

Abstract
Positive allosteric modulation of metabotropic glutamate receptor subtype 5 (mGlu5) is a promising novel approach for the treatment of schizophrenia and cognitive disorders. Allosteric binding sites are topographically distinct from the endogenous ligand (orthosteric) binding site, allowing for co-occupation of a single receptor with the endogenous ligand and an allosteric modulator. Negative allosteric modulators (NAMs) inhibit and positive allosteric modulators (PAMs) enhance the affinity and/or efficacy of the orthosteric agonist. The molecular determinants that govern mGlu5 modulator affinity versus cooperativity are not well understood. Focusing on the modulators based on the acetylene scaffold, we sought to determine the molecular interactions that contribute to PAM versus NAM pharmacology. Generation of a comparative model of the transmembrane-spanning region of mGlu5 served as a tool to predict and interpret the impact of mutations in this region. Application of an operational model of allosterism allowed for determination of PAM and NAM affinity estimates at receptor constructs that possessed no detectable radioligand binding as well as delineation of effects on affinity versus cooperativity. Novel mutations within the transmembrane domain (TM) regions were identified that had differential effects on acetylene PAMs versus 2-methyl-6-(phenylethynyl)-pyridine, a prototypical NAM. Three conserved amino acids (Y658, T780, and S808) and two nonconserved residues (P654 and A809) were identified as key determinants of PAM activity. Interestingly, we identified two point mutations in TMs 6 and 7 that, when mutated, engender a mode switch in the pharmacology of certain PAMs.
Introduction
Metabotropic glutamate receptors (mGlus) are seven-transmembrane-spanning (G protein-coupled) receptors (7TMRs) that include eight subtypes, mGlu1–mGlu8, for the major excitatory neurotransmitter, glutamate (Niswender and Conn, 2010). Historically, it has been difficult to develop highly mGlu-subtype-selective ligands due to the high sequence conservation of the endogenous ligand (i.e., glutamate) orthosteric binding site. This led to the search for compounds that interact at “allosteric” sites, topographically distinct from the orthosteric site. Referred to as allosteric modulators, such compounds can affect the affinity and/or efficacy of an orthosteric ligand, a property referred to as cooperativity. Modulators that inhibit orthosteric ligand binding and/or activity are negative allosteric modulators (NAMs) and those that enhance are positive allosteric modulators (PAMs); a third category, silent (or neutral) allosteric modulators (SAMs), includes compounds that bind but do not modulate receptor activity.
Efforts to develop mGlu allosteric modulators have been especially successful for mGlu5; a broad range of allosteric modulators as well as allosteric radioligands has been developed, including pure PAMs, PAMs with agonist activity, weak and full NAMs, and SAMs (Gasparini et al., 1999; Varney et al., 1999; Cosford et al., 2003; O'Brien et al., 2004; Kinney et al., 2005; Rodriguez et al., 2005, 2009, 2010; Ametamey et al., 2007; Chen et al., 2007, 2008; Honer et al., 2007; Treyer et al., 2007; Liu et al., 2008; Noetzel et al., 2012). mGlu5 PAMs have potential utility for treatment of cognitive disorders and schizophrenia, whereas NAMs are being pursued for treatment of fragile X syndrome, depression, anxiety, and l-DOPA–induced dyskinesia (Gregory et al., 2011).
In addition to improvements in receptor selectivity, allosteric modulators offer a number of theoretical advantages over their competitive counterparts (Melancon et al., 2012). Modulators that possess no intrinsic efficacy have potential for spatial and temporal modulation of receptor activity. This is an especially important consideration for potential central nervous system therapeutics, where "fine-tuning" neurotransmission is likely to yield a better therapeutic outcome than the sustained blockade or activation by an orthosteric ligand. Furthermore, the cooperativity between the two sites is saturable; thus, allosteric modulators have a built-in “ceiling level” to their effect, and may therefore have a larger therapeutic index in the case of overdose.
Structure-activity relationships (SARs) for mGlu modulators, particularly with respect to targeting mGlu5, are also notoriously difficult; the SAR is often "steep" or "flat" with minimal changes to the structure, resulting in a complete loss of activity (Zhao et al., 2007). Furthermore, numerous mGlu modulator chemotypes display "molecular switches" whereby a PAM or SAM arises from a NAM scaffold or vice versa (Wood et al., 2011), originally observed during discovery of the first mGlu5 PAM, difluorobenzaldazine (DFB) (O'Brien et al., 2004). This phenomenon continues to be a challenge for medicinal chemists, with PAMs being derived from NAM scaffolds (Sharma et al., 2009; Rodriguez et al., 2010; Zhou et al., 2010), SAMs from either NAM or PAM chemotypes (Rodriguez et al., 2005; Hammond et al., 2010), and NAMs from PAMs (Lamb et al., 2011). Furthermore, molecular switches have also been described with respect to unanticipated alterations in receptor selectivity (Sheffler et al., 2012). Steep or flat SARs and molecular switches may be attributed to changes in the affinity and/or cooperativity of an allosteric modulator. Therefore, we were interested in probing the determinants of allosteric modulator affinity and cooperativity, focusing on the common allosteric site of group I mGlus as previously identified for mGlu5-selective modulators such as 2-methyl-6-(phenylethynyl)-pyridine (MPEP) (Pagano et al., 2000; Malherbe et al., 2003, 2006; Muhlemann et al., 2006). Two classes of acetylene PAMs, picolinamides and nicotinamides, that originally evolved from a NAM high-throughput screening lead (Rodriguez et al., 2010) were selected for in-depth characterization in comparison with MPEP. We identified seven novel residues that, when mutated, significantly decrease MPEP affinity. Moreover, a single point mutation (W784A) reduced the cooperativity of MPEP, such that it no longer fully blocked the response to glutamate. PAMs were found to interact with the common allosteric site used by MPEP, although these compounds showed differential sensitivities to certain mutations. Two different point mutations were identified that conferred a molecular switch in the pharmacology of PAMs: T780A converted N-tert-butyl-6-[2-(3-fluorophenyl)ethynyl]pyridine-3-carboxamide (VU0415051) to a weak NAM, while S808A converted (5-((3-fluorophenyl)ethynyl)pyridin-2-yl)(3-hydroxyazetidin-1-yl)methanone (VU0405398) from a weak PAM to a full NAM and N-(tert-butyl)-5-((3-fluorophenyl)ethynyl)picolinamide (VU0405386) from a PAM to a neutral modulator. Our findings build on the existing understanding of the location of the common allosteric site. Quantification of the effect of mutations on modulator pharmacology has allowed delineation of determinants for cooperativity versus affinity.
Materials and Methods
Materials.
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and antibiotics were purchased from Invitrogen (Carlsbad, CA). [3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine ([3H]methoxyPEPy; 76.3 Ci/mmol) was custom-synthesized by PerkinElmer Life and Analytical Sciences (Waltham, MA). N-Cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide hydrochloride (VU0360172), VU0405398, VU0405386, and VU0415051 were all synthesized in-house using previously reported methodologies (Rodriguez et al., 2010; Gregory et al., 2012). (6-((3-Fluorophenyl)ethynyl)pyridin-3-yl)(3-hydroxyazetidin-1-yl)methanone (VU0360173) and (N-cyclobutyl-5-((3-fluorophenyl)ethynyl)ethynyl)picolinamide hydrochloride (VU0403602) were synthesized in-house (Supplemental Material 1). Unless otherwise stated, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of an analytical grade.
Cell Culture and Mutagenesis.
Mutations were introduced into the wild-type rat mGlu5 in pCI-Neo using site-directed mutagenesis (Quikchange II; Agilent Technologies, Santa Clara, CA) and verified by sequencing. Wild-type and mutant rat mGlu5 receptor constructs were transfected into human embryonic kidney (HEK) 293A cells, using Fugene6 (Promega, Madison, WI) as the transfection reagent. Polyclonal stable cell lines were derived for rat mGlu5 mutant constructs by maintaining the cells at subconfluence for a minimum of four passages in the presence of 1 mg/ml G418 (Mediatech, Manassas, VA). Stably transfected cell lines were subsequently maintained in complete DMEM supplemented with 10% FBS, 2 mM l-glutamine, 20 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, antibiotic-antimycotic, and 500 μg/ml G418 at 37°C in a humidified incubator containing 5% CO2, 95% O2.
Intracellular Ca2+ Mobilization.
The day prior to assay, HEK293A-rat mGlu5 cells were seeded at 50,000 cells/well in poly-d-lysine–coated, black-walled, clear-bottom, 96-well plates in assay medium (DMEM supplemented with 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate). On the day of assay, the cell-permeant Ca2+ indicator dye Fluo-4 (Invitrogen) was used to assay receptor-mediated Ca2+ mobilization as described previously (Hammond et al., 2010) using a Flexstation II (Molecular Devices, Sunnyvale, CA). A 5-point smoothing function was applied to the raw fluorescent Ca2+ traces and basal fluorescence of individual wells determined during the first 20 seconds. The peak increase in fluorescence over basal was determined prior to normalization to the maximal peak response elicited by glutamate.
Radioligand Binding.
Radioligand binding assays were performed on HEK293A cell membranes as described previously (Gregory et al., 2012). Briefly, for saturation binding experiments, membranes (20–50 μg/well) were incubated with a range of [3H]methoxyPEPy concentrations (0.5–60 nM) for 1 hour at room temperature with shaking in binding buffer (50 mM Tris-HCl, 0.9% NaCl, pH 7.4). MPEP (10 μM) was used to determine nonspecific binding. For inhibition binding experiments, membranes were incubated with ∼2 nM [3H]methoxyPEPy and a range of concentrations of test ligand (100 pM to 100 μM) for 1 hour at room temperature with shaking in Ca2+ assay buffer with 1% dimethyl sulfoxide final. Assays were terminated by rapid filtration through GF/B Unifilter plates (PerkinElmer Life and Analytical Sciences) using a Brandel 96-well plate harvester (Brandel Inc., Gaithersburg, MD) and three washes with ice-cold binding buffer, separating bound from free radioligand. Plates were allowed to dry overnight and radioactivity counted using a TopCount Scintillation Counter (PerkinElmer Life and Analytical Sciences).
Generation of an mGlu5 Comparative Model.
A comparative model of mGlu5 was constructed using the protein structure prediction software package Rosetta version 3.4 (Leaver-Fay et al., 2011). Based on its high similarity (e-value of 3e-15 with a sequence coverage of 90%) to mGlu5 according to a search using National Center for Biotechnology Information (NCBI) BLASTP on sequences from the Protein Data Bank (PDB), the X-ray crystal structure human β2-adrenergic receptor (PDB ID 2RH1) (Cherezov et al., 2007) was chosen as a template. Both β2-adrenergic receptor and mGlu5 also share a conserved disulfide bond between a cysteine at the top of transmembrane helix 3 and a cysteine in extracellular loop 2. Members of the Family C 7TMRs, namely the human mGlus and calcium-sensing receptor (CaSR) sequences, were first aligned with CLUSTALW. Alignment of transmembrane domains (TMs) between Family C 7TMRs and Family A crystal structure templates were directly adopted from Malherbe et al. (2006), with the exception of TMs 2, 4, and 7, which were based on the alignment of CaSR with Family A 7TMRs from Miedlich et al. (2004) (Supplemental Fig. 1). In the construction of the comparative models, the backbone coordinates of the β2-adrenergic receptor were retained in the comparative model of mGlu5 while the loop coordinates were built in Rosetta using Monte Carlo Metropolis fragment replacement combined with cyclic coordinate descent loop closure. Rosetta ensures that ϕ-ψ angles of backbone segments from homologous sequence fragments from the PDB are introduced into the loop regions. After the fragment substitution, small movements in the ϕ-ψ angles are performed to close breaks in the protein chain. The resulting full-sequence models were subjected to eight iterative cycles of side-chain repacking and gradient minimization of ϕ, ψ, and χ angles in Rosetta Membrane (Yarov-Yarovoy et al., 2006).
Docking of Allosteric Modulators.
The NAM MPEP and six acetylene PAMs (VU0360173, VU0405398, VU0360172, VU0403602, VU0415051, and VU0405386) were computationally docked into the comparative model of mGlu5 using RosettaLigand (Meiler and Baker, 2006; Davis and Baker, 2009; Lemmon and Meiler, 2012). Each modulator was allowed to sample docking poses in a 5-Å radius centered at the putative binding site for MPEP, determined by the residues known to affect modulator affinity and/or function. For MPEP, separate docking experiments were carried out, centered on two residues shown to greatly influence modulator affinity when mutated: P654 and S808. For the six acetylene PAMs, docking experiments were all centered on P654. Once a binding mode had been determined by the docking procedure, 10 low-energy conformations of the ligand created by MOE (Molecular Operating Environment; Chemical Computing Group, Montreal, QC, Canada) were tested within the site. Side-chain rotamers around the ligand were optimized simultaneously in a Monte Carlo minimization algorithm. The energy function used during the docking procedure contains terms for van der Waals attractive and repulsive forces, hydrogen bonding, electrostatic interactions between pairs of amino acids, solvation, and a statistical term derived from the probability of observing a side-chain conformation from the PDB. For each modulator, >2000 docked complexes were generated and clustered for structural similarity using bcl::Cluster (Alexander et al., 2011). The lowest-energy binding modes from the five largest clusters for each modulator were used for further analysis. A detailed protocol capture for protein modeling and ligand docking, including links to input and output files, is provided (Supplemental Materials 2 and 3).
Data Analysis.
All computerized nonlinear regression was performed using Prism 5.01 (GraphPad Software, La Jolla, CA). Inhibition of [3H]methoxyPEPy binding data sets were fitted to a one-site inhibition binding model and estimates of inhibitor dissociation constants (KI) were derived using the Cheng-Prusoff equation for competitive ligands (Cheng and Prusoff, 1973) and the following version of the allosteric ternary complex model for ligands that did not fully displace radioligand (Lazareno and Birdsall, 1995):
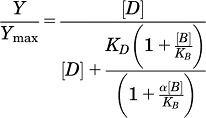 |
where Y/Ymax is the fractional specific binding, D is the radioligand concentration, B is the molar concentration of the allosteric modulator, KD is the radioligand equilibrium dissociation constant, and KB is the allosteric modulator equilibrium dissociation constant. α denotes the cooperativity factor, where values of α >1 describe positive cooperativity, values of α <1 (but >0) denote negative cooperativity, and α = 1 denotes neutral cooperativity.
Shifts of glutamate concentration-response curves by allosteric modulators were globally fitted to an operational model of allosterism (Leach et al., 2007):
where A is the molar concentration of the orthosteric agonist glutamate and B is the molar concentration of the allosteric modulator, KA is the equilibrium dissociation constant of the orthosteric agonist glutamate, and KB is the allosteric modulator equilibrium dissociation constant.
Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by β. The parameters τA and τB relate to the ability of the orthosteric and allosteric ligands, respectively, to engender receptor activation. Em and n denote the maximal possible system response and the transducer function that links occupancy to response, respectively.
Allosteric modulator and agonist concentration-response curves were fitted to a four-parameter logistic equation to determine potency estimates:
where bottom and top are the lower and upper plateaus, respectively, of the concentration-response curve; HillSlope is the Hill coefficient that describes the steepness of the curve; and EC50 is the molar concentration of modulator required to generate a response halfway between the top and bottom.
All affinity, cooperativity, and potency parameters were estimated as logarithms and are expressed as the mean ± S.E.M. (Christopoulos, 1998). Statistical analyses were performed where appropriate as indicated using one-way analysis of variance with Dunnett’s post-test when comparing to control or Tukey’s post-test when making multiple comparisons.
Results
Refining the Alignment of mGlu5 to Family A 7TMRs and Prediction of Amino Acids within the Common Allosteric “MPEP” Site.
In contrast to previous models of mGlu5 (Pagano et al., 2000; Malherbe et al., 2003, 2006), the alignment of mGlu5 presented here is based on a previously reported CaSR alignment (Miedlich et al., 2004) where the PKxY motif in TM7 of the human mGlus is aligned with the NPxxY motif in the Family A 7TMRs (Supplemental Fig. 1). This adjustment in the alignment shifts TM7 by seven residues, predicting that S806, S808, and T810 face the MPEP binding pocket. Indicated on the alignment of the human mGlus with bovine rhodopsin and human β2-adrenergic receptor (Supplemental Fig. 1) are point mutations included in the current study, which includes mutations previously reported to perturb NAM or PAM activity of mGlu5 modulators (Pagano et al., 2000; Malherbe et al., 2003, 2006; Muhlemann et al., 2006; Chen et al., 2008; Mølck et al., 2012) and mutations novel to this study (highlighted in gray). Based on the localization of these previously known residues, we elected to mutate additional residues predicted to be on the same inward-facing helical face of TMs 3, 5, and 6 (I650A, G651F, V739M, P742S, N746A, G747V, T779A, I783A, V788A, Y791F, and F792A). Conserved residues were substituted for Ala, while nonconserved residues were mutated to the corresponding amino acid in either group II or group III mGlus. To validate the alignment of TM7, seven residues were mutated that were predicted to line the inward-facing helical face of TM7: S806A, S808A, S808T, T810A, T810S, A812S, L813A, C815A, and M816A. As a negative control, a point mutation previously shown to affect PAM activity [by N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide (CPPHA)] at the second allosteric site on mGlu5, F585I, was also included (Chen et al., 2008).
Identification of Eight Novel Point Mutations That Perturb MPEP Inhibition of Glutamate.
To assess the contribution of both novel and previously identified residues to the MPEP binding pocket, MPEP (Fig. 1) was screened for its effect on the maximal response to glutamate in the Ca2+ mobilization assay at a single concentration. For screening purposes 10 nM MPEP was selected, as this concentration caused a significant decrease in glutamate Emax to ∼45% at mGlu5-wt, such that increases or decreases in the percent inhibition caused by MPEP could be detected (Fig. 1A). All mutations were functional and expressed at levels ranging from 0.4–3.8 pmol/mg (Supplemental Table 1; Table 1). Mutations that showed a lack of [3H]methoxyPEPy binding showed a similar range of expression levels (between that of the low-expressing mGlu5-wt cell line and the higher-expressing, polyclonal mGlu5-wt line) as confirmed by immunoblotting (Supplemental Fig. 2). Twelve point mutations, corresponding to nine different amino acids, significantly reduced inhibition of glutamate Emax by 10 nM MPEP (Fig. 1B). From the original 33 point mutations screened, 24 were selected for further characterization, including the negative control F585I.
Fig. 1.

Probing the common allosteric binding site on mGlu5 with the NAM MPEP. (A) At the wild-type rat mGlu5 receptor, MPEP inhibits glutamate-mediated mobilization of intracellular Ca2+, depressing the maximal response. (B) Single point mutations of mGlu5 were screened for their ability to impact inhibition of the maximal response to glutamate in the presence of 10 nM MPEP. (C) Comparison of MPEP affinity estimates at mutants with wild type. Data represent the mean ± S.E.M. of 3–6 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
TABLE 1.
Equilibrium binding parameters for [3H]methoxyPEPy and MPEP at mGlu5 mutations
Data represent the mean ± S.E.M. of a minimum of three independent determinations.
| Mutation | [3H]methoxyPEPy pKDa | Bmaxb | MPEP pKIc |
|---|---|---|---|
| M | pmol/mg | M | |
| R5-wt (low) | 8.24 ± 0.09 | 0.6 ± 0.0*# | 7.87 ± 0.04# |
| R5-wt (poly) | 8.23 ± 0.10 | 3.8 ± 0.8 | 7.77 ± 0.03 |
| F585I | 7.91 ± 0.19 | 1.8 ± 0.6 | 7.93 ± 0.09 |
| R647A | 8.22 ± 0.08 | 1.6 ± 1.3 | N.D. |
| I650A | 8.43 ± 0.11 | 1.0 ± 0.1* | 8.34 ± 0.11* |
| G651F | No appreciable binding | ||
| P654S | No appreciable binding | ||
| P654F | No appreciable binding | ||
| S657C | 8.35 ± 0.28 | 0.8 ± 0.3 * | 8.12 ± 0.09 |
| Y658V | No appreciable binding | ||
| V739M | 8.19 ± 0.02 | 1.6 ± 0.3 | 7.91 ± 0.07 |
| P742S | 7.71 ± 0.29 | 0.4 ± 0.2* | 7.67 ± 0.14 |
| L743V | 7.92 ± 0.09 | 1.1 ± 0.2* | 7.23 ± 0.10* |
| N746A | 7.72 ± 0.05 | 1.2 ± 0.4* | 7.60 ± 0.08 |
| G747V | 8.37 ± 0.07 | 1.9 ± 0.6 | 7.88 ± 0.06 |
| T779A | 8.17 ± 0.13 | 0.8 ± 0.2* | |
| T780A | No appreciable binding | ||
| W784A | No appreciable binding | ||
| V788A | 8.13 ± 0.08 | 1.0 ± 0.3* | 7.71 ± 0.13 |
| F792A | 8.11 ± 0.21 | 1.7 ± 0.6 | 7.90 ± 0.13 |
| S806A | 7.71 ± 0.07 | 1.2 ± 0.1* | 7.21 ± 0.07* |
| S808A | No appreciable binding | ||
| S808T | No appreciable binding | ||
| A809V | No appreciable binding | ||
| A809G | No appreciable binding | ||
| T810A | 7.86 ± 0.15 | 2.0 ± 0.4 | 7.28 ± 0.09* |
| C815A | 8.00 ± 0.19 | 0.8 ± 0.1* | 7.57 ± 0.02 |
N.D., not determined.
Negative logarithm of the equilibrium dissociation constant of [3H]methoxyPEPy.
Maximal number of binding sites.
Negative logarithm of the equilibrium dissociation constant of MPEP.
P < 0.05 vs. wild-type (polyclonal) value, one-way analysis of variance (ANOVA), Dunnett’s post-test; #Data previously reported (Gregory et al., 2012).
Delineation of Impact of Mutations on MPEP Affinity versus Cooperativity.
Progressive fold-shift analysis by MPEP of the glutamate concentration-response curve for Ca2+ mobilization was performed using the operational model of allosterism (Leach et al., 2007). This model has previously been validated for estimating affinity and cooperativity of mGlu5 allosteric modulators (Gregory et al., 2012). As expected, given that mutations were introduced into the TMs, little or no change was observed in the potency and efficacy (logτA) of glutamate across all point mutations compared with wild type (Supplemental Table 1). Furthermore, the assumption that glutamate affinity was unaffected by point mutations in the TMs had no effect on the estimates of modulator pKB (Supplemental Fig. 1A). The affinity (pKB) of MPEP was found to be significantly reduced compared with wild type at 16 point mutations (Fig. 1C; Table 1). MPEP affinity estimates were reduced 3- to 10-fold at P742S, L743V, G747V, T779A, V788A, and T810A; 10- to 30-fold at G651F, P654S, T780A, and A809G; and 30- to 100-fold at Y658V, S808A, S808T, and A809V. MPEP affinity >1000-fold lower was observed at P654F and W784A compared with wild type. Binding and functional affinity estimates showed good agreement. No appreciable [3H]methoxyPEPy binding was observed at G651F, P654S, P654F, Y658V, T780A, W784A, S808A, S808T, A809V, and A809G (Table 1), corresponding to mutations where MPEP affinity was estimated to be decreased 10-fold or greater compared with wild type. MPEP completely blocked the maximal response to glutamate at all constructs (unpublished data) with the exception of P654F and W784A (Fig. 2, A and B). At W784A (Fig. 2B), inhibition of glutamate by MPEP approached a limit, where logβ = −0.27 ± 0.03, indicating that MPEP negative cooperativity is weaker at W784A compared with wild type.
Fig. 2.
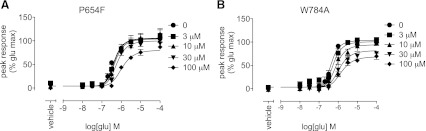
At two point mutations, MPEP did not fully depress the maximal response to glutamate. MPEP inhibition of glutamate-mediated mobilization of intracellular Ca2+ at mGlu5 P654F (A) and W784A (B). In the presence of MPEP at concentrations up to 100 μM, glutamate retained some activity in both cell lines. Data represent the mean ± S.E.M. of 3–5 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
Modeling MPEP Binding to mGlu5 Comparative Model.
MPEP was docked into the comparative model of mGlu5 to aid interpretation of these mutational data. Docking experiments were centered on two sites, represented by two residues that were demonstrated to reduce MPEP affinity when mutated, P654 and S808. Representative binding modes from each experiment demonstrate possible binding modes for MPEP interacting with mGlu5 (Fig. 3A). The lowest-energy MPEP binding poses from the largest clusters for docking to both sites are shown in Fig. 3, B and C. Docking to P654 identified a common binding site within the top three clusters, representing 37% of all models; five out of seven point mutations that reduce MPEP affinity are predicted to line the pocket depicted by these poses. However, given the linearity of MPEP, the orientation of the ligand proved more difficult for Rosetta to differentiate; in two of the top three clusters the pyridine ring of MPEP points toward the extracellular space. Interestingly, despite mutations causing 30- to 100-fold reduction in affinity, S808 and A809 were not predicted to interact with MPEP in the P654-based docking runs. The lowest-energy MPEP binding modes of the largest five clusters (accounting for 28% of models) with docking centered on S808 demonstrated greater diversity (Fig. 3B). In four out of these five binding modes, the nitrogen from the pyridine ring forms a hydrogen bond with S808. A similar pose for MPEP has recently been reported (Mølck et al., 2012). This interaction may also account for the impact of substitutions to A809, potentially influencing the conformation of its neighboring residue S808. Given the location of S808 at the top of TM7 closer to extracellular loop regions, the greater diversity in binding poses is not unreasonable. We hypothesize that S808 and A809 are important for the initial recognition of the receptor by MPEP, ultimately facilitating binding deeper within the pocket created by TMs 3, 5, 6, and 7. Residues that when mutated significantly reduced MPEP affinity are highlighted in Fig. 3 and color-coded based on their relative impact on MPEP affinity. Notably, residues that have the greatest effect on MPEP affinity are found in close proximity to MPEP, whereas those with less of an effect (3- to 10-fold reduction) are more remote. Hydrophobic interactions with MPEP are likely occurring with W784 and Y658. Given its placement in relation to MPEP, the P654F mutation may introduce steric clash into the MPEP binding site, preventing MPEP binding and consequently reducing affinity. Ser substitution of this same amino acid did not reduce affinity to the extent of the Phe mutation, with MPEP negative cooperativity retained. Thus it is also possible that replacing Pro with Phe both influences the helix conformation (by removing Pro-induced kink) and introduces a larger hydrophobic amino acid, thereby dramatically changing the geography of the MPEP binding pocket, perturbing MPEP affinity and potentially cooperativity. The effect of mutations to S806, S808, and T810 provides evidence in favor of the alignment of the PKxY motif in the mGlus with the NPxxY motif in the Family A 7TMRs. As predicted by the model, the S808A mutation affects MPEP affinity, likely because its position facing the binding pocket provides an interaction with MPEP. S806 and T810 face away from the binding pocket and are not predicted to affect affinity, which is in agreement with the functional data (Fig. 1C). This verification of the MPEP binding mode through point mutations encouraged the further analysis with positive allosteric modulators of mGlu5.
Fig. 3.

The NAM MPEP docked into the mGlu5 comparative model. MPEP was docked into the mGlu5 comparative model in two separate experiments, centered at P654 and S808. (A) The lowest-energy conformation for MPEP from the largest cluster docked at P654 is shown in green and at S808 is shown in cyan. Highlighted are the residues that caused decreases in MPEP affinity when mutated, colored by graded effect compared with wild type. (B) The lowest-energy models from the largest three clusters for MPEP docked at P654. (C) The lowest-energy models from the largest five clusters for MPEP docked at S808. Predicted hydrogen bonds between the nitrogen on the pyridine ring and S808 are depicted by dotted blue lines.
Identification of Point Mutations That Affect Positive Allosteric Modulation of Glutamate Activity.
Three pairs of picolinamide and nicotinamide acetylene mGlu5-selective PAMs were selected for investigation of the binding mode of these two chemical scaffolds in the common allosteric site of mGlu5 (Fig. 4). These six PAMs span varying degrees of affinity at the wild-type receptor from low (VU0360173, 8 μM) to high (VU0403602, 6 nM; VU0405386, 10 nM) (Tables 2 and 3). Picolinamide PAMs show ∼10-fold-higher affinity for the wild-type receptor than their nicotinamide counterparts (Tables 2 and 3). Furthermore, these PAMs show varying degrees of cooperativity, i.e., their ability to induce leftward shifts (fold-shifts) of the glutamate concentration-response curve (Fig. 4; Table 4). Similar to the approach used for MPEP above, activity of mutant receptors was assessed at a single concentration of PAM. Based on the ability of these compounds to potentiate glutamate Ca2+ mobilization at the wild-type receptor (Fig. 4), PAM concentrations were selected that caused submaximal, but reproducible, fold-shifts in the concentration-response curve to glutamate. The fold-shift at a single concentration of PAM was compared with the fold-shift at wild type (Fig. 5). Point mutations that significantly increased or decreased potentiation were selected for further characterization using analysis of progressive fold-shift of the glutamate concentration-response curve to delineate their effects on PAM affinity versus cooperativity.
Fig. 4.

Potentiation of glutamate-mediated Ca2+ mobilization by nicotinamide and picolinamide acetylene PAMs at wild-type mGlu5. The six PAMs included in this study potentiate the response to glutamate at mGlu5 wild type in a Ca2+ mobilization assay with varying degrees of cooperativity, as evidenced by increased glutamate potencies in the presence of PAMs. Nicotinamide acetylene PAMs are shown on the left (A–C), with the corresponding picolinamide acetylene PAM on the right (D–F). Data represent the mean ± S.E.M. of 3–7 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
TABLE 2.
Affinity estimates (pKI) for PAMs at mGlu5-wt and mutants from inhibition binding assays
Data represent the mean ± S.E.M. of a minimum of three independent determinations.
| Mutation | VU0360172 (Nicotinamide) | VU0403602 (Picolinamide) | VU0360173 (Nicotinamide) | VU0405398 (Picolinamide) | VU0415051 (Nicotinamide) | VU0405386 (Picolinamide) |
|---|---|---|---|---|---|---|
| R5-wt (low) | 6.57 ± 0.02# | 8.26 ± 0.15 | 5.12 ± 0.07 | 6.05 ± 0.12# | 6.88 ± 0.04# | 7.98 ± 0.05# |
| I650A | N.D. | 7.89 ± 0.10 | 5.30 ± 0.26 | 6.41 ± 0.14 | 6.62 ± 0.11 | 7.69 ± 0.15 |
| P742S | 5.90 ± 0.12* | 7.24 ± 0.23* | 5.12 ± 0.27 | 5.78 ± 0.14 | 6.35 ± 0.13* | 7.19 ± 0.12* |
| L743V | 6.32 ± 0.06 | 6.98 ± 0.10* | 4.86 ± 0.08 | 5.71 ± 0.01* | 6.25 ± 0.17* | 7.58 ± 0.04 |
| N746A | 6.22 ± 0.07 | 7.49 ± 0.07* | 4.63 ± 0.10 | 5.75 ± 0.03 | 6.35 ± 0.26* | 7.45 ± 0.30 |
| G747V | 6.76 ± 0.05 | N.D. | N.D. | N.D. | 7.06 ± 0.27 | 8.17 ± 0.35 |
| V788A | 7.08 ± 0.23 | 8.70 ± 0.02 | 6.10 ± 0.09* | 7.87 ± 0.05* | 8.15 ± 0.07* | 9.20 ± 0.10* |
| F792A | 7.06 ± 0.15 | 7.85 ± 0.04 | 5.68 ± 0.06 | 6.74 ± 0.08* | 7.09 ± 0.06 | 7.71 ± 0.19 |
| C815A | 6.74 ± 0.24 | 8.01 ± 0.03 | 5.01 ± 0.07 | 6.26 ± 0.04 | 6.92 ± 0.11 | 7.96 ± 0.16 |
N.D., not determined.
P < 0.05 vs. wild-type receptor, one-way analysis of variance (ANOVA), Dunnett’s post-test; #data previously reported (Gregory et al., 2012).
TABLE 3.
Affinity estimates (pKB) for allosteric modulators at mGlu5-wt and mutants derived from operational model analysis of interactions with glutamate
Data represent the mean ± S.E.M. from 4–8 independent experiments performed in duplicate.
| Mutation | MPEP | VU0360172 (Nicotinamide) | VU0403602 (Picolinamide) | VU0360173 (Nicotinamide) | VU0405398 (Picolinamide) | VU0415051 (Nicotinamide) | VU0405386 (Picolinamide) |
|---|---|---|---|---|---|---|---|
| R5-wt (poly) | 8.58 ± 0.17# | 6.68 ± 0.15 | 8.13 ± 0.26 | 5.45 ± 0.27 | 6.94 ± 0.17 | 7.34 ± 0.18 | 8.04 ± 0.26 |
| R647A | 8.89 ± 0.19 | 6.29 ± 0.12 | 7.99 ± 0.09 | 4.91 ± 0.14 | 6.94 ± 0.10 | 7.28 ± 0.25 | 7.74 ± 0.12 |
| I650A | 8.62 ± 0.03 | 7.12 ± 0.19 | 7.97 ± 0.13 | N.D. | 7.29 ± 0.15 | N.D. | 8.10 ± 0.15 |
| G651F | 7.53 ± 0.18* | 5.86 ± 0.18* | 6.50 ± 0.18* | No PAM | 5.99 ± 0.28* | 6.94 ± 0.19 | 7.14 ± 0.08 |
| P654S | 7.10 ± 0.06* | 6.39 ± 0.24 | 7.33 ± 0.16 | <5 | 5.80 ± 0.27* | 5.69 ± 0.46* | 7.81 ± 0.27 |
| P654F | 4.11 ± 0.21* | 5.91 ± 0.17 | 5.99 ± 0.26* | No PAM | 5.33 ± 0.27* | 5.25 ± 0.19* | 6.23 ± 0.22* |
| S657C | 8.32 ± 0.08 | N.D. | 7.41 ± 0.17 | N.D. | 6.99 ± 0.31 | 6.78 ± 0.12 | 7.68 ± 0.06 |
| Y658V | 6.57 ± 0.13*# | No PAM | No PAM | No PAM | 4.97 ± 0.35* | No PAM | No PAM |
| P742S | 8.07 ± 0.17* | 6.57 ± 0.10 | 7.50 ± 0.12 | 4.65 ± 0.17 | 6.83 ± 0.20 | 7.09 ± 0.04 | 7.96 ± 0.16 |
| L743V | 8.04 ± 0.10*# | 6.97 ± 0.15 | 7.93 ± 0.14 | 5.46 ± 0.12 | 6.77 ± 0.11 | 7.75 ± 0.20 | 8.36 ± 0.10 |
| N746A | 8.30 ± 0.06 | 6.66 ± 0.10 | 7.99 ± 0.11 | N.D. | 6.50 ± 0.23 | N.D. | 8.06 ± 0.17 |
| T780A | 7.36 ± 0.02* | No PAM | 6.03 ± 0.13* | No PAM | 5.59 ± 0.05* | 5.82 ± 0.40* | 5.32 ± 0.24* |
| W784A | 5.50 ± 0.29* | 6.76 ± 0.14 | 7.09 ± 0.34* | 5.03 ± 0.44 | 6.23 ± 0.20 | 7.14 ± 0.15 | 7.53 ± 0.14 |
| V788A | 7.89 ± 0.13* | 7.28 ± 0.17 | 8.43 ± 0.20 | 6.28 ± 0.18 | 7.84 ± 0.14 | 8.07 ± 0.19 | 8.79 ± 0.08 |
| F792A | 8.93 ± 0.04 | 7.25 ± 0.33 | 8.03 ± 0.02 | 4.80 ± 0.20 | 7.34 ± 0.20 | 8.06 ± 0.24 | 7.02 ± 0.47* |
| S806A | 8.36 ± 0.06 | N.D. | N.D. | N.D. | N.D. | N.D. | 7.29 ± 0.45 |
| S808A | 6.98 ± 0.18* | 6.15 ± 0.30 | 6.47 ± 0.10* | No PAM | 6.28 ± 0.09 | 7.20 ± 0.44 | 7.48 ± 0.20 |
| S808T | 6.90 ± 0.06* | 6.59 ± 0.20 | 7.15 ± 0.33* | 5.04 ± 0.33 | 6.40 ± 0.23 | 6.36 ± 0.26* | 8.05 ± 0.27 |
| A809V | 6.52 ± 0.12*# | 5.64 ± 0.19* | 6.52 ± 0.21* | No PAM | No PAM | 5.56 ± 0.12* | 6.22 ± 0.14* |
| A809G | 7.18 ± 0.04* | 5.93 ± 0.18 | 6.59 ± 0.05* | 5.13 ± 0.13 | 5.88 ± 0.05* | 6.28 ± 0.11* | 6.60 ± 0.24* |
| C815A | 8.35 ± 0.05 | 6.58 ± 0.22 | 7.29 ± 0.26* | 4.81 ± 0.13 | 6.73 ± 0.35 | 7.26 ± 0.11 | 7.63 ± 0.15 |
N.D., not determined; No PAM, no observed positive allosteric modulation.
P < 0.05 vs. wild-type value, one-way analysis of variance (ANOVA), Dunnett’s post-test; #data previously reported (Gregory et al., 2012).
TABLE 4.
Functional cooperativity factors (logβ) for allosteric modulators at mGlu5-wt and mutants derived from operational model analysis of interactions with glutamate
Data represent the mean ± S.E.M. from 4–8 independent experiments performed in duplicate.
| Mutation | VU0360172 (Nicotinamide) | VU0403602 (Picolinamide) | VU0360173 (Nicotinamide) | VU0405398 (Picolinamide) | VU0415051 (Nicotinamide) | VU0405386 (Picolinamide) |
|---|---|---|---|---|---|---|
| R5-wt (poly) | 0.37 ± 0.05 | 0.61 ± 0.08 | 0.17 ± 0.04 | 0.37 ± 0.04 | 0.40 ± 0.02 | 0.55 ± 0.07 |
| R647A | 0.44 ± 0.13 | 0.60 ± 0.09 | 0.26 ± 0.03 | 0.38 ± 0.05 | 0.41 ± 0.11 | 0.58 ± 0.06 |
| I650A | 0.58 ± 0.05 | 0.55 ± 0.08 | N.D. | 0.51 ± 0.08 | N.D. | 0.56 ± 0.08 |
| G651F | 0.54 ± 0.07 | 0.79 ± 0.05 | No PAM | 0.43 ± 0.04 | 0.32 ± 0.05 | 0.70 ± 0.07 |
| P654S | 0.26 ± 0.01 | 0.56 ± 0.05 | <5 | 0.34 ± 0.08 | 0.22 ± 0.10 | 0.32 ± 0.05 |
| P654F | 0.25 ± 0.04 | 0.58 ± 0.14 | No PAM | 0.17 ± 0.04 | 0.66 ± 0.09* | 0.53 ± 0.09 |
| S657C | N.D. | 0.82 ± 0.15 | N.D. | 0.43 ± 0.15 | 0.55 ± 0.01 | 0.56 ± 0.12 |
| Y658V | No PAM | No PAM | No PAM | NAM | No PAM | No PAM |
| P742S | 0.87 ± 0.10* | 1.23 ± 0.06* | 0.69 ± 0.07* | 0.70 ± 0.15 | 0.92 ± 0.08* | 1.12 ± 0.17* |
| L743V | 0.36 ± 0.14 | 0.60 ± 0.06 | 0.43 ± 0.03 | 0.54 ± 0.08 | 0.36 ± 0.04 | 0.80 ± 0.03 |
| N746A | 0.62 ± 0.04 | 0.67 ± 0.04 | N.D. | 0.57 ± 0.05 | N.D. | 0.63 ± 0.02 |
| T780A | No PAM | 0.77 ± 0.08 | No PAM | 0.19 ± 0.01 | −0.28 ± 0.02* | 0.39 ± 0.04 |
| W784A | 0.90 ± 0.14* | 1.05 ± 0.11* | 0.41 ± 0.10 | 0.48 ± 0.09 | 0.88 ± 0.10* | 0.66 ± 0.15 |
| V788A | 0.57 ± 0.10 | 0.68 ± 0.09 | 0.66 ± 0.12* | 0.47 ± 0.05 | 0.53 ± 0.06 | 0.56 ± 0.11 |
| F792A | 0.66 ± 0.12 | 0.97 ± 0.10 | 0.29 ± 0.11 | 0.73 ± 0.32 | 0.29 ± 0.02 | 0.85 ± 0.16 |
| S806A | N.D. | N.D. | N.D. | N.D. | N.D. | 0.79 ± 0.13 |
| S808A | 0.50 ± 0.06 | 0.57 ± 0.10* | No PAM | NAM | 0.22 ± 0.02 | Neutral |
| S808T | 0.46 ± 0.06 | 0.76 ± 0.12 | 0.28 ± 0.07 | 0.34 ± 0.04 | 0.41 ± 0.04 | 0.45 ± 0.06 |
| A809V | 0.60 ± 0.07 | 0.58 ± 0.09 | No PAM | No PAM | 0.37 ± 0.07 | 0.58 ± 0.11 |
| A809G | 0.55 ± 0.04 | 1.29 ± 0.07* | 0.41 ± 0.07 | 0.70 ± 0.03 | 0.78 ± 0.12* | 1.12 ± 0.17* |
| C815A | 1.17 ± 0.12* | 1.29 ± 0.10* | 0.66 ± 0.06* | 0.75 ± 0.24 | 0.86 ± 0.05* | 1.38 ± 0.14* |
N.D., not determined; No PAM, no observed positive allosteric modulation.
P < 0.05 vs. wild-type value, one-way analysis of variance (ANOVA), Dunnett’s post-test.
Fig. 5.

Effect of mutations on the fold-shift caused by a single concentration of PAM. Nicotinamide acetylene PAMs are shown on the left, with the corresponding picolinamide acetylene PAM on the right as indicated. The increase in glutamate potency in the presence of PAM, or fold-shift, at each mutant is expressed relative to that observed for the same concentration at the wild-type receptor. Specifically, PAM concentrations used were 10 μM VU0360173 (A), 1 μM VU0360172 (B), 100 nM VU0415051 (C), 100 nM VU0405398 (D), 10 nM VU0403602 (E), and 10 nM VU0405386 (F). #No detectable PAM activity; *P < 0.05 vs. wild type, one-way analysis of variance (ANOVA), Dunnett’s post-test. Data represent the mean ± S.E.M. of 3–7 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
Impact of Mutations on PAM Affinity: Interactions within the Common Allosteric Pocket.
To ensure that potential determinants were not missed in the initial screen, mutations that perturbed potentiation by at least one member of class (either picolinamide or nicotinamide) were assessed across all three members. In addition, mutations that affected one member of a picolinamide/nicotinamide PAM pair were further assessed at both. Affinity estimates (pKB) for PAMs, derived from modulation of glutamate-mediated Ca2+ mobilization, are shown in Table 3; for the most part, mutations influenced modulator affinity (Fig. 6). Where practical, PAM affinity was also assessed using inhibition of [3H]methoxyPEPy binding (Table 2). Functional (pKB) and binding (pKI) estimates of affinity showed strong correlation (Supplemental Fig. 3B). All mutations in TM3 that showed significant effects could not be assessed using radioligand binding–based approaches, relying instead on affinity estimates derived using the operational model of allosterism.
Fig. 6.

Effect of mutations on PAM affinity estimates. Nicotinamide acetylene PAMs are shown in the top three panels (A–C), with the corresponding picolinamide acetylene PAM in the bottom three panels (D–F). Affinity estimates (pKB) were derived using an operational model of allosterism (Leach et al., 2007; Gregory et al., 2012) from progressive fold-shifts of the glutamate concentration-response curve for Ca2+ mobilization. The difference between the pKB for the mutant versus wild type is plotted. #No detectable PAM activity; *P < 0.05 vs. wild type, one-way analysis of variance (ANOVA), Dunnett’s post-test. Data represent the mean ± S.E.M. of 3–7 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
No potentiation was evident for VU0360173 (up to 30 μM) at G651F, P654S, P654F, Y658V, T780A, S808A, and A809V. The corresponding picolinamide, VU0405398, also showed no detectable potentiation at A809V and 10- to 30-fold-decreased affinity at G651F, P654S, T780A, and A809G; 30- to 100-fold reductions were noted at P654F and Y658V. Collectively, these results suggest that the lack of potentiation observed for VU0360173 at these mutants is a result of decreased affinity.
VU0360172 affinity was reduced approximately 10-fold at G651F and A809V. No positive allosteric modulation was observed at Y658V or T780A. Interestingly, P654S had no significant effect on VU0360172 affinity, while at the Phe substitution VU0360172 affinity was also reduced (6-fold; this did not reach significance). The picolinamide counterpart of VU0360172, VU0403602, also showed no appreciable PAM activity at Y658V, alongside marked reductions in affinity (>100-fold) at P654F and T780A. VU0403602 affinity was also decreased 30- to 100-fold at G651F, S808A, A809V, and A809G and 10- to 30-fold at S808T. At P654S and C815A, VU0403602 pKB values were reduced 3- to 10-fold compared with wild type, although this did not reach significance at P654S.
Similar to that reported for the previous four PAMs, VU0415051 did not potentiate glutamate at Y658V (up to 30 μM). Decreased VU0415051 affinity was noted at S808T and A809G (10- to 30-fold); P654S, T780A, and A809V (30- to 100-fold); and P654F (>100-fold). VU0405386 also showed no discernible PAM activity at Y658V or S808A. The affinity of VU0405386 was decreased compared with wild type at F792A and A809G (10- to 30-fold); P654F and A809V (30- to 100-fold); and T780A (>300-fold). Interestingly, neither of the VU0415051/VU0405386 pair was unaffected by G651F.
Three point mutations in TM5—P742S, L743V, and N746A—showed no significant effects in functional assays; however, in inhibition binding assays, significant reductions in affinity were observed for some PAMs (Table 2). P742S significantly decreased pKI values 3- to 10-fold for VU0361072, VU0403602, VU0415051, and VU0405386. Significantly decreased pKI values were also noted for VU0403602 (19-fold), VU0405398 (2-fold), and VU0415051 (4-fold) at L743V and for VU0403602 (6-fold) and VU0415051 (3-fold) at N746A. In contrast, V788A, in TM6, showed a trend for increased pKB and pKI values (2- to 10-fold).
Docking of PAMs to mGlu5 Comparative Model.
Docking of the PAMs in the mGlu5 comparative model provides insight into the significant residues identified to reduce affinity in the binding pocket. Each of the six PAMs was allowed to explore a 5-Å radius around P654, and the lowest-energy binding modes of the largest five clusters were shown to bind in the same pocket as MPEP (Fig. 7, A–C). As seen with MPEP, mutation of P654 in TM3 to a bulky residue (Phe) likely introduces a steric clash, reducing the ability of PAMs to engage the common mGlu5 binding pocket, accounting for the reduced affinity for all six PAMs. As noted previously, the amino acid in this position is not conserved across the mGlu family, such that it is likely that P654 also contributes to the subtype selectivity of these PAMs. Introduction of steric bulk is also likely to underscore the impact of the G651F mutation on all modulators except for the VU0415051/VU0405386 pair. The impact of substitution of Y658 with Val can likely be attributed to disruption of key polar interactions between modulators and Y658, T780, and W784 (highlighted by dashed lines in Fig. 7, A–C). Similarly, T780 likely participates in a polar interaction with the carbonyl of the PAMs, with the exception of VU0403602 where the tertiary amide is predicted to interact with this residue, such that Ala substitution of this amino acid causes the drastic loss of affinity seen for all modulators. W784 likely contributes to hydrophobic interactions within the pocket, which may account for the trend for picolinamide PAM affinity to be perturbed by W784A; it is also noteworthy that the W784 may be involved in a hydrogen bonding network linking the modulators, TM3, and TM7. V788A shows a trend for increased affinity, which may be due to secondary effects on protein conformation and the previously implicated F787, where Ala substitution reduced NAM and PAM interactions (Malherbe et al., 2003, 2006; Muhlemann et al., 2006). S808 in TM7 may participate in hydrogen bonding with the fluorine of the modulators when in close proximity, although this was not evident in the lowest-energy binding poses. Substitution of A809 with Gly or Val reduced modulator activity across the board, pointing to the importance of the Ala in maintaining the correct helix conformation for binding. With respect to C815 and VU0403602, it is likely that there is an interaction with the cyclobutane of the modulator; however, it is not immediately apparent whether this is a direct or indirect effect.
Fig. 7.

Computational docking of three pairs of nicotinamide and picolinamide acetylene PAMs into mGlu5. (A) VU0360173 in blue and VU0405398 in green, (B) VU0360172 in blue and VU0403602 in green, and (C) VU0415051 in blue and VU0405386 in green. Residues that when mutated caused a significant decrease in modulator affinity in mGlu5 are highlighted in the respective color of the modulator. Residues that affect both the nicotinamide and picolinamde in the pair are highlighted in purple. A predicted hydrogen bond network involving the modulators, Y658, T780A, and W784 is represented by the dashed black lines. Highlighted in gray are residues that influence the cooperativity of certain modulators.
Quantifying Effects on Cooperativity: Identification of Mutations That Engender “Molecular Switches.”
When applying the operational model of allosterism, the interaction between glutamate and PAMs was assumed to be exclusively via efficacy modulation, an assumption previously validated for mGlu5 PAMs from this scaffold (Gregory et al., 2012). Cooperativity estimates (logβ) are summarized in Table 4 and comparisons with wild type are shown in Fig. 8. Alongside marked reductions in PAM affinity, P654F also significantly increased cooperativity of VU0415051 (∼2-fold). P742S in TM5 had no effect on PAM affinity, yet increased cooperativity (∼3-fold). In TM7, A809G significantly increased the cooperativity of the higher-affinity PAMs, VU0403602, VU0415051, and VU0405386 (2- to 5-fold), with a similar trend observed for VU0360172, VU0360173, and VU0405398. Also in TM7, C815A significantly increased the cooperativity of all PAMs (2- to 7-fold) with the exception of VU0405398. P742S, A809G, and C815A all represent mutations in which the glutamate potency and/or efficacy was lower than the wild type (Supplemental Table 1), such that these increases in PAM cooperativity may be attributable to PAMs stabilizing an unstable mutant receptor. Significantly increased cooperativity (3-fold) was also observed at W784A for VU0360172, VU0403602, and VU0415051, with VU0360173 showing higher, although not significant, cooperativity. Interestingly, this was not a global PAM phenomenon, as the cooperativity of VU0405386 and VU0405398 was unchanged. Given the putative involvement of W784 in a key hydrogen bonding network for the PAMs (dotted lines in Fig. 7, A–C), perhaps this differential effect is driven by the relative importance of this interaction over that of the PAM functional head group. Also in TM6, V788A increased the cooperativity of VU0360173 (3-fold) alone; no direct interaction is predicted from the docking between VU0360173 and V788, suggesting that this is an indirect effect on the geography of the binding pocket. The selective effect of V788A on VU0360173 cooperativity may be attributable to the fact that this PAM has the weakest cooperativity and lowest affinity.
Fig. 8.
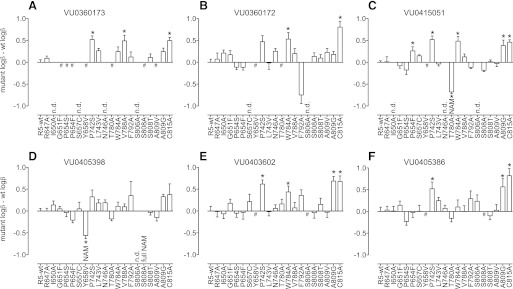
Effect of mutations on PAM cooperativity factors. Nicotinamide acetylene PAMs are shown in the top three panels (A–C), with the corresponding picolinamide acetylene PAM in the bottom three panels (D–F). Cooperativity estimates (logβ) were derived using an operational model of allosterism (Leach et al., 2007; Gregory et al., 2012) from progressive fold-shifts of the glutamate concentration-response curve for Ca2+ mobilization. The difference between the logβ for the mutant versus wild type is plotted. #No detectable PAM activity; *P < 0.05 vs. wild type, one-way analysis of variance (ANOVA), Dunnett’s post-test. Data represent the mean ± S.E.M. of 3–7 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
Three point mutations altered cooperativity drastically, such that PAMs behaved as NAMs. At Y658V, VU0405398 was a weak NAM, reducing the maximal response to glutamate by ∼25% at 30 μM (Fig. 9A). This same mutation resulted in a loss of potentiation by all other PAMs. At T780A, VU0415051 became a weak NAM or “partial antagonist,” where logβ = −0.28, corresponding to an ∼40% depression in the glutamate maximal response (Fig. 9B). This inhibition approached saturation, the hallmark feature of an allosteric interaction.
Fig. 9.

Characterization of mutations that engender a molecular switch in PAM pharmacology. (A) At mGlu5 Y658V, 10 μM and 30 μM VU0405398 inhibited the response to maximal glutamate. (B) The interaction between glutamate and VU0415051 at the T780A mutant is negative, with inhibition approaching a limit as defined by the cooperativity. (C) At S808A, VU0405398 causes a reduction in glutamate potency and depresses the maximal response to glutamate. (D) Concentration-response curves for VU0405398 inhibition of an ∼EC80 of glutamate in the absence and presence of the indicated concentrations of VU0405386. (E) Schild regression of the interaction between VU0405386 and VU0405398 at S808A. Data represent the mean ± S.E.M. of 3–6 experiments performed in duplicate. Error bars not shown lie within the dimensions of the symbol.
The most profound molecular switch engendered by a single point mutation was that of S808A, where VU0405398 behaved as a full NAM, abolishing the maximal response to glutamate (Fig. 9C). As mentioned earlier, at this same mutation VU0405386 showed no discernible PAM activity. Given that this mutation has little or no impact on the affinity of the other PAMs tested, we hypothesized that the lack of potentiation by VU0405386 was caused by a molecular switch from PAM to neutral. To test this hypothesis, concentration-response curves for VU0405398 inhibition of an EC80 glutamate concentration were performed at S808A in the presence of varied concentrations of VU0405386. As shown in Fig. 9D, VU0405386 has no effect on the response to glutamate, but causes parallel rightward shifts in the VU0405398 curve, in a manner consistent with a competitive interaction [Fig. 9E; where the Schild slope was not significantly different from unity (0.92 ± 0.09) and pKB = 7.48 ± 0.20].
Modeling PAM Molecular Switches.
To investigate the molecular cause of the PAM-to-NAM or PAM-to-neutral switches, modulators were docked into mGlu5 models containing the mutation engendering the switch. VU0405398 docked in wild-type mGlu5 was compared with VU0405398 docked into the S808A mutant (Fig. 10A). Interestingly, introducing this single point mutation resulted in movement of TM7, such that in the mutant receptor VU0405398 is further away from this helix. It is unclear why this substitution results in such a conformational change of the receptor. Although not evident in the docked results, it is possible that rotation of the side chain of S808 in the wild-type receptor would allow hydrogen bonding to occur between the fluorine of the modulator and S808, with direct interactions with TM7 being important for stabilizing active receptor conformations. At the mutant receptor, such an interaction is no longer available and VU0405398 instead stabilizes an inactive receptor conformation at the mutated receptor. From the docked poses it is also evident that the picolinamide functional group of VU0405398 adopts a strikingly different orientation within the pocket. Similarly, VU0415051 was docked into the T780A mutant receptor and binding modes compared with those in wild-type mGlu5 (Fig. 10B). At the T780A mutant, a polar interaction is no longer formed between the carbonyl of the modulator and the mutant A780; hydrogen bonding is no longer evident with the side-chain hydroxyl of Y658 and the modulator carbonyl, nor between the tertiary nitrogen and S657. In the mutant receptor construct, the nicotinamide tert-butyl moiety of VU0415051 adopts a different orientation with decreased affinity. Docking of VU0405386 in the S808A receptor shows subtle differences in the binding mode of the ligand when compared with wild type (Fig. 10C). The movement of TM7 observed with docking VU0405398 to this same receptor mutation is not evident, which may account for the switch from PAM to neutral rather than to a robust NAM as seen for VU0405386. Although not evident in the static docked pose, we hypothesize that the side chain of S808 may rotate to form a polar interaction with the fluorine of the modulator when interacting with the wild-type receptor that is not possible in the mutant. It is also clear that this point mutation at the top of TM7 results in the ligand adopting a different pose with respect to the picolinamide tert-butyl group, which may contribute to the observed switch to neutral cooperativity.
Fig. 10.

Mutations engendering a molecular switch for mGlu5 allosteric modulators. (A) VU0405398 docked into wild-type mGlu5 (green) and the S808A mutant (magenta). (B) VU0415051 docked into wild-type mGlu5 (blue) and the T780A mutant (magenta). (C) VU0405386 docked into wild-type mGlu5 (blue) and the S808A mutant (magenta). Mutated residues are colored by element. Key affinity determinants are highlighted to show conformational changes in the binding pocket.
Discussion
By utilizing an operational model of allosterism (Leach et al., 2007; Gregory et al., 2012), we have quantitatively assessed the interactions of PAMs within the common allosteric site of mGlu5, successfully delineating the impact of point mutations on cooperativity versus affinity. Seven novel point mutations were discovered that negatively impact the MPEP affinity, building on our understanding of the common allosteric binding pocket. Furthermore, Ala substitution of W784 reduced MPEP negative cooperativity. For the six PAMs studied, three conserved (Y658, T780, and S808) and two nonconserved residues (P654 and A809) were identified as critical determinants of PAM affinity. Interestingly, two point mutations engendered molecular switches in certain PAMs, changing their pharmacology to either NAMs or neutral modulators.
Close examination of the MPEP mutational data reveals a number of interesting observations. First, [3H]methoxyPEPy nonbinding mutants corresponded to mutations in which MPEP affinity estimates, as derived from functional assays, were decreased 10-fold or greater compared with wild type. Indeed, four previously reported mutations, Y658V, W784A, A809V, and A809G, that cause a loss of NAM binding to mGlu1 and mGlu5 and/or decreased MPEP potency for glutamate inhibition (Pagano et al., 2000; Malherbe et al., 2003, 2006; Muhlemann et al., 2006; Mølck et al., 2012) were confirmed herein and attributed to >30-fold reductions in MPEP affinity. Also in agreement with previous data (Pagano et al., 2000; Malherbe et al., 2003, 2006), P654S, L743V, and T780A all reduced MPEP affinity, although the effect of T780A was more pronounced in the current study than previously reported (17- versus 5-fold) (Malherbe et al., 2003). Second, in mutating residues predicted to line the helical face toward the binding site as shown in the comparative model, we have further validated utilization of Family A 7TMR crystal structures as templates for mGlu transmembrane-spanning domain comparative modeling. Our TM7 alignment, which aligns PKxY of mGlu5 to the conserved NPxxY motif, differs by seven residues compared with earlier reports (Pagano et al., 2000; Malherbe et al., 2006), agreeing with the recent report by Mølck et al. (2012). In support of this new alignment for TM7, S808 was hypothesized to contribute to the common allosteric site of mGlu5 as opposed to S806 and T810. Mutation of S808 to Ala perturbed MPEP affinity significantly more than S806A and T810A. S808T had a similar impact on MPEP affinity, suggesting that a polar interaction may occur between S808 and MPEP that is not achieved by Thr. Alternatively, S808 may be important for maintaining the allosteric binding pocket geography, perhaps via a hydrogen bonding network.
Molecular models of receptor-ligand complexes provide important tools for hypothesis generation, predicting binding modes where experimental structures are unavailable. In the current study, the model provides an orientation of transmembrane helices, known to be aligned well across G protein-coupled receptor templates, as well as the TM7 helix/loop transition, that reasonably explains experimental results demonstrated by S806, S808, and T810 mutations. The comparative model has less confidence predicting receptor loop regions, as these have <20% sequence homology to the template G protein-coupled receptor structures used. The predicted modulator binding site captures residues identified in experimental studies, lending confidence to the binding site location depth within the receptor. The long axes of the ligands were consistently aligned parallel with the helices; however, computationally it was difficult predicting the orientation of these linear ligands and distinguishing whether the functional group points toward the intracellular or extracellular space. Despite these challenges, the mGlu5 computational model with allosteric modulators has provided valuable hypotheses, validated experimentally herein.
Comparison of the PAM data with that of MPEP shows a number of marked differences in mutation susceptibility. Interestingly, more mutations perturbed MPEP affinity than the PAMs; however, with one exception (VU0403602 at C815A), there were no mutations that influenced PAM affinity without affecting MPEP. It is clear that these acetylene PAMs interact with the common allosteric site on mGlu5. These differences likely underscore the determinants that contribute to a NAM versus PAM interacting with the receptor. Interestingly, W784A caused an ∼1000-fold reduction in MPEP affinity; however, nicotinamide PAMs were insensitive to this mutation, while the picolinamides showed 3- to 10-fold-decreased affinity. W784A increased cooperativity of some PAMs, in agreement with the previous report that W784A enhanced DFB potentiation (Muhlemann et al., 2006). Ala substitution of the equivalent Trp in mGlu1 (W798) has differential effects on mGlu1 NAMs (Suzuki et al., 2007; Fukuda et al., 2009). W784 is analogous to the W of the CWxP motif in Family A 7TMRs that is involved in the well known rotamer-toggle activation switch (Shi et al., 2002; Visiers et al., 2002; Holst et al., 2010). The modeling herein predicts a network of polar interactions involving W784 and T780 in TM6, Y658 in TM3, and the PAMs. We hypothesize that the differential effect of W784A on mGlu5 PAMs versus MPEP is likely underscored by PAMs interacting with an active versus inactive receptor conformation.
In each case, picolinamide modulators have higher affinity than their corresponding nicotinamide modulators. From the studies herein, it is not entirely clear which interactions within the binding pocket drive this higher affinity for picolinamides. However, a trend was observed where a greater reliance upon residues in TM7 was observed for compounds, both nicotinamides and picolinamides, with higher affinity (<100 nM). Picolinamides also tended to be susceptible to W784A; however, this only reached significance for VU0403602.
For all PAMs, Y658V abolished potentiation, except for VU0405398, where a weak NAM switch was observed. Trp substitution of the equivalent residue in mGlu8 suppresses the activity of a constitutively active mutant receptor (Yanagawa et al., 2009), raising the possibility that this mGlu5 mutation may have a global receptor activation effect. However, previously DFB potentiation was reportedly unaffected by Y658V (Muhlemann et al., 2006), supporting the hypothetical modulator binding modes shown, where an interaction is predicted with this residue, rather than Y658V impacting active receptor states.
A number of previous studies have identified PAMs as competitive with the common mGlu5 allosteric (or “MPEP”) site on the basis of a single point mutation in TM7, A809V (Chen et al., 2008; Hammond et al., 2010). Previously, A809V was reported to cause an ∼30-fold decrease in the affinity of 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (VU29), an mGlu5 PAM from the 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB) series (Gregory et al., 2012). Validating the utilization of this single point mutation as a readout of interaction with this common allosteric site, all six PAMs exhibited decreased affinity for this mutant construct. Although this interaction appears to be crucial for all PAMs that are competitive with MPEP tested to date, it should be noted that this does not necessarily have to be the case. Notably, L743V and the equivalent mutation in mGlu1 were previously shown to enhance potentiation by PAMs (Knoflach et al., 2001; Muhlemann et al., 2006); however, for acetylene PAMs, L743V had no effect on cooperativity or affinity. Further studies are under way to probe interactions within the common allosteric site by modulators from distinct chemotypes to better inform our understanding of the molecular determinants of modulator affinity and cooperativity (Manka et al., 2012).
A key finding arising from this study was the identification of residues that, when mutated, engendered a mode switch in allosteric modulator pharmacology, specifically T780A in TM6 and S808A in TM7. Considering that these PAMs originated from a NAM high-throughput screening lead (Rodriguez et al., 2010), such drastic changes in modulator cooperativity are not altogether surprising. Indeed, the acetylene series of mGlu allosteric modulators is prone to “molecular switches” (Wood et al., 2011), the SAR plagued by unanticipated changes in the mode of pharmacology and selectivity (Wood et al., 2011; Sheffler et al., 2012). Muhlemann et al. (2006) previously reported a similar result for the early mGlu5 PAM DFB, where at the F787A mutant DFB behaved as a weak NAM. Pharmacological mode switches were also noted during the discovery of DFB and related compounds (O'Brien et al., 2003). As noted above, movements in TM6 have been implicated in the transition of Family A 7TMRs from inactive to active states. TM7 contains the NPxxY motif, also well known for its role in receptor activation (Barak et al., 1995; Prioleau et al., 2002; Fritze et al., 2003). Furthermore, a water-hydrogen bond network involving polar residues in TMs 1, 2, 6, and 7 is postulated to play an integral role in receptor activation (Nygaard et al., 2010). Given the importance of TMs 6 and 7 for the transitioning of receptors into active conformations, these mode switches may be attributed to either a loss of an important direct contact that facilitates receptor activation upon modulator binding or a global (secondary) effect on protein conformations that prevents some modulators, but not others, from engendering active conformations.
Collectively, these findings highlight the subtleties of interactions within the common mGlu allosteric binding pocket that determine allosteric modulator affinity and cooperativity. The identification of point mutations that engender a molecular switch in PAM pharmacology provides the first clues from the protein side of the equation as to the underlying determinants for this phenomenon. The prevalence of molecular switches raises concerns regarding metabolite pharmacology. A deeper understanding of the molecular basis of allosteric modulation has the potential to aid rational drug design efforts to predict and avoid undesirable pharmacology, including mode switches.
Supplementary Material
Acknowledgments
The authors acknowledge Kristian W. Kauffman and Eric S. Dawson for their contribution to the comparative modeling of mGlu5, and Colleen M. Niswender for critical reading of the manuscript.
Abbreviations
- CaSR
calcium-sensing receptor
- CDPPB
3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- CPPHA
N-[4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl]-2-hydroxybenzamide
- DFB
difluorobenzaldazine
- DMEM
Dulbecco’s modified Eagle's medium
- FBS
fetal bovine serum
- HEK
human embryonic kidney
- [3H]methoxyPEPy
[3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine
- mGlu
metabotropic glutamate receptor
- MPEP
2-methyl-6-(phenylethynyl)-pyridine
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- PDB
Protein Data Bank
- SAM
silent allosteric modulator
- SAR
structure-activity relationship
- TM
transmembrane domain
- 7TMR
seven-transmembrane-spanning (G-protein-coupled) receptor
- VU0360172
N-cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide hydrochloride, VU0360173, (6-((3-fluorophenyl)ethynyl)pyridin-3-yl)(3-hydroxyazetidin-1-yl)methanone
- VU0403602
(N-cyclobutyl-5-((3-fluorophenyl)ethynyl)ethynyl)picolinamide hydrochloride
- VU0405386
N-(tert-butyl)-5-((3-fluorophenyl)ethynyl)picolinamide
- VU0405398
(5-((3-fluorophenyl)ethynyl)pyridin-2-yl)(3-hydroxyazetidin-1-yl)methanone
- VU0415051
N-tert-butyl-6-[2-(3-fluorophenyl)ethynyl]pyridine-3-carboxamide
- VU29
4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
Authorship Contributions
Participated in research design: Gregory, Nguyen, Stauffer, Meiler, Conn.
Conducted experiments: Gregory, Nguyen, Reiff, Squire.
Contributed new reagents or analytic tools: Stauffer, Lindsley.
Performed data analysis: Gregory, Nguyen, Reiff, Squire.
Wrote or contributed to the writing of manuscript: Gregory, Nguyen, Meiler, Conn.
Footnotes
This work is supported by the National Institutes of Health National Institute of Mental Health [Grant 2R01-MH062646-13]; National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant 2R01-NS031373-16A2]; National Institutes of Health National Institute on Drug Abuse [Grant 1R01-DA023947]; and Molecular Libraries Probe Production Centers Network [Grants 5 u54 MH84659-03 and 5 u54 MH84659-03S1]. K.J.G. is a recipient of a National Alliance for Research on Schizophrenia and Depression (NARSAD)–Maltz Young Investigator Award, an American Australian Association Merck Foundation Fellowship, and a National Health and Medical Research Council (Australia) Overseas Biomedical Postdoctoral Training Fellowship. Work in J.M.'s laboratory is supported through the National Institutes of Health [Grants R01-GM080403, R01-MH090192, and R01-GM099842] and the National Science Foundation [Career 0742762]. P.J.C. is a consultant for Seaside Therapeutic and receives research support from Seaside Therapeutics and Johnson and Johnson/Janssen Pharmaceutica.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Alexander N, Woetzel N, and Meiler J (2011) Bcl::Cluster: a method for clustering biological molecules coupled with visualization in the Pymol Molecular Graphics System, in Proceedings of the 2011 IEEE 1st International Conference on Computational Advances in Bio and Medical Sciences; 2011 February 3–5; Orlando, FL. pp 13–18, IEEE Computer Society, Los Alamitos. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ametamey SM, Treyer V, Streffer J, Wyss MT, Schmidt M, Blagoev M, Hintermann S, Auberson Y, Gasparini F, Fischer UC, et al. (2007) Human PET studies of metabotropic glutamate receptor subtype 5 with 11C-ABP688. J Nucl Med 48:247–252 [PubMed] [Google Scholar]
- Barak LS, Ménard L, Ferguson SS, Colapietro AM, Caron MG. (1995) The conserved seven-transmembrane sequence NP(X)2,3Y of the G-protein-coupled receptor superfamily regulates multiple properties of the beta 2-adrenergic receptor. Biochemistry 34:15407–15414 [DOI] [PubMed] [Google Scholar]
- Chen Y, Goudet C, Pin JP, Conn PJ. (2008) N-4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 73:909–918 [DOI] [PubMed] [Google Scholar]
- Chen Y, Nong Y, Goudet C, Hemstapat K, de Paulis T, Pin JP, Conn PJ. (2007) Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol Pharmacol 71:1389–1398 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A. (1998) Assessing the distribution of parameters in models of ligand-receptor interaction: to log or not to log. Trends Pharmacol Sci 19:351–357 [DOI] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, et al. (2003) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem 46:204–206 [DOI] [PubMed] [Google Scholar]
- Davis IW, Baker D. (2009) RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol 385:381–392 [DOI] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. (2003) Role of the conserved NPxxY(x)5,6F motif in the rhodopsin ground state and during activation. Proc Natl Acad Sci USA 100:2290–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda J, Suzuki G, Kimura T, Nagatomi Y, Ito S, Kawamoto H, Ozaki S, Ohta H. (2009) Identification of a novel transmembrane domain involved in the negative modulation of mGluR1 using a newly discovered allosteric mGluR1 antagonist, 3-cyclohexyl-5-fluoro-6-methyl-7-(2-morpholin-4-ylethoxy)-4H-chromen-4-one. Neuropharmacology 57:438–445 [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, et al. (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38:1493–1503 [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Dong EN, Meiler J, Conn PJ. (2011) Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 60:66–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, et al. (2012) Investigating metabotropic glutamate receptor 5 allosteric modulator cooperativity, affinity, and agonism: enriching structure-function studies and structure-activity relationships. Mol Pharmacol 82:860–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, Conn PJ. (2010) Discovery of a novel chemical class of mGlu(5) allosteric ligands with distinct modes of pharmacology. ACS Chem Neurosci 1:702–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B, Nygaard R, Valentin-Hansen L, Bach A, Engelstoft MS, Petersen PS, Frimurer TM, Schwartz TW. (2010) A conserved aromatic lock for the tryptophan rotameric switch in TM-VI of seven-transmembrane receptors. J Biol Chem 285:3973–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honer M, Stoffel A, Kessler LJ, Schubiger PA, Ametamey SM. (2007) Radiolabeling and in vitro and in vivo evaluation of [18F]-FE-DABP688 as a PET radioligand for the metabotropic glutamate receptor subtype 5. Nucl Med Biol 34:973–980 [DOI] [PubMed] [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther 313:199–206 [DOI] [PubMed] [Google Scholar]
- Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. (2001) Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci USA 98:13402–13407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb JP, Engers DW, Niswender CM, Rodriguez AL, Venable DF, Conn PJ, Lindsley CW. (2011) Discovery of molecular switches within the ADX-47273 mGlu5 PAM scaffold that modulate modes of pharmacology to afford potent mGlu5 NAMs, PAMs and partial antagonists. Bioorg Med Chem Lett 21:2711–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. (1995) Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol 48:362–378 [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389 [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon G, Meiler J. (2012) Rosetta Ligand docking with flexible XML protocols. Methods Mol Biol 819:143–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, et al. (2008) ADX47273 [S-(4-fluoro-phenyl)-3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839 [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Mühlemann A, Zenner M-T, Fischer C, Stahl M, Gerber PR, Jaeschke G, Porter RHP. (2006) Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J Neurochem 98:601–615 [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. (2003) Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol 64:823–832 [DOI] [PubMed] [Google Scholar]
- Manka JT, Vinson PN, Gregory KJ, Zhou Y, Williams R, Gogi K, Days E, Jadhav S, Herman EJ, Lavreysen H, et al. (2012) Optimization of an ether series of mGlu5 positive allosteric modulators: molecular determinants of MPEP-site interaction crossover. Bioorg Med Chem Lett 22:6481–6485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J, Baker D. (2006) ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins 65:538–548 [DOI] [PubMed] [Google Scholar]
- Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. (2012) Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem 55:1445–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miedlich SU, Gama L, Seuwen K, Wolf RM, Breitwieser GE. (2004) Homology modeling of the transmembrane domain of the human calcium sensing receptor and localization of an allosteric binding site. J Biol Chem 279:7254–7263 [DOI] [PubMed] [Google Scholar]
- Mølck C, Harpsøe K, Gloriam DE, Clausen RP, Madsen U, Pedersen LO, Jimenez HN, Nielsen SM, Mathiesen JM, Bräuner-Osborne H. (2012) Pharmacological characterization and modeling of the binding sites of novel 1,3-bis(pyridinylethynyl)benzenes as metabotropic glutamate receptor 5-selective negative allosteric modulators. Mol Pharmacol 82:929–937 [DOI] [PubMed] [Google Scholar]
- Mühlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, Jaeschke G, Malherbe P, Porter RH. (2006) Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol 529:95–104 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, et al. (2012) Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81:120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Valentin-Hansen L, Mokrosinski J, Frimurer TM, Schwartz TW. (2010) Conserved water-mediated hydrogen bond network between TM-I, -II, -VI, and -VII in 7TM receptor activation. J Biol Chem 285:19625–19636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, et al. (2003) A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol 64:731–740 [DOI] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, et al. (2004) A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther 309:568–577 [DOI] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezèau L, Carroll F, Pin JP, et al. (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275:33750–33758 [DOI] [PubMed] [Google Scholar]
- Prioleau C, Visiers I, Ebersole BJ, Weinstein H, Sealfon SC. (2002) Conserved helix 7 tyrosine acts as a multistate conformational switch in the 5HT2C receptor. Identification of a novel “locked-on” phenotype and double revertant mutations. J Biol Chem 277:36577–36584 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, et al. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 78:1105–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol 68:1793–1802 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Williams R, Zhou Y, Lindsley SR, Le U, Grier MD, Weaver CD, Conn PJ, Lindsley CW. (2009) Discovery and SAR of novel mGluR5 non-competitive antagonists not based on an MPEP chemotype. Bioorg Med Chem Lett 19:3209–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kedrowski J, Rook JM, Smith RL, Jones CK, Rodriguez AL, Conn PJ, Lindsley CW. (2009) Discovery of molecular switches that modulate modes of metabotropic glutamate receptor subtype 5 (mGlu5) pharmacology in vitro and in vivo within a series of functionalized, regioisomeric 2- and 5-(phenylethynyl)pyrimidines. J Med Chem 52:4103–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler DJ, Wenthur CJ, Bruner JA, Carrington SJS, Vinson PN, Gogi KK, Blobaum AL, Morrison RD, Vamos M, Cosford NDP, et al. (2012) Development of a novel, CNS-penetrant, metabotropic glutamate receptor 3 (mGlu3) NAM probe (ML289) derived from a closely related mGlu5 PAM. Bioorg Med Chem Lett 22:3921–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. (2002) Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem 277:40989–40996 [DOI] [PubMed] [Google Scholar]
- Suzuki G, Kimura T, Satow A, Kaneko N, Fukuda J, Hikichi H, Sakai N, Maehara S, Kawagoe-Takaki H, Hata M, et al. (2007) Pharmacological characterization of a new, orally active and potent allosteric metabotropic glutamate receptor 1 antagonist, 4-[1-(2-fluoropyridin-3-yl)-5-methyl-1H-1,2,3-triazol-4-yl]-N-isopropyl-N-methyl-3,6-dihydropyridine-1(2H)-carboxamide (FTIDC). J Pharmacol Exp Ther 321:1144–1153 [DOI] [PubMed] [Google Scholar]
- Treyer V, Streffer J, Wyss MT, Bettio A, Ametamey SM, Fischer U, Schmidt M, Gasparini F, Hock C, Buck A. (2007) Evaluation of the metabotropic glutamate receptor subtype 5 using PET and 11C-ABP688: assessment of methods. J Nucl Med 48:1207–1215 [DOI] [PubMed] [Google Scholar]
- Varney MA, Cosford ND, Jachec C, Rao SP, Sacaan A, Lin FF, Bleicher L, Santori EM, Flor PJ, Allgeier H, et al. (1999) SIB-1757 and SIB-1893: selective, noncompetitive antagonists of metabotropic glutamate receptor type 5. J Pharmacol Exp Ther 290:170–181 [PubMed] [Google Scholar]
- Visiers I, Ballesteros JA, Weinstein H. (2002) Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol 343:329–371 [DOI] [PubMed] [Google Scholar]
- Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. (2011) “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry 50:2403–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa M, Yamashita T, Shichida Y. (2009) Activation switch in the transmembrane domain of metabotropic glutamate receptor. Mol Pharmacol 76:201–207 [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Schonbrun J, Baker D. (2006) Multipass membrane protein structure prediction using Rosetta. Proteins 62:1010–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Wisnoski DD, O’Brien JA, Lemaire W, Williams DL, Jr, Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, et al. (2007) Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg Med Chem Lett 17:1386–1391 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Manka JT, Rodriguez AL, Weaver CD, Days EL, Vinson PN, Jadhav S, Hermann EJ, Jones CK, Conn PJ, et al. (2010) Discovery of N-aryl piperazines as selective mGlu(5) potentiators with efficacy in a rodent model predictive of anti-psychotic activity. ACS Med Chem Lett 1:433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.