 10* | 935 ± 166*† | 197 ± 16 | 269 ± 7† | |
| Tg-CYP2D6 | Cmax (µM) | 0.406 ± 0.096 | 1.91 ± 0.14*# | 10.1 ± 0.8*† | 2.46 ± 0.51 | 10.0 ± 1.2† |
| AUC (µΜ⋅min)a | 17.1 ± 3.6# | 109 ± 13*# | 416 ± 66*#† | 111 ± 7# | 234 ± 22#† | |
AUC15 min→∞ for the treatments of harmaline plus 2 mg/kg 5-MeO-DMT, AUC15→105min for the treatments of 5 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, and AUC15→45min for the treatments of15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT.
P < 0.05 compared with wild-type mice under the same treatment; *P < 0.05 compared with the same genotype mice treated with the same dose of 5-MeO-DMT plus 2 mg/kg harmaline; †P < 0.05 compared with the same genotype mice treated with the same dose of 5-MeO-DMT plus 5 mg/kg harmaline.
Coadministration of MAOI Harmaline Significantly Increases the Systemic Exposure to 5-MeO-DMT, Which May be Further Affected by CYP2D6 Status at Some Dose Combinations.
To delineate the PK interactions between harmaline and 5-MeO-DMT as well as the potential impact of CYP2D6, various dose combinations of harmaline (2, 5, or 15 mg/kg) and 5-MeO-DMT (2 or 10 mg/kg) were tested in wild-type and Tg-CYP2D6 mice (Figs. 2 and 3), in addition to i.v. and i.p. administration of 5-MeO-DMT alone (Fig. 4). Our data revealed that coadministration of MAOI harmaline reduced 5-MeO-DMT elimination, leading to a significantly greater systemic exposure (AUC) to 5-MeO-DMT (Table 2). For instance, the AUC15 minute→∞ values of 5-MeO-DMT were 21.1 ± 2.3 and 25.1 ± 1.3 μM⋅min in Tg-CYP2D6 and wild-type mice, respectively. When 2 mg/kg MAOI harmaline was coadministered, these values increased to 71.1 ± 13.3 and 97.6 ± 12.2 μM⋅min, respectively (Table 2). The results highlight the critical role of MAO-A in 5-MeO-DMT PK.
Fig. 4.

Pharmacokinetics of 5-MeO-DMT in wild-type and Tg-CYP2D6 mice after i.v. (A and B) and i.p. (C and D) administration of 2 (○), 10 (▵), and 20 mg/kg (◇) 5-MeO-DMT alone (Shen et al., 2011b). The formation of bufotenine (E and F) after i.p. administration of 5-MeO-DMT was also shown. 5-MeO-DMT was dosed at 0 min. Values are mean ± S.D. (n = 3 at each time point). The dashed, solid, and dotted lines represent the fitted profiles obtained from simultaneous estimation using the final integrated PK model (Fig. 1).
TABLE 2.
Noncompartmental analysis of serum 5-MeO-DMT PK in wild-type and Tg-CYP2D6 mice coadministered with harmaline and 5-MeO-DMT
Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively.
| Mice | Parameters | 5-MeO-DMT 2 mg/kg |
5-MeO-DMT 10 mg/kg |
|||||
|---|---|---|---|---|---|---|---|---|
| Harmaline 0 mg/kg | Harmaline 2 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | Harmaline 0 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | ||
| Wild-type | Cmax (µM) | 1.36 ± 0.16 | 1.88 ± 0.29* | 1.97 ± 0.15* | 1.93 ± 0.14* | 6.66 ± 1.13 | 13.3 ± 1.0* | 11.7 ± 1.5* |
| AUC (µM⋅min)a | 25.1 ± 1.3 | 97.6 ± 12.2* | 109.5 ± 14.3* | 134.0 ± 5.0* | 227 ± 41 | 759 ± 91* | 275 ± 27* | |
| Tg-CYP2D6 | Cmax (µM) | 1.19 ± 0.33 | 1.90 ± 0.29* | 1.68 ± 0.39 | 2.26 ± 0.14*# | 8.42 ± 1.03 | 10.4 ± 0.8*# | 12.0 ± 1.8* |
| AUC (µM⋅min)a | 21.1 ± 2.3# | 71.1 ± 13.3*# | 76.3 ± 12.6*# | 120.6 ± 5.1*# | 249 ± 46 | 571 ± 11*# | 264 ± 41* | |
AUC15→105min for the treatments of 5 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, AUC15→45min for the treatments of15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, and AUC15 min→∞ for all other treatments.
P < 0.05 compared with corresponding values for the same genotype mice treated with the same dose of 5-MeO-DMT alone; #P < 0.05 compared with wild-type mice under the same treatment.
The impact of harmaline on 5-MeO-DMT PK can be further influenced by CYP2D6 that determines harmaline PK, although CYP2D6 has minimal effect on the PK of 5-MeO-DMT administered alone (Fig. 4). For instance, coadministration of 5 mg/kg harmaline increased the exposure to 5-MeO-DMT (10 mg/kg) (AUC15 minute→∞ for 5-MeO-DMT alone versus AUC15→105 minute for 5-MeO-DMT plus harmaline) to a greater extent in wild-type mice (227 ± 41 versus 759 ± 91 μM⋅min) than Tg-CYP2D6 mice (249 ± 46 versus 571 ± 11 μM⋅min) (Table 2). This is also manifested by the Cmax values (Table 2). Together, our results indicate a possible complication of harmaline-5-MeO-DMT DDI by CYP2D6, likely through its action on harmaline PK (Figs. 2, A and B, and 3, A and B).
Harmaline Shows a Dose-Dependent Biphasic Effect on CYP2D6-Catalyzed 5-MeO-DMT O-Demethylation, Due to the Capacity of Harmaline in Inhibition of CYP2D6 Enzymatic Activity.
Serum bufotenine concentrations were also determined to define the impact of harmaline on 5-MeO-DMT O-demethylation that is mediated by CYP2D6 enzyme (Yu et al., 2003a; Shen et al., 2010b). In both wild-type and Tg-CYP2D6 mice, coadministration of a lower dose (5 mg/kg) of harmaline significantly increased bufotenine production from 5-MeO-DMT. This is manifested by a greater Cmax and AUC values in mice coadministered with 5 mg/kg harmaline compared with 5-MeO-DMT alone (Table 3). Surprisingly, further increase of harmaline dose did not show a dose-dependent increase in bufotenine formation. Instead, bufotenine concentrations and overall AUC were decreased in all mice when the harmaline dose was increased from 5 to 15 mg/kg (Fig. 2, E and F; Table 3). For instance, the systemic exposure to bufotenine produced from 2 mg/kg 5-MeO-DMT was reduced from 16.3 ± 1.4 to 5.16 ± 0.76 µM/min in wild-type mice and from 18.0 ± 1.5 to 6.86 ± 1.33 µM/min in Tg-CYP2D6 mice, respectively, when harmaline dose was increased from 5 to 15 mg/kg (Table 3). This suggests a dose-dependent biphasic influence of harmaline on bufotenine formation from 5-MeO-DMT.
TABLE 3.
Noncompartmental analysis of serum bufotenine PK in wild-type and Tg-CYP2D6 mice coadministered with harmaline and 5-MeO-DMT
Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively.
| Mice | Parameters | 5-MeO-DMT 2 mg/kg |
5-MeO-DMT 10 mg/kg |
||||
|---|---|---|---|---|---|---|---|
| Harmaline 0 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | Harmaline 0 mg/kg | Harmaline 5 mg/kg | Harmaline 15 mg/kg | ||
| Wild-type | Cmax (µM) | 0.051 ± 0.009 | 0.132 ± 0.009* | 0.075 ± 0.015*& | 0.114 ± 0.023 | 0.185 ± 0.014* | 0.131 ± 0.037*& |
| AUC (µM⋅min)a | 2.69 ± 1.21 | 16.3 ± 1.4* | 5.16 ± 0.76*& | 5.66 ± 0.88 | 12.9 ± 1.2* | 2.72 ± 0.34*& | |
| Tg-CYP2D6 | Cmax (µM) | 0.053 ± 0.013 | 0.137 ± 0.014* | 0.090 ± 0.010*& | 0.151 ± 0.085 | 0.300 ± 0.030*# | 0.170 ± 0.066*& |
| AUC (µM⋅min)a | 1.82 ± 0.74 | 18.0 ± 1.5* | 6.86 ± 1.33*& | 7.30 ± 4.21 | 18.2 ± 1.9*# | 3.59 ± 0.94*& | |
AUC15→105min for the treatments of 5 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, AUC15→45min for the treatments of 15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, and AUC15 min→∞ for all other treatments.
P < 0.05 compared with corresponding values for the same genotype mice treated with the same dose of 5-MeO-DMT alone; &P < 0.05 compared with corresponding values for the same genotype mice treated with the same dose of 5-MeO-DMT plus 5 mg/kg harmaline; #P < 0.05 compared with wild-type mice under the same treatment.
Working with the hypothesis that higher concentrations of harmaline might inhibit 5-MeO-DMT O-demethylation, we conducted an enzymatic study to assess whether harmaline inhibits CYP2D6 activity in vitro in the production of bufotenine from 5-MeO-DMT. We tested 2 and 10 µM 5-MeO-DMT in the absence and presence of 2 or 10 µM harmaline because such concentrations were relevant to the blood drug concentrations (Figs. 2 and 3). Our data showed that indeed harmaline was able to inhibit bufotenine formation from 5-MeO-DMT in a concentration-dependent manner (Fig. 5). Coincubation of 2 µM harmaline showed approximately 50 and 25% reduction in bufotenine production from 2 and 10 µM 5-MeO-DMT, respectively. When harmaline concentration was increased to 10 µM, the extent of inhibition was about 75 and 50%, respectively (Fig. 5). The results support the hypothesis that harmaline is not only a CYP2D6 substrate but also an inhibitor, and its inhibitory action on CYP2D6 enzyme shown in vitro (Fig. 5) might explain the in vivo suppression of bufotenine formation from 5-MeO-DMT by higher dose levels of harmaline (15 mg/kg) (Table 3).
Fig. 5.

Concentration-dependent inhibitory effect of harmaline (HAR) against CYP2D6-catalyzed 5-MeO-DMT O-demethylation. 5-MeO-DMT (2 or 10 µM) was incubated with purified CYP2D6.1 enzyme for 20 minutes, in the absence (white bars) or presence of 2 (gray) or 10 (black) µM harmaline. Values represent mean ± S.E.M. *P < 0.05 compared with incubations in the absence of harmaline; #P < 0.05 compared with incubations with 2 µM harmaline.
A Unified Compartmental Model Is Developed to Describe the PK Interaction between Harmaline and 5-MeO-DMT.
A PK model was thus developed to better understand the in vivo harmaline-5-MeO-DMT interactions and the mechanistic roles for MAO-A and CYP2D6. The PK properties of blood harmaline and 5-MeO-DMT administered alone have been defined with compartmental models (Wu et al., 2009; Shen et al., 2011b). Those models were adopted with modifications in this study (Fig. 1), which described reasonably well the PK of 5-MeO-DMT and harmaline as well as the metabolite bufotenine in both DDI (Figs. 2 and 3) and single drug administration studies (Fig. 4). Based on the fitting criteria (e.g., Akaike’s information criterion, Schwarz criterion, coefficient variation of the estimates), a two-compartment model with linear elimination by CYP2D6-dependent (CLCYP2D6-H; present only in Tg-CYP2D6 mice) and independent (CLother-H; present in both wild-type and Tg-CYP2D6 mice) pathways from central compartment (Wu et al., 2009) nicely captured harmaline PK under the DDI condition in this study.
Harmaline interacts with 5-MeO-DMT (Fig. 1) through competitive inhibition of both MAO-A–mediated deamination elimination with a Ki(M) value, which was fixed to 0.048 μM as reported (Kim et al., 1997), and CYP2D6-catalyzed O-demethylation to produce bufotenine with an estimated Ki(D) value of 7.13 μM (Table 4). The estimated VC-M, VP-M, CLD-M, Ka-M, and FM values for 5-MeO-DMT in the final model were 0.460 l/kg, 2.29 l/kg, 0.301 l/min per kilogram, 0.0748 l/min, and 74.8%, respectively (Table 4). The estimated systemic clearance related values were 2.69 μmol/min per kilogram, 16.7 μM, 0.0610 μmol/min per kilogram, 0.446 μM, 0.0334 μmol/min per kilogram, and 1.27 μM for Vmax(M)–M, Km(M)–M, Vmax(O)–M, Km(O)–M, Vmax(D)–M, and Km(D)–M, respectively (Table 4). The fmCYP2D6(D)-M value (30.1%) indicates the extent of additional 5-MeO-DMT O-demethylase capacity by the CYP2D6 enzyme in Tg-CYP2D6 mice. The fitted values for bufotenine PK produced from 5-MeO-DMT were 0.606 l/kg, 0.203 1/min, 0.143 1/min, and 0.0453 1/min for VC′-B, K10-B, K12-B and K21-B, respectively.
TABLE 4.
Model-estimated parameters for harmaline, 5-MeO-DMT, and bufotenine PK
| Parameter | Unit | Definition | Fitted Values | CV% |
|---|---|---|---|---|
| F2mg-WT | % | Bioavailability of harmaline (2 mg/kg) in wild-type mice | 34.6 | 6.44 |
| F2mg-Tg-CYP2D6 | % | Bioavailability of harmaline (2 mg/kg) in Tg-CYP2D6 mice | 36.8 | 6.82 |
| Ki(M)-H | µM | Harmaline inhibition constant of MAO-A activity | 0.048 | Fixed |
| Ki(D)-H | µM | Harmaline inhibition constant of 5-MeO-DMT O-demethylase activity | 7.13 | 19.0 |
| Vmax(M)-M | µmol/min per kilogram | Maximum metabolic rate of 5-MeO-DMT by MAO-A | 2.69 | 0.0977 |
| Km(M)-M | µM | Concentration of 5-MeO-DMT at 50% of Vmax(M)-M | 16.7 | 0.175 |
| Vmax(O)-M | µmol/min per kilogram | Maximum elimination rate of 5-MeO-DMT by other murine elimination pathway | 0.0610 | 8.81 |
| Km(O)-M | µM | 5-MeO-DMT concentration at 50% of Vmax(O)-M | 0.446 | 8.52 |
| Vmax(D)-M | µmol/min per kilogram | Maximum metabolic rate of 5-MeO-DMT by O-demethylation | 0.0334 | 19.1 |
| Km(D)-M | µM | 5-MeO-DMT concentration at 50% of Vmax(D)-M | 1.27 | 5.876 |
| fmCYP2D6(D) | % | Fraction of CYP2D6 mediated 5-MeO-DMT O-demethylation | 30.1 | 18.9 |
| VC-M | l/kg | Central volume of distribution of 5-MeO-DMT | 0.460 | 0.0484 |
| VP-M | l/kg | Peripheral volume of distribution of 5-MeO-DMT | 2.29 | 0.0498 |
| CLD-M | l/min per kilogram | Distribution clearance of 5-MeO-DMT | 0.301 | 0.0483 |
| Ka-M | 1/min | First-order absorption rate constant of 5-MeO-DMT | 0.0748 | 0.0356 |
| FM | % | Bioavailability of 5-MeO-DMT | 74.8 | 0.0344 |
| K10-B | 1/min | First-order elimination rate constant of bufotenine | 0.203 | 104 |
| VC′-B | l/kg | Apparent central volume of distribution of bufotenine | 0.606 | 20.3 |
| K12-B | 1/min | First-order intercompartmental rate constant of bufotenine | 0.143 | 135 |
| K21-B | 1/min | First-order intercompartmental rate constant of bufotenine | 0.0453 | 212 |
CV%, coefficient variation of the estimates.
To evaluate the final model (Fig. 1), blood harmaline, 5-MeO-DMT, and bufotenine PK profiles (Fig. 3) were simulated using the final PK parameters (Table 4) for the treatments of 5 or 15 mg/kg harmaline plus 10 mg/kg 5-MeO-DMT, which induced an obvious toxicity in mice. The model (Fig. 1) nicely predicted the PK profiles at early time points (15–45 minutes; Fig. 3), but showed some bias at later time points (75–105 minutes; Fig. 3), which might be attributed to the physiologic changes under such toxic dose combinations of 5-MeO-DMT and harmaline. These results suggest that this DDI model may be employed to define the PK interactions of harmaline and 5-MeO-DMT at various dose levels and identify the potential influence of CYP2D6 pharmacogenetics.
Coadministration of MAOI Harmaline Sharply Elevates 5-MeO-DMT and Bufotenine Accumulation in Mouse Brain.
Given the fact that 5-MeO-DMT acts on the central nervous system, we further examined the impact of MAOI harmaline on brain 5-MeO-DMT accumulation. Our data showed that concurrent harmaline substantially increased brain 5-MeO-DMT concentrations (Fig. 6) as well as the metabolite bufotenine concentrations (Fig. 7) at various time points compared with treatment with 5-MeO-DMT alone (Shen et al., 2011b). For instance, a small dose (5 mg/kg) of harmaline led to a more than 10-fold increase in 5-MeO-DMT accumulation in mouse brain and a prolonged cerebral exposure to the drug (Fig. 6). Consistent with the findings on systemic drug exposure (Table 2), brain 5-MeO-DMT concentrations in wild-type mice could be significantly greater (e.g., about 40%) than those in Tg-CYP2D6 mice treated with certain dose levels of harmaline (e.g., 15 mg/kg) and 5-MeO-DMT (e.g., 2 mg/kg) (Fig. 6), indicating a potential influence by CYP2D6 status.
Fig. 6.

Brain 5-MeO-DMT concentrations in wild-type (open bars) and Tg-CYP2D6 (filled bars) mice at 45 minutes (A and D), 75 minutes (B and E), and 105 minutes (C and F) after coadministration of harmaline (HAR; 0, 5, or 15 mg/kg) and 5-MeO-DMT (5 or 10 mg/kg). Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively. Data are mean ± S.D. (n = 4 in each group). *P < 0.05 compared with the same genotype of mice treated with vehicle (harmaline 0 mg/kg) plus 5-MeO-DMT (Shen et al., 2011b); #P < 0.05 compared with wild-type mice under the same treatment.
Fig. 7.

Brain bufotenine concentrations in wild-type (open bars) and Tg-CYP2D6 (filled bars) mice at 45 minutes (A), 75 minutes (B), and 105 minutes (C) after coadministration of harmaline (HAR; 0 or 5 mg/kg) and 5-MeO-DMT (10 mg/kg). Harmaline and 5-MeO-DMT were administered i.p. at 0 and 15 minutes, respectively. Data are mean ± S.D. (n = 4 in each group). *P < 0.05 compared with the same genotype of mice treated with vehicle (harmaline 0 mg/kg) plus 5-MeO-DMT; #P < 0.05 compared with wild-type mice under the same treatment.
Discussion
This study systemically evaluates the PK interactions between harmaline and 5-MeO-DMT at multiple dose levels. The mechanistic effects of harmaline on 5-MeO-DMT PK are composed of the inhibitions of MAO-A–mediated deamination and CYP2D6-catalyzed O-demethylation, which together result in a dose-dependent biphasic effect on the production of active metabolite bufotenine. The inhibition of MAO-A–controlled metabolic elimination inevitably leads to a significantly greater and prolonged systemic and brain exposure to 5-MeO-DMT, which can be further influenced by CYP2D6 status at certain dose combinations. In addition, a mathematical model is developed to describe the PK interactions of harmaline and 5-MeO-DMT including the PK of the active metabolite bufotenine and the impact of CYP2D6 status.
Harmaline is a competitive inhibitor to MAO-A, with an in vitro Ki value of 0.048 µM in suppression of MAO-A enzyme activity (Kim et al., 1997). Data from the present study show that in both wild-type and Tg-CYP2D6 mice, blood harmaline concentrations are generally much higher than the Ki value (Figs. 2, A and B, and 3, A and B). Given the fact that the lowest dose (2 mg/kg) of harmaline tested in this study is actually lower than the human equivalent dose (5 mg/kg) (Ott, 2001; Riba et al., 2003; Yuruktumen et al., 2008), a complete inhibition of MAO-A by harmaline might occur and it is expected to alter the PK of 5-MeO-DMT or other MAO-A substrate drugs in humans. The complete blockage of MAO-A activity in mice treated with various doses of harmaline (e.g., 2, 5, and 15 mg/kg) plus the same dose of 5-MeO-DMT (e.g., 2 mg/kg) is also supported by the observation of similar blood 5-MeO-DMT Cmax values (e.g., about 1.9 µM; Table 2). Nevertheless, there is a harmaline dose-dependent increase of systemic exposure to the same dose of 5-MeO-DMT (Table 2) that is likely attributed to the prolonged inhibition of MAO-A by higher doses of harmaline (Figs. 2 and 3). This observation also agrees with our finding that the increase of systemic exposure to 5-MeO-DMT is more pronounced in wild-type mice than that in Tg-CYP2D6 mice (Table 2), consistent with a longer and greater exposure to the MAO-A inhibitor harmaline itself in wild-type mice lacking CYP2D6 activity (Figs. 2 and 3; Table 2).
This study also presents an interesting observation of in vivo inhibition of bufotenine formation from 5-MeO-DMT with the increase of harmaline dose from 5 to 15 mg/kg. The suppression of bufotenine production from 5-MeO-DMT by harmaline is supported by in vitro enzymatic study using purified CYP2D6 (Fig. 5). The inhibitory effect of harmaline on CYP2D6-catalyzed 5-MeO-DMT O-demethylation is incorporated in the final model (Fig. 1) that describes harmaline-5-MeO-DMT PK interactions. The estimated in vivo inhibition potency for harmaline inhibition of CYP2D6-catalyzed 5-MeO-DMT O-demethylation is 7.13 µM (Table 4), which is relatively lower than the Ki value (26.2 µM) for harmaline competitive inhibition of CYP2D6-mediated dextromethorphan O-demethylation (Zhao et al., 2011). This might be due to the difference in experimental systems and substrates. Nevertheless, harmaline itself is also metabolized by CYP2D6, which is considered in the final PK model that likely provides a more accurate estimation of the in vivo Ki value.
The integrated PK model (Fig. 1) recognizes the dual mechanisms of action of harmaline on 5-MeO-DMT metabolism, namely the inhibition of MAO-A and CYP2D6-catalyzed deamination and O-demethylation, respectively. The PK of 5-MeO-DMT administered alone is nonlinear, and the estimated Vmax, Km, and CLCYP2D6 values are 2.76 µmol/min per kilogram, 13.2 µM, and 0.0256 l/min per kilogram, respectively (Shen et al., 2011b). In this study, the nonlinearity of 5-MeO-DMT clearance is separated into MAO-A dependent (intrinsic clearance Vmax/Km, 0.161 l/min per kilogram) and independent (0.136 l/min per kilogram) pathways to clearly delineate the impact of MAOI harmaline. Although O-demethylation produces the active metabolite bufotenine (Fig. 1), it is a minor route for 5-MeO-DMT elimination, as indicated by a small estimated intrinsic clearance value (0.0264 and 0.0343 l/min per kilogram in wild-type and Tg-CYP2D6 mice, respectively). Based on the estimated Km(MAO-A)-M, Km(other)-M, and Km(D)-M values (16.7, 0.446, and 1.27 µM, respectively), both MAO-A independent and O-demethylation elimination routes should be easily saturable (Fig. 2). When greater doses of 5-MeO-DMT (e.g., 10 or 20 mg/kg; Figs. 3 and 4) are used, the drug is mainly eliminated through MAO-A–mediated metabolism (i.e., approximately 50% of the total clearance). Therefore, coadministration of even a relatively lower dose (e.g., 2 or 5 mg/kg) of harmaline readily blocks MAO-A–dependent elimination (Figs. 2 and 3) of 5-MeO-DMT and largely enhances the systemic and cerebral exposure to 5-MeO-DMT (Figs. 3 and 6) as well as hyperserotonergic effects (unpublished data) and behavioral changes (Halberstadt et al., 2008; Winter et al., 2011).
Since bufotenine acts primarily on the 5-HT2A receptor, whereas 5-MeO-DMT is more selective for 5-HT1A (Roth et al., 1997; Halberstadt and Geyer, 2011), the active metabolite bufotenine might complicate the interpretation of apparent 5-MeO-DMT pharmacological and toxicological effects. The incorporation of bufotenine in the PK model (Fig. 1) should enable the detection of bufotenine and 5-MeO-DMT specific effects. First, MAOI harmaline (e.g., 5 mg/kg) could shunt 5-MeO-DMT metabolism to alternative pathways such as O-demethylation, leading to a greater exposure to bufotenine (Table 3). However, further increases in harmaline dose (e.g., 15 mg/kg) actually reduce the production of bufotenine as harmaline itself inhibits CYP2D6 activity (Fig. 5) (Zhao et al., 2011). Second, comparison of the serum PK profiles of 5-MeO-DMT and bufotenine from multiple harmaline-5-MeO-DMT dose combinations indicates that bufotenine generally accounts for less than 10% of the parent drug 5-MeO-DMT (Table 2 and 3). Third, compared with the pronounced 5-MeO-DMT penetration through the blood-brain barrier (McBride, 2000) that is indicated by a 2- to 4-fold higher 5-MeO-DMT concentration in mouse brain (Fig. 6) than that in blood (Figs. 2–4), bufotenine has a poor capacity to cross the blood-brain barrier (McBride, 2000) which is manifested by a 10–25% of blood bufotenine concentration (Fig. 2–4) appearing in mouse brain (Fig. 7). Therefore, it may be concluded that the contribution of active metabolite bufotenine to the overall effects of harmaline-5-MeO-DMT DDI is likely limited to its high affinity to the peripheral 5-HT2A receptor (Cook et al., 1994; Lesurtel et al., 2006).
With the increase of 5-MeO-DMT concentrations after the inhibition of MAO-A by harmaline, blood (Figs. 2–4) and brain (Fig. 7) bufotenine concentrations are generally increased. The production of bufotenine from 5-MeO-DMT is determined by CYP2D6 enzyme (Yu et al., 2003a). Indeed, bufotenine concentrations are typically greater in Tg-CYP2D6 mice that exhibit additional CYP2D6 activity, and the difference reaches statistical significance at certain dose combinations such as 10 mg/kg 5-MeO-DMT plus 5 mg/kg harmaline (Fig. 7; Table 3) even with a noticeable murine O-demethylase activity in wild-type animals. Our findings on the impact of CYP2D6 status on in vivo bufotenine production from 5-MeO-DMT as well as harmaline PK and harmaline-5-MeO-DMT DDI in this study support the utility of Tg-CYP2D6 animal models in investigation of CYP2D6 pharmacogenetics (Jiang et al., 2011; Shen et al., 2011a).
Consistent with our previous observation (Wu et al., 2009), harmaline exhibits a dose-dependent bioavailability (Table 4). A greater bioavailability with increasing harmaline dose levels might be explained by the substrate inhibition of its own O-demethylation elimination (Fig. 5) (Zhao et al., 2011) that contributes to first-pass metabolism. Comparing the observed harmaline data (Table 1) in the present DDI study and the simulated profiles (Figs. 2 and 3) according to our previous PK model of harmaline administered alone (Wu et al., 2009), a slightly greater systemic exposure to harmaline under the DDI condition is noticeable. In particular, harmaline concentrations at the early time points after the administration of 5-MeO-DMT (Figs. 2 and 3) are slightly different from those when harmaline is administered alone, which might be caused by the difference in study design (e.g., additional animal handling for coadministration of 5-MeO-DMT; LC-MS/MS versus HPLC quantification) and/or an obvious change in mouse physiologic conditions (e.g., body temperature) by coadministration of 5-MeO-DMT with harmaline. Nevertheless, a minor difference in harmaline concentration at this range (>1 µM), which is much greater than the Ki value 0.048 μM (Kim et al., 1997) and likely causes a complete inhibition of MAO-A, does not have any significant effect on harmaline–5-MeO-DMT DDI or PK modeling.
In summary, our data demonstrate that coadministration of MAOI harmaline significantly increases and extends the systemic and cerebral exposure to 5-MeO-DMT as well as the active metabolite bufotenine. The PK interactions between harmaline and 5-MeO-DMT may be further influenced by CYP2D6 status under certain dose combinations. These findings would improve the understanding of risks of indolealkylamine intoxication. In addition, the unified PK model is consistent with mechanistic actions of harmaline on 5-MeO-DMT PK, namely the inhibitions of MAO-A–mediated deamination and CYP2D6-catalyzed O-demethylation. This model well describes the PK profiles of harmaline, 5-MeO-DMT, and bufotenine in wild-type and Tg-CYP2D6 mice, and provides a solid basis for further investigations of harmaline–5-MeO-DMT PK interactions and understanding the resultant complex pharmacological and toxicological effects.
Abbreviations
- 5-HT
serotonin
- 5-Me-DMT
5-methyl-N,N-dimethyltryptamine
- 5-MeO-DMT
5-Methoxy-N,N-dimethyltryptamine
- AUC
area under the concentration-time curve
- DDI
drug-drug interaction
- HPLC
high-performance liquid chromatography
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- MAO-A
monoamine oxidase A
- MAOI
MAO inhibitor
- PK
pharmacokinetics
- Tg-CYP2D6
CYP2D6-humanized
Authorship Contributions
Participated in research design: Jiang, Shen, Mager, Yu.
Conducted experiments: Jiang, Shen, Yu.
Contributed new reagents or analytic tools: Shen, Yu.
Performed data analysis: Jiang, Shen, Mager, Yu.
Wrote or contributed to the writing of the manuscript: Jiang, Mager, Yu.
Footnotes
This research was supported by the National Institutes of Health National Institute on Drug Abuse [Grant R01-DA021172]. X.-L.J. was supported by a Pfizer fellowship.
References
- Barker SA, McIlhenny EH, Strassman R. (2012) A critical review of reports of endogenous psychedelic N, N-dimethyltryptamines in humans: 1955-2010. Drug Test Anal 4:617–635 [DOI] [PubMed] [Google Scholar]
- Brush DE, Bird SB, Boyer EW. (2004) Monoamine oxidase inhibitor poisoning resulting from Internet misinformation on illicit substances. J Toxicol Clin Toxicol 42:191–195 [DOI] [PubMed] [Google Scholar]
- Cook EH, Jr, Fletcher KE, Wainwright M, Marks N, Yan SY, Leventhal BL. (1994) Primary structure of the human platelet serotonin 5-HT2A receptor: identify with frontal cortex serotonin 5-HT2A receptor. J Neurochem 63:465–469 [DOI] [PubMed] [Google Scholar]
- Corchero J, Granvil CP, Akiyama TE, Hayhurst GP, Pimprale S, Feigenbaum L, Idle JR, Gonzalez FJ. (2001) The CYP2D6 humanized mouse: effect of the human CYP2D6 transgene and HNF4alpha on the disposition of debrisoquine in the mouse. Mol Pharmacol 60:1260–1267 [DOI] [PubMed] [Google Scholar]
- Drug Enforcement Administration (DEA), Department of Justice (2004) Schedules of controlled substances: placement of alpha-methyltryptamine and 5-methoxy-N,N-diisopropyltryptamine into schedule I of the Controlled Substances Act. Final rule. Fed Regist 69:58950–58953 [PubMed] [Google Scholar]
- Drug Enforcement Administration (DEA), Department of Justice (2010) Schedules of controlled substances: placement of 5-methoxy-N,N-dimethyltryptamine into Schedule I of the Controlled Substances Act. Final rule. Fed Regist 75:79296–79300 [PubMed] [Google Scholar]
- Halberstadt AL, Buell MR, Masten VL, Risbrough VB, Geyer MA. (2008) Modification of the effects of 5-methoxy-N,N-dimethyltryptamine on exploratory behavior in rats by monoamine oxidase inhibitors. Psychopharmacology (Berl) 201:55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberstadt AL, Geyer MA. (2011) Multiple receptors contribute to the behavioral effects of indoleamine hallucinogens. Neuropharmacology 61:364–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SL, Thomas SH. (2011) Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila) 49:705–719 [DOI] [PubMed] [Google Scholar]
- Jiang XL, Gonzalez FJ, Yu AM. (2011) Drug-metabolizing enzyme, transporter, and nuclear receptor genetically modified mouse models. Drug Metab Rev 43:27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XL, Shen HW, Yu AM. (2009) Pinoline may be used as a probe for CYP2D6 activity. Drug Metab Dispos 37:443–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley NE, Tepper DE. (2012) Rescue therapy for acute migraine, part 1: triptans, dihydroergotamine, and magnesium. Headache 52:114–128 [DOI] [PubMed] [Google Scholar]
- Khor SP, Bozigian H, Mayersohn M. (1991) Potential error in the measurement of tissue to blood distribution coefficients in physiological pharmacokinetic modeling. Residual tissue blood. II. Distribution of phencyclidine in the rat. Drug Metab Dispos 19:486–490 [PubMed] [Google Scholar]
- Kim H, Sablin SO, Ramsay RR. (1997) Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch Biochem Biophys 337:137–142 [DOI] [PubMed] [Google Scholar]
- Krebs-Thomson K, Ruiz EM, Masten V, Buell M, Geyer MA. (2006) The roles of 5-HT1A and 5-HT2 receptors in the effects of 5-MeO-DMT on locomotor activity and prepulse inhibition in rats. Psychopharmacology (Berl) 189:319–329 [DOI] [PubMed] [Google Scholar]
- Lesurtel M, Graf R, Aleil B, Walther DJ, Tian Y, Jochum W, Gachet C, Bader M, Clavien PA. (2006) Platelet-derived serotonin mediates liver regeneration. Science 312:104–107 [DOI] [PubMed] [Google Scholar]
- McBride MC. (2000) Bufotenine: toward an understanding of possible psychoactive mechanisms. J Psychoactive Drugs 32:321–331 [DOI] [PubMed] [Google Scholar]
- McKenna DJ. (2004) Clinical investigations of the therapeutic potential of ayahuasca: rationale and regulatory challenges. Pharmacol Ther 102:111–129 [DOI] [PubMed] [Google Scholar]
- Ott J. (1999) Pharmahuasca: human pharmacology of oral DMT plus harmine. J Psychoactive Drugs 31:171–177 [DOI] [PubMed] [Google Scholar]
- Ott J. (2001) Pharmepéna-Psychonautics: Human intranasal, sublingual and oral pharmacology of 5-methoxy-N,N-dimethyl-tryptamine. J Psychoactive Drugs 33:403–407 [DOI] [PubMed] [Google Scholar]
- Riba J, Valle M, Urbano G, Yritia M, Morte A, Barbanoj MJ. (2003) Human pharmacology of ayahuasca: subjective and cardiovascular effects, monoamine metabolite excretion, and pharmacokinetics. J Pharmacol Exp Ther 306:73–83 [DOI] [PubMed] [Google Scholar]
- Roth BL, Choudhary MS, Khan N, Uluer AZ. (1997) High-affinity agonist binding is not sufficient for agonist efficacy at 5-hydroxytryptamine2A receptors: evidence in favor of a modified ternary complex model. J Pharmacol Exp Ther 280:576–583 [PubMed] [Google Scholar]
- Sanders E, Bush MT. (1967) Distribution, metabolism and excretion of bufotenine in the rat with preliminary studies of its O-methyl derivative. J Pharmacol Exp Ther 158:340–352 [PubMed] [Google Scholar]
- Shen HW, Jiang XL, Yu AM. (2009) Development of a LC-MS/MS method to analyze 5-methoxy-N,N-dimethyltryptamine and bufotenine, and application to pharmacokinetic study. Bioanalysis 1:87–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Jiang XL, Winter JC, Yu AM. (2010a) Psychedelic 5-methoxy-N,N-dimethyltryptamine: metabolism, pharmacokinetics, drug interactions, and pharmacological actions. Curr Drug Metab 11:659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Wu C, Jiang XL, Yu AM. (2010b) Effects of monoamine oxidase inhibitor and cytochrome P450 2D6 status on 5-methoxy-N,N-dimethyltryptamine metabolism and pharmacokinetics. Biochem Pharmacol 80:122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Jiang XL, Gonzalez FJ, Yu AM. (2011a) Humanized transgenic mouse models for drug metabolism and pharmacokinetic research. Curr Drug Metab 12:997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Jiang XL, Yu AM. (2011b) Nonlinear pharmacokinetics of 5-methoxy-N,N-dimethyltryptamine in mice. Drug Metab Dispos 39:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklerov J, Levine B, Moore KA, King T, Fowler D. (2005) A fatal intoxication following the ingestion of 5-methoxy-N,N-dimethyltryptamine in an ayahuasca preparation. J Anal Toxicol 29:838–841 [DOI] [PubMed] [Google Scholar]
- Tanaka E, Kamata T, Katagi M, Tsuchihashi H, Honda K. (2006) A fatal poisoning with 5-methoxy-N,N-diisopropyltryptamine, Foxy. Forensic Sci Int 163:152–154 [DOI] [PubMed] [Google Scholar]
- Winter JC, Amorosi DJ, Rice KC, Cheng K, Yu AM. (2011) Stimulus control by 5-methoxy-N,N-dimethyltryptamine in wild-type and CYP2D6-humanized mice. Pharmacol Biochem Behav 99:311–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter JC, Filipink RA, Timineri D, Helsley SE, Rabin RA. (2000) The paradox of 5-methoxy-N,N-dimethyltryptamine: an indoleamine hallucinogen that induces stimulus control via 5-HT1A receptors. Pharmacol Biochem Behav 65:75–82 [DOI] [PubMed] [Google Scholar]
- Wu C, Jiang XL, Shen HW, Yu AM. (2009) Effects of CYP2D6 status on harmaline metabolism, pharmacokinetics and pharmacodynamics, and a pharmacogenetics-based pharmacokinetic model. Biochem Pharmacol 78:617–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Kneller BM, Rettie AE, Haining RL. (2002) Expression, purification, biochemical characterization, and comparative function of human cytochrome P450 2D6.1, 2D6.2, 2D6.10, and 2D6.17 allelic isoforms. J Pharmacol Exp Ther 303:1291–1300 [DOI] [PubMed] [Google Scholar]
- Yu AM. (2008) Indolealkylamines: biotransformations and potential drug-drug interactions. AAPS J 10:242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AM, Idle JR, Gonzalez FJ. (2004) Polymorphic cytochrome P450 2D6: humanized mouse model and endogenous substrates. Drug Metab Rev 36:243–277 [DOI] [PubMed] [Google Scholar]
- Yu AM, Idle JR, Herraiz T, Küpfer A, Gonzalez FJ. (2003a) Screening for endogenous substrates reveals that CYP2D6 is a 5-methoxyindolethylamine O-demethylase. Pharmacogenetics 13:307–319 [DOI] [PubMed] [Google Scholar]
- Yu AM, Idle JR, Krausz KW, Küpfer A, Gonzalez FJ. (2003b) Contribution of individual cytochrome P450 isozymes to the O-demethylation of the psychotropic beta-carboline alkaloids harmaline and harmine. J Pharmacol Exp Ther 305:315–322 [DOI] [PubMed] [Google Scholar]
- Yuruktumen A, Karaduman S, Bengi F, Fowler J. (2008) Syrian rue tea: a recipe for disaster. Clin Toxicol (Phila) 46:749–752 [DOI] [PubMed] [Google Scholar]
- Zanger UM, Raimundo S, Eichelbaum M. (2004) Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol 369:23–37 [DOI] [PubMed] [Google Scholar]
- Zhang WY, Tu YB, Haining RL, Yu AM. (2009) Expression and functional analysis of CYP2D6.24, CYP2D6.26, CYP2D6.27, and CYP2D7 isozymes. Drug Metab Dispos 37:1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, He YQ, Wang J, Ding KM, Wang CH, Wang ZT. (2011) Inhibition of human cytochrome P450 enzymes 3A4 and 2D6 by β-carboline alkaloids, harmine derivatives. Phytother Res 25:1671–1677 [DOI] [PubMed] [Google Scholar]