

Abstract
Current antipsychotic medications do little to improve real-life function in most schizophrenia patients. A dispassionate view of the dispersed and variable neuropathology of schizophrenia strongly suggests that it is not currently, and may never be, correctable with drugs. In contrast, several forms of cognitive therapy have been demonstrated to have modest but lasting positive effects on cognition, symptoms, and functional outcomes in schizophrenia patients. To date, attempts to improve clinical outcomes in schizophrenia by adding pro-cognitive drugs to antipsychotic regimens have had limited success, but we propose that a more promising strategy would be to pair drugs that enhance specific neurocognitive functions with cognitive therapies that challenge and reinforce those functions. By using medications that engage spared neural resources in the service of cognitive interventions, it might be possible to significantly enhance the efficacy of cognitive therapies. We review and suggest several laboratory measures that might detect potential pro-neurocognitive effects of drugs in individual patients, using a “test dose” design, aided by specific biomarkers predicting an individual’s drug sensitivity. Lastly, we argue that drug classes viewed as “counter-intuitive” based on existing models for the pathophysiology of schizophrenia—including pro-catecholaminergic and NMDA-antagonistic drugs—might be important candidate “pro-cognitive therapy” drugs.
Keywords: Antipsychotics, Cognitive training, Cognitive therapy, Cognitive remediation, Neurocognition, Schizophrenia
1 Introduction
Schizophrenia (SZ) is a severe brain disorder affecting 1% of the world population. Its cost to society in terms of health care demands and lost productivity is well documented (Rice 2009), as are the personal stories of lifelong anguish and suffering among SZ patients and their families. While both genetic and epigenetic factors are associated with a risk for developing this disorder (Dick et al. 2010), the etiology and pathophysiology of SZ remain incompletely understood. More than 50 years after the introduction of drugs that target its symptoms, the standard medications for SZ are at best modestly effective.
Pharmacotherapy of SZ is dominated by drugs that functionally reduce dopamine (DA) neurotransmission, and primarily target “positive” symptoms of this disorder (hallucinations and delusions). Although antipsychotics can blunt the most severe psychotic symptoms, they do not have a meaningful impact on the course of SZ or on real-life function (Leucht et al. 2009; Lieberman et al. 2005). It is clear that cognitive deficits are major factors in the functional disability of SZ patients (Keefe and Harvey 2012). At a time when treatments based on old paradigms have resulted in only modest gains in the function and wellbeing of SZ patients, we must look for new approaches to make substantial improvements in patients’ lives.
In contrast to antipsychotic drugs—which are primarily developed to overcome pathologically elevated levels of dopaminergic neurotransmission in SZ—several forms of cognitive therapies (CTs; broadly including cognitive-behavioral and cognitive remediation or training) may both reduce symptoms and improve function in schizophrenia by engaging healthy neural systems to learn adaptive cognitive and behavioral strategies (Demily and Franck 2008; Klingberg et al. 2009; McGurk et al. 2007a; Medalia and Choi 2009; Tai and Turkington 2009). A number of meta-analyses document clear safety, feasibility, acceptance, and efficacy of cognitive interventions in SZ, with sustained benefits in many cases lasting years (Eack et al. 2009; Granholm et al. 2007; McGurk et al. 2009; Sellwood et al. 2007). Response predictors are being identified (Brabban et al. 2009; Kumari et al. 2009; Kurtz et al. 2009); treatments that target specific functional outcomes measures [e.g., vocation (McGurk et al. 2007a)] and specific symptoms [e.g., hallucinations (Penn et al. 2009)] are also being developed. Despite findings from several meta-analyses (e.g., Wykes et al. 2011), some individual studies have failed to detect significant benefits of CTs in SZ patients (e.g., Lynch et al. 2010), suggesting that important determinants of study outcome (e.g., patient characteristics, study design, forms of CT being used) may not yet be fully understood.
Although the neurobiological basis for therapeutic effects of CTs in SZ is not fully known, the biology underlying learning-based neuroplasticity has been elaborated at levels extending from molecules to systems, and studies are now identifying neural changes accompanying clinical benefits of these specialized “learning therapies.” Conceivably, these neural changes and their corresponding therapeutic impact might be augmented via medications resulting in an additive effect. Such a use of medications would depart substantially from the traditional approach to pharmacotherapy for SZ. However, This does not discount the importance of controlling psychosis: most psychotherapeutic interventions are complicated by severe psychiatric symptoms, and it is clear that controlling psychosis should benefit ongoing cognitive interventions in SZ. Compared to antipsychotic medications, drugs with pro-cognitive effects might more directly, and perhaps synergistically, enhance the clinical benefits of specific “learning therapies.” For example, drugs that enhance specific components of neurocognition, e.g., attention, might be predicted to yield clinical benefits in SZ when paired with interventions that access those components by placing demands on enhanced attention.
2 Distributed Neuropathology of SZ and Failures of the Simple “Medication Model”
Some prevailing models for the pharmacotherapy of SZ have been based on the misconception that this disorder reflects pathology that is restricted in scope, both in terms of the neurotransmitters that are dysregulated (e.g., dopamine, glutamate) and the brain region(s)—and neuronal element(s) within those regions—that are abnormal. It is now clear—as briefly reviewed below—that the neuropathology of schizophrenia is substantial in scope and complexity. In patients, structural abnormalities in about 20 brain regions span wide swaths of cortical and subcortical tissue, reflecting processes presumably well-advanced at birth. Roughly half as many regions are abnormal in unaffected relatives (cf. Swerdlow 2011). Within any region, laminar synaptic and cellular arrangements may be perturbed, replacing “intended” spatial and chemical connections with dysfunctional alternatives. The likelihood that medications will functionally untangle these dispersed aberrant connections in schizophrenia seems unlikely.
Evidence for distributed neural dysfunction in SZ is compelling, even when considering only the areas where structural abnormalities are reported (and not, for example, areas activated abnormally under experimental or symptomatic conditions [cf. Brown and Thompson 2010; Dolan et al. 1995; Heckers et al. 1998; Heckers and Konradi 2010; Kumari et al. 2003; Silbersweig et al. 1995; Volz et al. 1999)]. Findings document significant volumetric and/or morphometric abnormalities in over 20 brain regions in SZ patients (cf. Levitt et al. 2010; Swerdlow 2011). These abnormalities reflect perturbations in the number, size or shape of cells, fibers, or extra-parenchymal elements. Medline lists numerous papers reporting laminar- and subregion-specific reductions and other abnormalities in the number of neurons, length of their dendrites, density of their dendritic spines and varicosities, and levels of cellular proteins and mRNA in prefrontal, mesial temporal, and auditory cortex, striatum and thalamus, and even the cerebellum and midbrain DA nuclei, among other regions. Studies also document abnormalities in the number or distribution of neurotransmitter receptors in these and other brain regions, which may reflect a primary loss of cells that support them, a secondary response to abnormalities of the fibers that innervate them or the chemicals they deliver, or combinations thereof (cf. Abi-Dargham et al. 1998; Akil et al. 1999; Aparacio-Legarza et al. 1997; Cruz et al. 2009; Dean et al. 2009; Gur et al. 2007; Howes et al. 2009; Kessler et al. 2001, 2009; Laruelle 1998; Lee and Seeman 1980; Lewis et al. 2008; Roberts et al. 2009; Urban and Abi-Dargham 2010; Volk and Lewis 2010; Wong et al. 1986).
For several reasons, we can be confident that the neural disturbances in many SZ patients impact brain circuitry extending well beyond the neural disturbances listed in published reports from large samples of patients. First, studies of the neural circuit disturbances in SZ have been circumscribed in their targets, but findings of cortical abnormalities that extend well beyond the prefrontal and mesial temporal regions (Sweet et al. 2007, 2009) suggest more generalized neurodevelopmental disturbances. Second, disturbances in neuronal number, size, shape, and connectivity perturb neurotransmission, cellular metabolism, signal transduction molecules, gene expression, and other levels of the machinery required for normal neural function (cf. Benes 2010; Kvajo et al. 2010). Such a “cascade” of disturbances will inevitably intersect in time and space with a wide range of neurobiological processes. Third, identifiable disturbances in one neuronal element translate into widely distributed dysfunction within “intact” brain circuits efferent from, or projecting to, the “damaged” element. For example, pathology that impairs normal “γ-band” synchronization of discharges from large populations of cortical neurons can disrupt information processing among those “normal” cells and the circuits that they form (cf. Uhlhaas and Singer 2006). Thus, disturbances in one cell type can have multiplier effects downstream, even among circuits that—in postmortem analyses or resting state imaging—have normal structural and morphological properties. Fourth, variance across and within studies for each abnormality is substantial. In two individuals with SZ, the same brain region may be relatively normal in one and grossly abnormal in another. Furthermore, among the list of regions that are statistically different in cohorts of patients versus controls, any given patient might exhibit some but not all of the regional abnormalities. And with any given cortico-striato-pallido-thalamic locus, reduced volumes in two different patients might reflect disturbances in different cell populations, resulting in different patterns of abnormal efferent projections and innervation.
Perhaps most important, as it relates to therapeutic approaches to this disorder, is the fact that neuropathology in SZ evolves across early life, and likely reflects failures of early cell development and migration. These early developmental failures disrupt the tightly choreographed processes that lead to the proper population of forebrain nuclei, and formation of the synaptic connectivity both within [e.g., prefrontal laminar connectivity (Volk and Lewis 2010)] and between [e.g., hippocampal-frontal synchronization (Heckers and Konradi 2010)] these regions. By the time that SZ symptoms emerge, treatment that merely antagonizes or augments receptors at the molecular level cannot reasonably be expected to normalize function within the proper connections that did not form, nor the improper ones that did, across 20 different brain regions and their substantially larger “fall-out field.”
3 Cognitive Therapies for Schizophrenia
While it is not classically viewed as a “biological” intervention, it is now clear that psychotherapy (particularly cognitive and behavioral therapy) changes the brain (Baxter et al. 1992; Saxena et al. 2009; Schwartz et al. 1996). How psychotherapy changes the brain, and the extent to which these changes reflect processes from gene expression up to the organization of circuits and systems, are questions of ongoing investigation (de Lange et al. 2008; Fox 2009; Keller and Just 2009; Korosi and Baram 2009; Porto et al. 2009; Saxena et al. 2009).
While some forms of psychotherapy are considered to be suboptimal, and even potentially harmful for patients with psychotic disorders—e.g., psychoanalytic or other “regressive” forms of psychotherapy—a variety of therapies based on cognitive constructs and behavioral theories have been found to be helpful for SZ patients. Most frequently studied and commonly cited is Cognitive Behavioral Therapy (CBT), a manualized therapy in which maladaptive thoughts and beliefs (cognitions) that affect the patient’s function are identified and explored with the patient to examine how they affect the patient’s interpretations of their experiences and the resulting behaviors; behavioral techniques are then applied to help modify maladaptive patterns. Other evidence-based psychosocial treatments for SZ patients include social skills training (SST) and supported employment (SE), which target the psychosocial deficits and occupational impairments commonly found in patients in order to help them reach their functional goals. SST teaches skills to help patients communicate with others and understand both verbal and nonverbal cues; these classes provide a setting for patients to discuss challenges they encounter and to practice their newly learned skills. SE interventions take an individualized approach to teaching the skills a client needs to get and keep competitive work in the community. While the types of cognitive processes and “learning” engaged varies widely across these different forms of therapy, they each have both primary and secondary consequences on brain function, i.e., the neurobiological changes produced by the therapy-specific learning, and those resulting from the positive social and functional consequences that are based on the learned adaptive behaviors.
As cognitive deficits in SZ patients have been found to reduce response to psychosocial rehabilitation (McGurk and Mueser 2004; Mueser et al. 1991; Wykes et al. 1990), and to impact functional outcome far more than the more prominent positive symptoms of hallucinations and delusions (Green et al. 2000), the development of strategies and programs to improve cognitive functioning has been a focus in SZ therapies. Cognitive interventions use repeated drills, compensatory strategies, or a blend of both approaches, to help patients with basic neurocognitive processes such as attention, information processing, problem solving, decision-making, and memory. Cognitive training differs from CBT interventions in both focus and methodology. While CBT targets the form and content of thought, such as attributional style and core beliefs, cognitive training targets the neurocognitive processes that underlie thought. Notably, although many studies have shown effectiveness of CBT in schizophrenia, some studies have shown that, when compared to some other control interventions, CBT was not necessarily better at reducing symptoms or preventing relapse (e.g., Lynch et al. 2010). Given that 70–80% of SZ patients are 1–2 standard deviations below normal populations in relevant neurocognitive measures (Heinrichs and Zakzanis 1998; Reichenberg and Harvey 2007) and that cognitive deficits correlate highly with life functioning and ability to meet functional goals (Green et al. 2004), the premise of cognitive training is that when cognitive function improves, these gains will generalize to functioning in the community. Indeed, substantial evidence indicates that cognitive training reduces symptoms and improves functioning in SZ patients (Klingberg et al. 2009; Kurtz et al. 2001; McGurk et al. 2007b; Medalia and Choi 2009; Wykes et al. 2011) with sustained benefits often lasting years (Eack et al. 2009; Granholm et al. 2007; McGurk et al. 2007a; Sellwood et al. 2007).
Also referred to as cognitive remediation or cognitive rehabilitation, cognitive training derives much of its background from the rehabilitation of brain injury patients (Twamley et al. 2008b). While the approach can be classified into three strategies—compensatory (strategies to work around deficits) versus restorative (correcting the deficits) versus environmental (modifying environment to accommodate deficits)—most cognitive training programs integrate the three approaches into various programs. In fact, which of the three approaches is most beneficial depends on each individual circumstance, and hence, there is significant range in cognitive training approaches. Some programs utilize a primarily restorative approach, which attempts to “repair” impairments by drill and practice exercises. These can be executed using paper–pencil worksheets, computer programs, or therapist-based interactions. Other programs use a compensatory approach that attempts to circumvent deficits by relying on other skills or environmental modifications. These programs tend to use a strategies-teaching approach, conveyed either by individual didactics or group discussions, followed by practice of strategies and planning for implementation in the community (McGurk et al. 2007a; Twamley et al. 2008b).
Even within a specific modality, such as the use of computer programs for restorative training, there is variation in the particular skills developed (e.g., memory vs. attention vs. other cognitive skills), the number of skills targeted simultaneously, the methods of developing the skill, how contextualized the exercises are (e.g., a dot in the center of the screen vs. a dot representing an oncoming train), the degree of engagement, interest and motivation incorporated in the practicing, the level of difficulty, and immediacy of feedback, as well as the type of feedback (Medalia and Choi 2009). Programs can be provided one-on-one or in groups; some regimens are manualized and allow more measured learning, while others utilize personalized and tailored curricula that allow for more flexibility. Some programs are centralized: patients come to the institution for training; others offer separate or integrated psychosocial programs to help transfer and generalize the acquired skills to real-life functioning, and still others use “coaches,” who help organize living and work environments, and help patients apply the newly acquired skills to particular situations. The duration of programs can range from several weeks to 2 years (most last about 3–6 months), with most requiring from 1 to 4 hr sessions per week (Medalia and Choi 2009).
Despite early studies suggesting no clear evidence for the effect of social skills training and cognitive remediation (Pilling et al. 2002), recent studies show the effectiveness of cognitive training for both measures of cognitive test performance and real life functioning. Six meta-analytic studies showed effect sizes (d) ranging from 0.2 to 1.2, with greatest improvement in neuropsychological measures, followed by psychosocial functioning, and lastly symptom reduction (Medalia and Choi 2009). A meta-analysis of 40 randomized, controlled trials of 2,104 patients (Wykes et al. 2011) showed a medium effect size (0.5) for cognitive performance as measured by standardized cognitive tasks and a medium effect size (0.42) for psychosocial functioning as measured by the ability to obtain and work competitive jobs, quality of and satisfaction with interpersonal relationships, and ability to solve interpersonal problems. Effect sizes for cognition and functioning were maintained at follow-up. Notably, the effects on the more generalized psychosocial functioning were stronger in studies that provided adjunctive psychosocial rehabilitation as part of the program and when compensatory strategy training was provided in the context of psychosocial rehabilitation. For symptoms, a small effect size (0.18) was found, perhaps reflecting, in part, the positive training experiences that improved self-esteem, thereby improving mood. Notably, in all studies, there is variability in effectiveness in various domains, and the difficulty of knowing which therapeutic approach will work for which patient likely reflects the heterogeneity of the disorder.
Given the potential for cognitive training to improve real life functioning, efforts are underway to identify response predictors. While factors such as instructional technique, patient’s motivation, and the control of psychiatric symptoms including psychosis and mood changes are key to effective cognitive training, neurocognitive abilities also significantly impact outcome. The type and extent of neurocognitive impairment has been found to consistently affect the therapeutic impact of cognitive training (Fiszdon et al. 2006). Delayed verbal memory impairs training (Medalia and Richardson 2005), while sustained attention, working memory, and verbal learning are also important determinants of outcome, even in the context of crystallized verbal intelligence (Fiszdon et al. 2005; Kurtz et al. 2008, 2009). However, Twamley et al. (2011) found that in a compensatory cognitive training intervention, lower baseline cognitive and functional abilities predicted greater improvement, possibly because lower functioning participants had more room to improve or because those with higher abilities had a narrower scope of dysfunction and the intervention did not match their needs. For example, improvement in attention (forward digit span) at post-treatment was associated with lower baseline attention (r = −0.73, p < 0.001, n = 20), and improvement in functional capacity (UPSA) at follow-up was associated with lower functional capacity scores at baseline (r = −0.56, p = 0.007, n = 22). It is thus rational to consider whether medications that enhance these basic neurocognitive functions in patients with SZ might increase the therapeutic impact of cognitive training or other cognitive therapies. By bolstering a patient’s abilities to engage spared neural substrates for memory, attention, learning, etc., these medications would maximize their ability to meet the demands of a variety of cognitive interventions.
4 Pro-Cognitive Agents in the Treatment of SZ: The Bad News and the Good News
The concerted effort by our field to develop and apply pro-cognitive agents in SZ, however, has not been based on their potential ability to enhance the therapeutic impact of cognitive interventions. Thus far, most of these efforts have used a “stand-alone” drug strategy similar to that used to justify standard antipsychotic therapy: if we can make a pill that normalizes a neurochemical abnormality in an adult SZ patient, it should help normalize their neurocognitive function, and this should automatically translate into improved life function. For example, based on emerging models for NMDA receptor hypofunction in SZ, a number of putative pro-cognitive agents have been tested that directly or indirectly enhance forebrain glutamate neurotransmission. To date, however, trials of potential pro-cognitive glutamatergic agents in SZ have largely yielded negative results (Barch 2010; Buchanan et al. 2007; Goff et al. 1996, 1999, 2007, 2008; Green 2007). A large, multicenter study of the glycine-site agonist, D-cycloserine (DCS) showed no benefit on negative symptoms or cognition in SZ (Buchanan et al. 2007). A small study reported in 2007 with the mGluR2/3 agonist, LY2140023, suggested some modest clinical benefit (Patil et al. 2007) that has yet to be reproduced in larger samples. Studies are in progress with a number of compounds, including glycine transport inhibitors and nicotinic agonists, but preliminary findings reported as Conference Proceedings suggest minimal, if any benefit from these agents (e.g., Marder and Buchanan 2009).
In fact, in their comprehensive review of clinical trials of putative pro-cognitive agents in SZ, Barch (2010) concluded that well-controlled, double-blind studies have, to date, failed to yield any particularly encouraging results; and Keefe noted that the majority of completed trials thus far were underpowered and short in duration, but that ongoing trials have larger samples sizes and longer durations, and may provide insight into the subject characteristics that have greatest potential for cognition enhancing drugs (Keefe et al. 2011a). Clearly, the use of cognition enhancing medications in SZ requires further studies with validated measures to illuminate how these medications can be used to improve function in SZ patients. Interestingly, some single-dose studies with nicotine and amphetamine have demonstrated enhanced performance on specific neurocognitive measures in SZ patients, but it is not known whether these changes are “of a clinically significant magnitude.” It is important to recognize, however, that of the many (>100) trials of potential pro-cognitive agents in SZ—almost all yielding negative or inconclusive results—until very recently, none were conducted within the context of systematic cognitive interventions (Barch 2010; Buchanan et al. 2007; Goff et al. 1996, 1999, 2007, 2008; cf. Green 2007). In fact, given the prevailing state of outpatient care for SZ patients, it is likely that—absent specific experimental designs to do otherwise—most SZ patients in trials of putative pro-cognitive agents took these medications within an environment relatively void of constructive cognitive challenges. It is not surprising that little benefit was gleaned from pro-cognitive agents among patients whose daily activities are often dominated by social isolation, and at best passive cognitive engagement by television and sedentary “board and care” surroundings: drugs designed to enhance specific components of neurocognition might not be beneficial unless paired with interventions that access, utilize, and place demands on those components. Without a structure for acquiring reparative or compensatory thoughts or behaviors, any gains in neurocognitive capacity would be wasted. Analogous reasoning underlies the use of anabolic steroids to promote exercise-increased muscle mass, and perhaps more importantly (as discussed below), the use of pro-extinction drugs to enhance therapeutic benefits of CBT for anxiety disorders (Ressler et al. 2004). By adding pro-cognitive drugs that engage spared neural resources and enhance specific neurocognitive functions to CT regimens, it might be possible to significantly enhance the efficacy of the CTs.
Had these studies been designed differently, how might pro-cognitive medications have enhanced the therapeutic impact of an ongoing cognitive intervention in SZ? Let’s pick a specific form of cognitive training to provide a concrete example. As reviewed earlier, compensatory cognitive training is a cognitive intervention that encourages patients to develop compensatory strategies—both internal (e.g., acronyms or visual imagery) and external (e.g., writing information down to remember it later)—for learning and remembering information. In so doing, it specifically activates prefrontal regions subserving working memory and attention (Haut et al. 2010). One form of cognitive training includes four modules (Twamley et al. 2008a, b), addressing: (1) prospective memory (i.e., remembering to do things in the future); (2) conversational and task vigilance; (3) learning and memory; and (4) cognitive flexibility and problem-solving (i.e., executive functioning). Drugs that enhance working memory, attention and vigilance, cognitive flexibility, and problem-solving will enhance a SZ patient’s ability to develop and utilize new strategies for learning and remembering; this should then translate to improved function in addressing the demands of daily life.
In truth, most studies of CT effects in SZ patients have been conducted using CT as an “add-on” to “standard” medication therapy; as a result, the beneficial effects of CTs reported in many studies (e.g., Grant et al. 2012) may have reflected, indirectly or directly, the positive impact of antipsychotics on CT. Though the pseudospecificity of the results is difficult to tease out, as in the case of Grant et al. (2012), the antipsychotic class or dose has not been determined to significantly impact the benefits of CT. It seems highly likely that the control of overt psychotic symptoms with antipsychotic medication will, for the foreseeable future, be a prerequisite for any effective cognitive intervention; this issue is orthogonal to the question of whether pro-neurocognitive agents can directly enhance the benefits of CTs.
One recent report did pair a putative pro-cognitive agent with a cognitive intervention—in this case, CBT for delusions—to examine their potential synergistic effects in SZ patients. In this case (Gottlieb et al. 2011), D-cycloserine (DCS) was selected both based on its potential ability to compensate for a proposed NMDA receptor hypofunction in SZ, and because DCS has been reported to facilitate extinction (Davis et al. 2006), and to enhance memory consolidation in SZ patients after a single dose (Goff et al. 2008). DCS also has complexities related to its propensity to produce rapid tolerance (Parnas et al. 2005; Quartermain et al. 1994), though once-weekly dosing for 8 weeks has been reported to reduce negative symptoms in SZ patients (Goff et al. 2008). In their study, Gottlieb et al. examined the effects of DCS (placebo or 50 mg) on the ability of two CBT sessions to reduce the intensity of delusions in 20 SZ patients; in this design, the active drug (DCS 50 mg) was administered prior to only one training session. The findings of this small study were complex—there was no overall effect of DCS on the beneficial effects of CBT for delusions, but a potential order effect with some DCS benefits among subjects receiving DCS before placebo (Gottlieb et al. 2011). Conceivably, a larger study, or one designed differently, might have detected more robust effects of DCS. One might also question whether the choice of DCS based on its neurochemical properties (to compensate for NMDA hypofunction) or primary neurocognitive effects (enhancing fear extinction) makes sense, in the search for a drug to enhance CBT effects in SZ. Conceivably, pro-extinction agents might be useful for reducing the perceived “threat” posed by a specific paranoid object, but it is not clear that such extinction would impact an underlying frontal dysfunction that is manifested more generally in a propensity for developing fixed, irrational beliefs.
5 Medication-Enhancement of Therapeutic Learning and Neurocognition: “Proof of Concept”
Cognitive deficits predict poor outcomes in a number of cognitive and vocationally oriented therapies (Becker et al. 1998; Green 1996; McGurk and Meltzer 2000; McGurk and Mueser 2004), and it thus seems parsimonious to suggest that patients will benefit the most if they are able to meet the cognitive demands of CTs. It would then follow that interventions that enhance a patient’s cognitive abilities should enable them to glean the most clinical benefit from CT. While there is ample experience from everyday clinical practice to support the notion that psychotherapeutic agents that reduce cognitively impairing symptoms—e.g., severe depression, anxiety, or psychosis—can enhance the benefits of psychotherapies, there has not yet been a robust “test” of whether drugs that specifically enhance neurocognition can enhance the benefits of CT in SZ patients.
Perhaps the closest “proof of concept” comes again from the use of DCS for its pro-extinction properties, but in this example, it was not used to help “extinguish” psychotic thinking, but rather to facilitate extinction of a specific phobia (Ressler et al. 2004). In this study, acrophobic subjects were treated with two brief virtual reality-based exposure sessions, separated by 1–2 weeks. Prior to each session, subjects took one pill of either placebo (n = 10), 50 mg DCS (n = 8), or 500 mg DCS (n = 9). When tested 1–2 weeks or 3 months later, height-related distress—both experimental and “real-world”—was significantly reduced in both active dose groups compared to the placebo group. Importantly, the ability of DCS to enhance the effects of CT on acrophobia was not thought to reflect a hypoglutamatergic basis of acrophobia, or even an “extinction deficit” in acrophobia. Rather, they were attributed to the ability of DCS to enhance the function of intact, healthy “extinction circuitry” in acrophobics, which is specifically “exercised” by a symptom-directed cognitive intervention. By combining a drug that enhances a specific cognitive process with a therapy that demands that process, patients were able to benefit more from that therapy. Certainly, we cannot assume that SZ patients will benefit from a therapeutic algorithm that reduced agoraphobia, or even that the type of “learning” enhanced by DCS in reducing height-related distress is relevant to the type of “learning” engaged during CT in SZ patients. Nonetheless, the findings of Ressler et al. (2004) support the concept that drugs that enhance a specific neurocognitive process can enhance the benefits of a psychotherapeutic intervention that places demands on that process.
6 Predicting Medication Effects on Neurocognitive Function in Individual Patients
It is apparent that many different forms of cognitive interventions might be useful in treating individuals with SZ, and that each of these different forms of therapy likely places “demands” on different neurocognitive and psychological processes. Furthermore, we can anticipate that the mechanisms of action will differ substantially across many different putative “pro-CT” agents. Because of the time and resources required to complete a full program of CT, it will be particularly valuable to be able to predict which patients will benefit most from which therapy, and which pro-CT medication. It is thus worth considering how one might make such predictions.
At a neurobiological level, the model for the efficacy of cognitive interventions in SZ comes primarily from its use in treating stroke syndromes: these interventions engage the normal physiological and anatomical properties of healthy brain circuits (e.g., in neighboring regions or parallel circuits) to restore or subsume the function of damaged circuit elements (cf. Taub et al. 2002). In fact, schizophrenia patients with the largest “reserve” of cortical function—particularly temporal lobe—are most likely to benefit from CT (Keshavan et al. 2011). An implication of the variability in neuroimaging and neuropathological findings in schizophrenia is that in many patients, portions of the cortico-striato-pallido-thalamic circuitry may remain relatively intact. The model proposed herein suggests that medications that enhance specific cognitive functions (e.g., attention, memory, reasoning, or processing speed) by acting on remaining healthy brain circuits (not on areas of neural dysfunction per se) might reasonably be expected to amplify the clinical benefits of cognitive interventions, even if these medications are clinically ineffective when administered without the demands of cognitive interventions (Fig. 1).
Fig. 1.

Schematic of theoretical and empirical rationale for predictive value of laboratory measures in identifying potential “pro-CT” drugs
One key step in predicting whether a patient might benefit from a particular “pro-CT” agent would be to identify evidence for medication-responsive, healthy or “spared” brain circuitry within any individual or biomarker-identified subgroup of SZ patients. Towards this end, specific neurophysiological, psychophysiological, or neurocognitive changes in response to a drug challenge may provide indication that “spared” healthy neural circuitry exists and can be a target for medication-enhanced CT. There is both a theoretical and an empirical basis for making such a prediction. Theoretically, if specific laboratory measures are regulated by elements of the cortico-striato-pallido-thalamic circuitry that also regulates neurocognitive functions important for CT, then drug-induced enhancement of laboratory-based performance would be expected to translate into a real-world enhancement of CT. In fact, independent of their specific neural substrates, if laboratory measures index basic psychological functions of value to CT—e.g., attention, vigilance, sensorimotor gating, etc.—one might expect that drug-enhanced laboratory performance might predict pro-CT effects of these drugs. As with any predictive model, this strategy would have limits of sensitivity and specificity, and drugs yielding “positive” findings might be clinically irrelevant based on many factors, including the propensity for tolerance, a potential negative impact on other cognitive processes (e.g., impulsivity), symptoms, or other forms of toxicity.
There is also empirical support that greater performance on specific laboratory-based measures should predict enhanced CT effects. For example, higher levels of mismatch negativity (MMN; discussed below) are associated with a positive therapeutic response to 12 weeks of social skills/cognitive rehabilitative therapy in SZ (Kawakubo et al. 2007). In many [but not all (e.g., Hasenkamp et al. 2011)] studies, higher levels of prepulse inhibition of startle (PPI; discussed below) are associated with higher performance on measures of executive functioning (Bitsios et al. 2006; Giakoumaki et al. 2006; Greenwood et al. 2012; Light et al. 2007a; van der Linden et al. 2006), which in turn predict greater benefit from some forms of CTs (Becker et al. 1998; Green 1996; McGurk and Meltzer 2000; McGurk and Mueser 2004). In fact, a recent study determined that pre-therapy levels of PPI were strongly correlated with symptomatic improvement after CBT in schizophrenia patients (Kumari et al. 2012). Thus, empirical evidence suggests an association between performance on specific laboratory measures, basic psychological processes of relevance to CTs, and clinical improvement resulting from CT in schizophrenia. There remains a conceptual gap, though we submit one well worth exploring: whether medications that increase an individual’s performance on these laboratory measures or in the basic neurocognitive processes of relevance to CTs, will improve that individual’s ability to benefit clinically from CTs. Certainly, given the complex neural regulation of even simple laboratory measures, neurocognition, CTs and SZ, one would anticipate that studies not specifically designed to test these relationships would identify both false positive and negative outcomes. For example, PPI appears to be enhanced in SZ patients by atypical APs (e.g., Swerdlow et al. 2006a), which may—or may not—directly enhance the therapeutic effects of CTs (discussed above).
7 Biomarkers?
As with any therapeutic intervention in complex disorders, it will be very important to identify biomarkers that predict an increased sensitivity to the ability of a drug to enhance CT. This biomarker profile might inform both the choice of drug and the predictive measure. For example, high levels of dopamine D3 receptor expression are associated with working memory-enhancing effects of the D3 agonist, pramipexole (Ersche et al. 2011), while individuals carrying the Val/Val alleles of the Val158Met COMT polymorphism exhibit greater sensitivity to the ability of tolcapone (Roussos et al. 2009) to enhance sensorimotor gating, and individuals with low basal levels of PPI are most sensitive to the PPI-enhancing effects of memantine (Swerdlow et al. 2009b). A patient carrying one or more of these predictive biomarkers could then be tested in a within-subject “challenge dose” design (placebo vs. active dose), and findings of drug-enhanced performance in one or more predictive measure would suggest that the requisite neural circuitry for such a drug effect is “spared,” and could be drug-activated in the service of CT. The patient might then be entered in a structured CT program with daily drug augmentation. While one might imagine reasons for continuing such “pro-CT” drugs beyond the course of the cognitive intervention, it is also conceivable that their therapeutic value would be maximized by administration concurrent with CT, and thus the value of limited versus long-term dosing of such medications would need to be assessed empirically (Fig. 2).
Fig. 2.
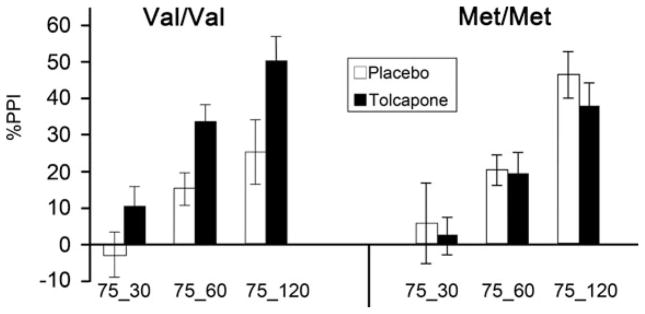
Biomarker predicting drug-enhanced PPI in normal subjects (figure modified from Roussos et al. 2009). %PPI is shown on Y-Axis; trial type (intensity in dB and prepulse interval in ms) is shown on X-axis. As in SZ patients, clinically healthy subjects carrying the Val allele of the Val158Met COMT polymorphism exhibit lower basal levels of PPI; in this important report, the authors demonstrated that the COMT inhibitor, tolcapone, increases PPI selectively among Val/Val individuals
Conceivably, some forms of CT might benefit most from enhanced performance in specific neurocognitive or neurophysiological processes, and might be matched according to such drug effects identified in any given patient. Parallel translational research activities might identify the neural mechanisms for drug effects on specific neurophysiological processes; prospective trials would identify the strongest biomarker- and laboratory measure-predictors of positive drug effects on specific forms of CT.
An overview of several “candidate” laboratory measures for identifying potential “pro-CT” drugs is provided below. This overview is not meant to be comprehensive, and it is clear that many other measures might have predictive value for such drugs.
7.1 PPI
Prepulse inhibition of startle (PPI) might be a good neurophysiological “biomarker” for predicting positive drug effects on neurophysiological processes and the therapeutic impact of CT. PPI is regulated by the cortico-striato-pallido-thalamic circuitry (cf. Swerdlow et al. 2008), reduced in schizophrenia patients (Braff et al. 1978; Swerdlow et al. 2006a), correlated with CT-relevant executive functions including working memory, attention, strategy formation, execution times, and degree of mental fatigue (Bitsios et al. 2006; Giakoumaki et al. 2006; Light et al. 2007a; van der Linden et al. 2006) and sensitive to acute drug effects in a manner that might be useful for predicting individualized drug sensitivities in patients (e.g., Swerdlow et al. 2006b, 2009a, b; Talledo et al. 2009; Vollenweider et al. 2006). Also, as noted above, PPI levels prior to CBT correlate significantly with symptomatic improvement in SZ patients (Kumari et al. 2012); this finding suggests that it is rational to speculate that drugs that increase basal PPI levels might enhance CBT outcome in SZ.
In PPI, a weak lead stimulus (prepulse) inhibits the magnitude of a startle response to an intense, abrupt stimulus occurring 30–120 ms later. On average, the amount of reflex inhibition generated by the prepulse is diminished in SZ patients, as reported by more than a dozen independent research groups on 4 continents (cf. Braff et al. 1978; Grillon et al. 1992; Kumari et al. 1999; Kunugi et al. 2007; Quednow et al. 2010; Swerdlow et al. 2006a; Weike et al. 2000). This PPI deficit appears to be particularly marked among patients carrying the Val158Met polymorphism (homozygous Val/Val) conferring high activity of the enzyme, catechol-O-methyl transferase (COMT) (Quednow et al. 2010), suggesting that a Val/Val COMT genotype might be a useful biomarker predicting pro-CT effects for PPI-enhancing agents.
With only 10–120 ms separating the prepulse and startling stimulus in the “uninstructed” PPI paradigm, PPI is generally viewed as a measure of automatic, preattentional inhibition (Swerdlow 1996). Surprisingly, in several [but not all (e.g., Hasenkamp et al. 2011)] studies, the amount of this “automatic” inhibition correlates significantly with higher cognitive measures, including measures of working memory (Fig. 1) and strategy formation, as well as measures of cognitive efficiency (Bitsios et al. 2006; Giakoumaki et al. 2006; Light et al. 2007a; van der Linden et al. 2006). The PPI-enhancing effects of tolcapone among Val/Val healthy control subjects are accompanied by significant increases in n-back and letter–number sequencing performance (Giakoumaki et al. 2008). Perhaps most surprising is the finding that higher levels of PPI among SZ outpatients significantly predict higher global functioning (Swerdlow et al. 2006a), though we view this relationship as correlative and not causal (i.e., greater neural dysfunction is associated both with more global functional impairment, and with reduced PPI) (Fig. 3).
Fig. 3.
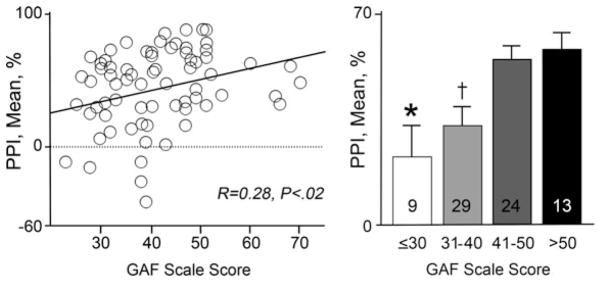
Relationship between levels of sensorimotor gating, as measured by %PPI, and global functioning, as measured by GAF in male schizophrenia outpatients (from Swerdlow et al. 2006a). “N” indicated by numbers inside bars; *P<.004 vs “41–50” and vs “>50.” †P<.01 vs “41–50” and vs “>50” by Fisher protected least-significant difference. Clearly, the causal pathway from higher PPI to higher GAF is indirect, but these findings suggest that the ability of a drug to enhance PPI in schizophrenia patients might be one rational “signal” for selecting compounds with the potential for enhancing the functional impact of cognitive interventions
PPI is regulated in both laboratory animals and humans by neural circuits connecting portions of the prefrontal cortex and mesial temporal lobes, with subcortical systems extending from the basal ganglia to the primary startle circuit in the pons (cf. Swerdlow et al. 2001). Drugs acting at prominent nodes in this circuitry, particularly via changes in DA, 5-HT, and NMDA neurotransmission, have very potent effects on PPI that have been studied extensively in rodents, and more recently in humans (cf. Swerdlow et al. 2008).
In healthy individuals under specific conditions, some drugs increase PPI; of these, clozapine (Vollenweider et al. 2006) and quetiapine (Swerdlow et al. 2006b) are atypical antipsychotics, which also increase PPI in SZ patients (Swerdlow et al. 2006a). Other PPI-increasing drugs come from drug classes not intuitively associated with SZ therapeutics: NMDA antagonists and catecholamine agonists. In this regard, it is important to not categorically reject candidate drug classes based on hypotheses for the pathogenesis of SZ. For example, amantadine has both DA agonist and NMDA antagonist properties, and has been safely used in SZ patients for over four decades (Kelly and Abuzzahab 1971), despite prevailing hypotheses linking SZ to excessive DA activity and deficient NMDA activity. The inverse is also true: as noted above, atypical antipsychotics increase PPI in healthy subjects and SZ patients, yet it is not known whether these drugs contribute to the benefits of CTs, and if so, whether they do so via a specific effect on neurocognition versus a secondary result of reduced psychosis.
7.2 Electro-Encephalographic Measures
Two electro-encephalographic (EEG)-based neurophysiological phenotypes—mismatch negativity (MMN) and gamma band synchronization (GBS)—may also serve as “biomarkers” to predict sensitivity to pro-cognitive and therapeutic drug effects, and/or therapeutic response to CT in SZ patients.
MMN is an auditory ERP component elicited 50–150 ms after a sequence of repetitive standard sounds is interrupted infrequently by deviant, “oddball” stimuli. MMN is the first measurable brain response component that differentiates between frequent and deviant auditory stimuli and reflects the properties of an automatic, memory-based comparison process (cf. Turetsky et al. 2007). It is rapidly assessed, impaired in SZ and highly stable; MMN deficits in SZ patients are of large effect size (d > 1.0), and after 1 year, MMN reliability coefficients (ICC’s) in patients are ~0.90 (Light and Braff 2005a). While MMN is predominantly automatic and “preattentional,” MMN is strongly associated with neurocognition and psychosocial functioning in healthy subjects (Light et al. 2007b) and SZ patients (Kawakubo et al. 2007; Kawakubo and Kasai 2006; Light and Braff 2005a, b; Wynn et al. 2010). MMN is regulated by forebrain circuitry, and may be particularly sensitive to NMDA activity: MMN is disrupted in nonhuman primates by phencyclidine, and in healthy subjects by ketamine, but existing evidence suggests that MEM is associated with increased MMN in healthy subjects (Korostenskaja et al. 2007). In turn, increased MMN is associated with a positive therapeutic response to 12 weeks of social skills/cognitive rehabilitative therapy in SZ (Kawakubo et al. 2007).
Synchronous neural oscillations (GBS) in the 30–80 Hz range (centered near 40 Hz) appear to reflect a fundamental brain resonance frequency that is critical for cortico-cortical communication and the large-scale integration of distributed neural functions (Uhlhaas and Singer 2006). EEG evidence demonstrates that the automatic entrainment (“synchronization”) of gamma band oscillations to 40 Hz auditory stimuli is deficient and correlates with working memory in SZ patients (Light et al. 2006). Relatively little is known regarding the neural regulation of GBS; gamma frequency oscillations appear to be enhanced by ketamine in both mice (Lazarewicz et al. 2010) and healthy humans (Hong et al. 2010).
MMN and GBS are EEG-based measures of processes underlying automatic, preattentive information processing that are impaired in SZ, and associated with neurocognition in HCS (MMN) and SZ patients (MMN and GBS). There is evidence that NMDA regulates both MMN and GBS, that memantine increases MMN, and that greater MMN predicts a positive therapeutic response to cognitive rehabilitation in SZ.
7.3 MATRICS Consensus Cognitive Battery (MCCB)
With the recognition of cognitive deficits as a core feature of SZ and its major impact on function, an NIMH initiative brought together a group of experts from the Neurocognition Subcommittee for the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) to identify the most important domains of cognitive deficits in SZ. Their consensus opinion was that working memory, attention/vigilance, verbal learning and memory, visual learning and memory, reasoning and problem solving, speed of processing, and social cognition comprised the primary domains of cognitive deficits in SZ (Green et al. 2000).
The MATRICS Consensus Cognitive Battery was developed to evaluate these key cognitive domains relevant to SZ (Kern et al. 2008; Nuechterlein et al. 2008). It was designed as an outcome measure for clinical trials of cognition-enhancing drugs for SZ, an outcome measure for studies of cognitive remediation, a measure of cognitive change in repeated testing applications, and a cognitive reference point for nonintervention studies of SZ and related disorders, and is accepted by the FDA as a primary endpoint measure for clinical trials targeting cognition in schizophrenia.
The MCCB includes ten tests that assess seven cognitive domains: speed of processing, attention/vigilance, working memory (verbal and nonverbal), verbal learning, visual learning, reasoning and problem solving, and social cognition. In a variety of multi-site clinical trials, it has demonstrated sensitivity to cognitive deficits in all domains, excellent test–retest reliability and inter-site reliability, and is highly correlated with measures of functional capacity (Buchanan et al. 2011; Keefe et al. 2011b). The use of the MCCB as a validated measure for studies evaluating cognition-enhancing drugs is well underway and the use of this standardized measure will be critical for the comparison of results between studies evaluating the ability of a drug to enhance the therapeutic effects of CT.
8 Examples of Candidate “Pro-CT” Drugs
A variety of candidate pro-cognitive agents might warrant assessment for their ability to enhance the functional impact of cognitive interventions in SZ. Two classes of drugs will be discussed here as examples. Importantly, many other classes of compounds might certainly warrant investigation, but as reviewed by Barch (2010), compelling support for one drug class over another at this point is lacking.
8.1 Direct and Indirect Catecholaminergic Agents
Several direct and indirect catecholaminergic agonists enhance neurocognitive performance in clinically normal subjects. For example, methylphenidate (Clark et al. 1986; Elliott et al. 1997; Mehta et al. 2000), amphetamine (Mattay et al. 2000, 2003), bromocriptine (Kimberg et al. 1997; Luciana et al. 1992, 1998; Luciana and Collins 1997), and pergolide (Kimberg and D’Esposito 2003; Müller et al. 1998) enhance working memory in healthy subjects. Perhaps the most-studied of the catecholaminergic drugs is the indirect DA agonist, amphetamine; it has been reported to enhance performance on several neurocognitive measures both in medicated SZ patients (e.g., Barch and Carter 2005) and in unmedicated individuals with schizotypal personality disorder (Kirrane et al. 2000; Siegel et al. 1996; Wonodi et al. 2006). Amphetamine and other DA agonists may be particularly effective in enhancing neurocognitive performance in individuals with low basal performance levels (Kimberg et al. 1997; Kimberg and D’Esposito 2003; Mattay et al. 2000, 2003; Mehta et al. 2001; Swerdlow et al. 2011) and in individuals carrying certain genetic biomarkers or related phenotypes (Ersche et al. 2011; Fleming et al. 1995; Giakoumaki et al. 2008; Mattay et al. 2003; Roussos et al. 2009).
Among other laboratory measures, PPI is sensitive to the positive effects of pro-catecholamine agents, in a manner that might predict CT-enhancing effects of these drugs. Catecholaminergic drugs that increase PPI in healthy subjects include amphetamine (Talledo et al. 2009); the COMT inhibitor, tolcapone (Giakoumaki et al. 2008; Roussos et al. 2009) and the D3 agonist, pramipexole (Swerdlow et al. 2009a). Similar to what is observed with several neurocognitive measures, the PPI-enhancing effects of catecholaminergic drugs can be either subgroup—or biomarker-sensitive—e.g., in individuals carrying the Val/Val alleles of the Val158Met COMT polymorphism (Giakoumaki et al. 2008; Roussos et al. 2009) or its associated phenotypes (Talledo et al. 2009)—or most marked among individuals with low basal performance levels (Fig. 4). Theoretically, this might suggest that amphetamine would be most effective in increasing PPI (and its associated neurocognitive functions) in SZ patients, who as a group exhibit low basal PPI, particularly among those patients carrying a Val allele in the COMT polymorphism (Quednow et al. 2010).
Fig. 4.
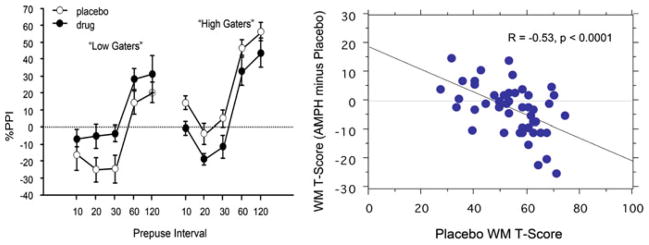
“Rate-dependent” effects of AMPH (20 mg po) on measures of %PPI (left; Talledo et al. 2009) and working memory (MCCB domain T-scores; Swerdlow et al. 2011) in clinically normal adults. AMPH-enhanced PPI was detected in specific subgroups of healthy women with PPI- or personality-based phenotypes associated with the COMT Val/Val allele. Working memory-enhancing effects of AMPH were detected among a subgroup of 50 healthy men and women characterized by low-basal working memory performance. Schizophrenia patients as a group have reduced PPI and impaired working memory; thus, based solely on the rate-dependent effects detected in normal adults, schizophrenia patients would be predicted to show PPI- and working memory-enhancing effects of acute AMPH treatment. The latter finding was already reported by Barch and Carter (2005)
Certainly, there are rational arguments against the indiscriminate use of pro-catecholaminergic drugs in SZ, and some drugs [e.g., tolcapone (cf. Haasio 2010)] carry other medical contraindications. However, DA agonists have been used safely in schizophrenia for many years (e.g., Benkert et al. 1995; Kasper et al. 1997), and—as suggested by Barch (2010) and others—their use in concert with antipsychotics might prevent potentially deleterious effects of subcortical D2 activation, while permitting potentially beneficial activation of cortical D1 receptors. Whether there is a role for some of these agents in biomarker-identified subpopulations, in time-limited combinations with CT and antipsychotic agents, remains an empirical question.
8.2 Low-Potency NMDA Antagonists
NMDA antagonists increase PPI in humans. While the potent competitive NMDA antagonist phencyclidine (PCP) disrupts PPI in both rodents and nonhuman primates (Linn and Javitt 2001; Mansbach and Geyer 1989), low-potency NMDA antagonists that increase PPI in healthy human subjects include amantadine (Swerdlow et al. 2002) and memantine (Swerdlow et al. 2009b) (Fig. 5). Amantadine’s PPI-enhancing effects were detected only under conditions where subjects were instructed to rate the intensity of the startling stimulus (Swerdlow et al. 2002), suggesting that the mechanisms engaged by amantadine were attentionally dependent. Consistent with this, when studied in a sample of 19 healthy men, memantine enhanced PPI only at relatively long prepulse intervals (120 ms), which are known to be attentionally sensitive, compared with intervals below 60 ms (Swerdlow et al. 2009b).
Fig. 5.
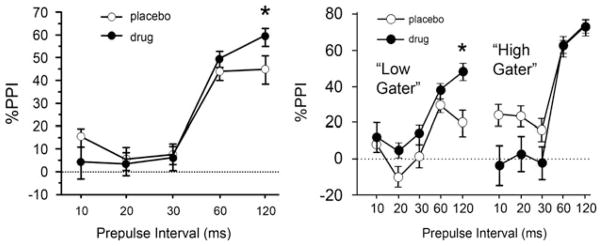
In healthy men, PPI was significantly increased by 20 mg memantine at 120 ms prepulse intervals (left). When subjects were divided into those in the upper or lower 50% of baseline PPI values (right, “high” and “low”, respectively), ANOVA revealed PPI-enhancing effects of memantine only among the “low gaters” (Swerdlow et al. 2009b)
Interestingly, memantine’s PPI-enhancing effects appear to be most potent among individuals with phenotypes linked to the Val/Val alleles of the Val158Met COMT polymorphism (Giakoumaki et al. 2008; Golimbet et al. 2007; Roussos et al. 2009), suggesting a potential biomarker for identifying an enriched treatment cohort. In addition to PPI, memantine challenge in healthy subjects enhances other markers of cortico-striato-pallido-thalamic and functional deficits in schizophrenia, including MMN (Korostenskaja et al. 2007; Light and Braff 2005a, b), and in preliminary studies appears to increase working memory performance in some individuals (Swerdlow et al. 2010). Consistent with the model presented herein, the ability of a memantine “challenge” to enhance PPI or other specific neurophysiological measures in a patient could provide evidence for residual, healthy circuitry that could be recruited to enhance the effectiveness of CT.
As with other putative “pro-cognitive” agents, memantine does not appear to have dramatic positive effects on symptoms or function in SZ patients, without the concomitant use of cognitive interventions. A recent double-blind study did report large reductions in positive and negative symptoms (d = 1.38–3.33) and improved Mini-Mental State Exam scores after 12 weeks of memantine (20 mg/d; n = 10) versus placebo (PBO; n = 11) added to clozapine (de Lucena et al. 2009). However, an earlier 8-week double-blind, study of memantine (20 mg/d; n = 70) vs. PBO (n = 68) added to atypical antipsychotics detected no change in positive or negative symptoms or Brief Assessment of Cognition in Schizophrenia scores (Lieberman et al. 2009), but found that memantine was associated with more adverse events (adverse events memantine vs. PBO = 8.7% vs. 6.0%) and treatment discontinuation due to AEs (11.6% vs. 3.0%). A smaller, 6-week, open label study of memantine (5–20 mg/d) in symptomatic SZ inpatients reported a significant improvement in positive and negative symptoms but not cognitive performance, with no adverse events (Krivoy et al. 2008). Three case reports (cf. Zdanys and Tampi 2008) described beneficial effects of memantine (5–10 mg/d) in SZ patients, with reductions in negative symptoms and functional impairment and no adverse events; in two reports, symptoms returned on memantine discontinuation and resolved again after restarting memantine.
As with catecholaminergic agents, the utility of an NMDA antagonist in SZ patients would seem counterintuitive, based on prevailing models for NMDA hypofunction in SZ. In truth, we can only speculate about the neural mechanisms that might underlie such utility. Memantine enhances cortical metabolic efficiency (Willenborg et al. 2011) and protects cerebral function under conditions of hypoglycemic challenge in healthy adults, and increased frontal and parietal cortical glucose utilization after memantine correlates with increased Mini-Mental State performance among individuals with traumatic brain injury (Kim et al. 2010). Conceivably, such properties might represent a basis for bolstering frontal cortical function—and thereby the neurocognitive resources necessary to successfully engage in CTs—particularly among patients whose frontal cortical efficiency is already taxed by risk factors related to COMT status or schizophrenia-related pathological changes. While it is always wise to consider whether drugs with NMDA antagonist properties might have deleterious consequences in SZ patients, there is evidence that, in fact, memantine is neuroprotective (Kornhuber et al. 1994; Lipton 2006; Rogawski and Wenk 2003), well-tolerated by SZ patients (de Lucena et al. 2009; Krivoy et al. 2008; Lieberman et al. 2009; Zdanys and Tampi 2008) and has been safely used in many millions of patients, including elderly, frail clinical populations (cf. Jones 2010). Conceivably, in addition to memantine, “next generation” low-affinity NMDA antagonists would warrant investigation in studies of neurophysiological and neurocognitive measures of relevance to cortico-striato-pallido-thalamic function and SZ, to assess their potential as pro-CT candidates.
9 Summary
Current pharmacotherapy for SZ primarily targets positive symptoms but does not significantly impact either the substantial neurocognitive or life functional impairments associated with this disorder. It is not surprising that current medications yield limited symptomatic relief, in a disorder caused by disturbances in early brain development producing pervasive, widely distributed and variable patterns of neuropathology. In contrast, controlled studies of a variety of cognitive interventions in SZ patients have demonstrated modest yet significant neurocognitive and functional gains. In the absence of such CTs, drugs with putative “pro-cognitive” properties have failed to benefit patients with SZ. We propose here that a rational strategy for future therapeutic development in SZ is to identify drugs that can enhance the therapeutic impact of CTs in SZ; such “pro-CT effects” would result from the ability of these drugs to engage spared, healthy brain circuits in SZ patients, in the service of basic neurocognitive demands of particular forms of CT. We propose an experimental approach for identifying such drugs via the use of single drug challenges in concert with laboratory-based neurophysiological and neurocognitive measures, in patients with particular biomarkers predicting drug sensitivity. Among the candidate “pro-CT” drugs are ones with properties—e.g., pro-catecholaminergic or NMDA-antagonistic—that, on the surface, would seem counter-productive in the treatment of SZ. A rationale is discussed—supported by convergent empirical findings—that suggests that evolving strategies for new SZ therapeutics should not be based on imprecise models for the widespread and variable brain dysfunction that occurs because of this disorder, but should be based instead on models that identify and engage spared, healthy brain functions that persists despite it.
Acknowledgments
The authors acknowledge the valuable assistance of Ms. Maria Bongiovanni and Ms. Jo Talledo in the preparation of this manuscript. NRS was supported by MH093453, MH059803; MH042228 and a PALA Award from the VISN-22 MIRECC; HHC was supported by 5T32MH018399 – 25 and MH093453; ET was supported by MH080150.
References
- Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edger CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Aparacio-Legarza MI, Cutts AJ, Davis B, Reynolds GP. Deficits in [3H]D-aspartate binding to glutamate uptake sites in striatal and accumbens tissue in patients with schizophrenia. Neurosci Lett. 1997;232:13–16. doi: 10.1016/s0304-3940(97)00563-6. [DOI] [PubMed] [Google Scholar]
- Barch DM. Pharmacological strategies for enhancing cognition in schizophrenia. In: Swerdlow N, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Vol. 4. Springer; Heidelberg: 2010. pp. 43–96. [DOI] [PubMed] [Google Scholar]
- Barch DM, Carter CS. Amphetamine improves cognitive function in medicated individuals with schizophrenia and in healthy volunteers. Schizophr Res. 2005;77:43–58. doi: 10.1016/j.schres.2004.12.019. [DOI] [PubMed] [Google Scholar]
- Baxter LR, Schwartz JM, Bergman KS, Szuba MP, Guze BH, Mazziotta JC, Alazraki A, Selin CE, Ferng HK, Munford P, Phelps ME. Caudate glucose metabolic rate changes with both drug and behavior therapy for obsessive-compulsive disorder. Arch Gen Psychiatry. 1992;49:681–689. doi: 10.1001/archpsyc.1992.01820090009002. [DOI] [PubMed] [Google Scholar]
- Becker DR, Drake RE, Bond GR, Xie H, Dain BJ, Harrison K. Job terminations among persons with severe mental illness participating in supported employment. Commun Ment Health J. 1998;34:71–82. doi: 10.1023/a:1018716313218. [DOI] [PubMed] [Google Scholar]
- Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology. 2010;35:239–257. doi: 10.1038/npp.2009.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert O, Müller-Siecheneder F, Wetzel H. Dopamine agonists in schizophrenia: a review. Eur Neuropsychopharmacol. 1995;5(Suppl):43–53. doi: 10.1016/0924-977x(95)00022-h. [DOI] [PubMed] [Google Scholar]
- Bitsios P, Giakoumaki SG, Theou K, Frangou S. Increased prepulse inhibition of the acoustic startle response is associated with better strategy formation and execution times in healthy males. Neuropsychologia. 2006;44:2494–2499. doi: 10.1016/j.neuropsychologia.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Brabban A, Tai S, Turkington D. Predictors of outcome in brief cognitive behavior therapy for schizophrenia. Schizophr Bull. 2009;35:859–864. doi: 10.1093/schbul/sbp065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15:339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Brown GG, Thompson WK. Functional brain imaging in schizophrenia: Selected results and methods. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 181–214. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Javitt DC, Marder SR, Schooler NR, Gold JM, McMahon RP, Heresco-Levy U, Carpenter WT. The cognitive and negative symptoms in schizophrenia trial (CONSIST): the efficacy of glutamatergic agents for negative symptoms and cognitive impairments. Am J Psychiatry. 2007;164:1593–1602. doi: 10.1176/appi.ajp.2007.06081358. [DOI] [PubMed] [Google Scholar]
- Buchanan RW, Keefe RS, Umbricht D, Green MF, Laughren T, Marder SR. The FDA-NIMH-MATRICS guidelines for clinical trial design of cognitive-enhancing drugs: What do we know 5 years later? Schizophr Bull. 2011;37:1209–1017. doi: 10.1093/schbul/sbq038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CR, Geffen GM, Geffen LB. Role of monoamine pathways in the control of attention: effects of droperidol and methylphenidate on normal adult humans. Psychopharmacology. 1986;90:28–34. doi: 10.1007/BF00172867. [DOI] [PubMed] [Google Scholar]
- Cruz DA, Weaver CL, Lovallo EM, Melchitzky DS, Lewis DA. Selective alterations in postsynaptic markers of chandelier cell inputs to cortical pyramidal neurons in subjects with schizophrenia. Neuropsychopharmacology. 2009;34:2112–2124. doi: 10.1038/npp.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Ressler K, Rothbaum BO, Richardson R. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol Psychiatry. 2006;60:369–375. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- de Lange FP, Koers A, Kalkman JS, Bleijenberg G, Hagoort P, van der Meer JW, Toni I. Increase in prefrontal cortical volume following cognitive behavioural therapy in patients with chronic fatigue syndrome. Brain. 2008;131:2172–2180. doi: 10.1093/brain/awn140. [DOI] [PubMed] [Google Scholar]
- de Lucena D, Fernandes BS, Berk M, Dodd S, Medeiros DW, Pedrini M, Kunz M, Gomes FA, Giglio LF, Lobato MI, Belmonte-de-Abreu PS, Gama CS. Improvement of negative and positive symptoms in treatment-refractory schizophrenia: a double-blind, randomized, placebo-controlled trial with memantine as add-on therapy to clozapine. J Clin Psychiatry. 2009;70:1416–1423. doi: 10.4088/JCP.08m04935gry. [DOI] [PubMed] [Google Scholar]
- Dean B, Boer S, Gibbons A, Money T, Scarr E. Recent advances in postmortem pathology and neurochemistry in schizophrenia. Curr Opin Psychiatry. 2009;22:154–160. doi: 10.1097/YCO.0b013e328323d52e. [DOI] [PubMed] [Google Scholar]
- Demily C, Franck N. Cognitive remediation: a promising tool for the treatment of schizophrenia. Expert Rev Neurother. 2008;8:1029–1036. doi: 10.1586/14737175.8.7.1029. [DOI] [PubMed] [Google Scholar]
- Dick DM, Riley B, Kendler KS. Nature and nurture in neuropsychiatric genetics: where do we stand? Dialogues Clin Neurosci. 2010;12:7–23. doi: 10.31887/DCNS.2010.12.1/ddick. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan RJ, Fletcher P, Frith CD, Friston KJ, Frackowiak RS, Grasby PM. Dopaminergic modulation of impaired cognitive activation in the anterior cingulate cortex in schizophrenia. Nature. 1995;378:180–182. doi: 10.1038/378180a0. [DOI] [PubMed] [Google Scholar]
- Eack SM, Greenwald DP, Hogarty SS, Cooley SJ, DiBarry AL, Montrose DM, Keshavan MS. Cognitive enhancement therapy for early-course schizophrenia: effects of a two-year randomized controlled trial. Psychiatr Serv. 2009;60:1468–1476. doi: 10.1176/appi.ps.60.11.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R, Sahakian BJ, Matthews K, Bannerjea A, Rimmer J, Robbins TW. Effects of methylphenidate on spatial working memory and planning in healthy young adults. Psychopharmacology. 1997;131:196–206. doi: 10.1007/s002130050284. [DOI] [PubMed] [Google Scholar]
- Ersche KD, Roiser JP, Lucas M, Domenici E, Robbins TW, Bullmore ET. Peripheral biomarkers of cognitive response to dopamine receptor agonist treatment. Psychopharmacology (Berl) 2011;214:779–789. doi: 10.1007/s00213-010-2087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszdon JM, Cardenas AS, Bryson GJ, Bell MD. Predictors of remediation success on a trained memory task. J Nerv Ment Dis. 2005;193:602–608. doi: 10.1097/01.nmd.0000177790.23311.ba. [DOI] [PubMed] [Google Scholar]
- Fiszdon JM, Choi J, Bryson GJ, Bell MD. Impact of intellectual status on response to cognitive task training in patients with schizophrenia. Schizophr Res. 2006;87:261–269. doi: 10.1016/j.schres.2006.04.011. [DOI] [PubMed] [Google Scholar]
- Fleming K, Bigelow LB, Weinberger DR, Goldberg TE. Neuropsychological effects of amphetamine may correlate with personality characteristics. Psychopharmacol Bull. 1995;31:357–362. [PubMed] [Google Scholar]
- Fox K. Experience-dependent plasticity mechanisms for neural rehabilitation in somatosensory cortex. Philos Trans R Soc Lond Series B Biol Sci. 2009;364:369–381. doi: 10.1098/rstb.2008.0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giakoumaki SG, Bitsios P, Frangou S. The level of prepulse inhibition in healthy individuals may index cortical modulation of early information processing. Brain Res. 2006;1078:168–170. doi: 10.1016/j.brainres.2006.01.056. [DOI] [PubMed] [Google Scholar]
- Giakoumaki SG, Roussos P, Bitsios P. Improvement of prepulse inhibition and executive function by the COMT inhibitor tolcapone depends on COMT Val158Met polymorphism. Neuropsychopharmacology. 2008;33:3058–3068. doi: 10.1038/npp.2008.82. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. D-cycloserine added to clozapine for patients with schizophrenia. Am J Psychiatry. 1996;153:1628–1630. doi: 10.1176/ajp.153.12.1628. [DOI] [PubMed] [Google Scholar]
- Goff DC, Tsai G, Levitt J, Amico E, Manoach D, Schoenfeld DA, Hayden DL, McCarley R, Coyle JT. A placebo-controlled trial of D-cycloserine added to conventional neuroleptics in patients with schizophrenia. Arch Gen Psychiatry. 1999;56:21–27. doi: 10.1001/archpsyc.56.1.21. [DOI] [PubMed] [Google Scholar]
- Goff DC, Keefe R, Citrome L, Davy K, Krystal JH, Large C, Thompson TR, Volavka J, Webster EL. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol. 2007;27:582–589. doi: 10.1097/jcp.0b013e31815abf34. [DOI] [PubMed] [Google Scholar]
- Goff DC, Cather C, Gottlieb JD, Evins AE, Walsh J, Raeke L, Otto MW, Schoenfeld D, Green MF. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr Res. 2008;106:320–327. doi: 10.1016/j.schres.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golimbet VE, Alfimova MV, Gritsenko IK, Ebstein RP. Relationship between dopamine system genes and extraversion and novelty seeking. Neurosci Behav Physiol. 2007;37:601–606. doi: 10.1007/s11055-007-0058-8. [DOI] [PubMed] [Google Scholar]
- Gottlieb JD, Cather C, Shanahan M, Creedon T, Macklin EA, Goff DC. D-cycloserine facilitation of cognitive behavioral therapy for delusions in schizophrenia. Schizophr Res. 2011;131:69–74. doi: 10.1016/j.schres.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm E, McQuaid JR, McClure FS, Link PC, Perivoliotis D, Gottlieb JD, Patterson TL, Jeste DV. Randomized controlled trial of cognitive behavioral social skills training for older people with schizophrenia: 12-month follow-up. J Clin Psychiatry. 2007;68:730–737. doi: 10.4088/jcp.v68n0510. [DOI] [PubMed] [Google Scholar]
- Grant PM, Huh GA, Perivoliotis D, Stolar NM, Beck AT. Randomized trial to evaluate the efficacy of cognitive therapy for low-functioning patients with schizophrenia. Arch Gen Psychiatry. 2012;69:121–127. doi: 10.1001/archgenpsychiatry.2011.129. [DOI] [PubMed] [Google Scholar]
- Green MF. What are the functional consequences of neurocognitive deficits in schizophrenia? Am J Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green MF. Cognition, drug treatment, and functional outcome in schizophrenia: A tale of two transitions. Am J Psychiatry. 2007;164:992–994. doi: 10.1176/ajp.2007.164.7.992. [DOI] [PubMed] [Google Scholar]
- Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26:119–136. doi: 10.1093/oxfordjournals.schbul.a033430. [DOI] [PubMed] [Google Scholar]
- Green MF, Kern RS, Heaton RK. Longitudinal studies of cognition and functional outcome in schizophrenia: implications for MATRICS. Schizophr Res. 2004;72:41–51. doi: 10.1016/j.schres.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Greenwood TA, Light GA, Swerdlow NR, Radant AD, Braff DL. Association analysis of 94 candidate genes and schizophrenia-related endophenotypes. PLoS One. 2012;7(1):e29630. doi: 10.1371/journal.pone.0029630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Ameli R, Charney DS, Krystal J, Braff D. Startle gating deficits occur across prepulse intensities in schizophrenic patients. Biol Psychiatry. 1992;32:939–943. doi: 10.1016/0006-3223(92)90183-z. [DOI] [PubMed] [Google Scholar]
- Gur RE, Keshavan MS, Lawrie SM. Deconstructing psychosis with human brain imaging. Schizophr Bull. 2007;33:921–931. doi: 10.1093/schbul/sbm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenkamp W, Kelley M, Egan G, Green A, Wilcox L, Boshoven W, Lewison B, Duncan E. Lack of relationship between acoustic startle and cognitive variables in schizophrenia and control subjects. Psychiatry Res. 2011;187:324–328. doi: 10.1016/j.psychres.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haasio K. Toxicology and safety of COMT inhibitors. Int Rev Neurobiol. 2010;95:163–189. doi: 10.1016/B978-0-12-381326-8.00007-7. [DOI] [PubMed] [Google Scholar]
- Haut KM, Lim KO, MacDonald A. Prefrontal cortical changes following cognitive training in patients with chronic schizophrenia: effects of practice, generalization and specificity. Neuropsychopharmacology. 2010;35:1850–1859. doi: 10.1038/npp.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C. Hippocampal pathology in schizophrenia. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 529–553. [DOI] [PubMed] [Google Scholar]
- Heckers S, Rauch SL, Goff D, Savage CR, Schacter DL, Fischman AJ, Alpert NM. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998;12:426–445. doi: 10.1037//0894-4105.12.3.426. [DOI] [PubMed] [Google Scholar]
- Hong LE, Summerfelt A, Buchanan RW, O’Donnell P, Thaker GK, Weiler MA, Lahti AC. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35:632–640. doi: 10.1038/npp.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Egerton A, Allan V, McGuire P, Stokes P, Kapur S. Mechanisms underlying psychosis and antipsychotic treatment response in schizophrenia: insights from PET and SPECT imaging. Curr Pharm Des. 2009;15:2550–2559. doi: 10.2174/138161209788957528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RW. A review comparing the safety and tolerability of memantine with the acetyl-cholinesterase inhibitors. Int J Geriatr Psychiatry. 2010;25:547–553. doi: 10.1002/gps.2384. [DOI] [PubMed] [Google Scholar]
- Kasper S, Barnas C, Heiden A, Volz HP, Laakmann G, Zeit H, Pfolz H. Pramipexole as adjunct to haloperidol in schizophrenia. Safety and efficacy. Eur Neuropsychopharmacol. 1997;7:65–70. doi: 10.1016/s0924-977x(96)00393-8. [DOI] [PubMed] [Google Scholar]
- Kawakubo Y, Kasai K. Support for an association between mismatch negativity and social functioning in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1367–1368. doi: 10.1016/j.pnpbp.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Kawakubo Y, Kamio S, Nose T, Iwanami A, Nakagome K, Fukuda M, Kato N, Rogers MA, Kasai K. Phonetic mismatch negativity predicts social skills acquisition in schizophrenia. Psychiatry Res. 2007;152:261–265. doi: 10.1016/j.psychres.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Keefe RSE, Harvey PD. Cognitive impairment in schizophrenia. In: Geyer MA, Gross G, editors. Novel anti-schizophrenia treatments. Handbook of experimental pharmacology. 2012. [DOI] [PubMed] [Google Scholar]
- Keefe RSE, Buchanan RW, Marder SR, Schooler NR, Dugar A, Zivkov M, Stewart M. Clinical trials of potential cognitive-enhancing drugs in schizophrenia: what have we learned so far? Schizophr Bull. 2011a doi: 10.1093/schbul/sbr153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe RSE, Fox KH, Harvey PD, Cucchiaro J, Siu C, Loebel A. Characteristics of the MATRICS consensus cognitive battery in a 29-site antipsychotic schizophrenia clinical trial. Schizophr Res. 2011b;125:161–168. doi: 10.1016/j.schres.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Keller TA, Just MA. Altering cortical connectivity: remediation-induced changes in the white matter of poor readers. Neuron. 2009;64:624–631. doi: 10.1016/j.neuron.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly JT, Abuzzahab FS., Sr The antiparkinson properties of amantadine in drug-induced parkinsonism. J Clin Pharmacol New Drugs. 1971;11:211–214. [PubMed] [Google Scholar]
- Kern RS, Nuechterlein KH, Green MF, Baade LE, Fenton WS, Gold JM, Keefe RS, Mesholam-Gately R, Mintz J, Seidman LJ, Stover E, Marder SR. The MATRICS consensus cognitive battery: part 2. Co-norming and standardization. Am J Psychiatry. 2008;165:214–220. doi: 10.1176/appi.ajp.2007.07010043. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Eack SM, Wojtalik JA, Prasad KM, Francis AN, Bhojraj TS, Greenwald DP, Hogarty SS. A broad cortical reserve accelerates response to cognitive enhancement therapy in early course schizophrenia. Schizophr Res. 2011;130:123–129. doi: 10.1016/j.schres.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler LP, Walker E, Vega EM. Dopamine receptors in the brains of schizophrenia patients: a meta-analysis of the findings. Behav Pharmacol. 2001;12:355–371. doi: 10.1097/00008877-200109000-00007. [DOI] [PubMed] [Google Scholar]
- Kessler RM, Woodward ND, Riccardi P, Li R, Ansari MS, Anderson S, Dawant B, Zald D, Meltzer HY. Dopamine D2 receptor levels in striatum, thalamus, substantia nigra, limbic regions, and cortex in schizophrenic subjects. Biol Psychiatry. 2009;65:1024–1031. doi: 10.1016/j.biopsych.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YW, Shin JC, An YS. Changes in cerebral glucose metabolism in patients with posttraumatic cognitive impairment after memantine therapy: a preliminary study. Ann Nuclei Med. 2010;24:363–369. doi: 10.1007/s12149-010-0360-3. [DOI] [PubMed] [Google Scholar]
- Kimberg DY, D’Esposito M. Cognitive effects of the dopamine receptor agonist pergolide. Neuropsychologia. 2003;41:1020–1027. doi: 10.1016/s0028-3932(02)00317-2. [DOI] [PubMed] [Google Scholar]
- Kimberg DY, D’Esposito M, Farah MJ. Effects of bromocriptine on human subjects depend on working memory capacity. NeuroReport. 1997;8:381–385. doi: 10.1097/00001756-199711100-00032. [DOI] [PubMed] [Google Scholar]
- Kirrane RM, Mitropoulou V, Nunn M, New AS, Harvey PD, Schopick F, Silverman J, Siever LJ. Effects of amphetamine on visuospatial working memory performance in schizophrenia spectrum personality disorder. Neuropsychopharmacology. 2000;22:14–18. doi: 10.1016/S0893-133X(99)00075-5. [DOI] [PubMed] [Google Scholar]
- Klingberg S, Wittorf A, Herrlich J, Wiedemann G, Meisner C, Buchkremer G, Frommann N, Wölwer W. Cognitive behavioural treatment of negative symptoms in schizophrenia patients: study design of the TONES study, feasibility and safety of treatment. Eur Arch Psychiatry Clin Neurosci. 2009;259:S149–S154. doi: 10.1007/s00406-009-0047-8. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Weller M, Schoppmeyer K, Riederer P. Amantadine and memantine are NMDA receptor antagonists with neuroprotective properties. J Neural Transm Suppl. 1994;43:91–104. [PubMed] [Google Scholar]
- Korosi A, Baram TZ. The pathways from mother’s love to baby’s future. Front Behav Neurosci. 2009;3:27. doi: 10.3389/neuro.08.027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostenskaja M, Nikulin VV, Kicić D, Nikulina AV, Kähkönen S. Effects of NMDA receptor antagonist memantine on mismatch negativity. Brain Res Bull. 2007;72:275–283. doi: 10.1016/j.brainresbull.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Krivoy A, Weizman A, Laor L, Hellinger N, Zemishlany Z, Fischel T. Addition of memantine to antipsychotic treatment in schizophrenia inpatients with residual symptoms: A preliminary study. Eur Neuropsychopharmacol. 2008;18:117–121. doi: 10.1016/j.euroneuro.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Kumari V, Soni W, Sharma T. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999;156:1046–1051. doi: 10.1176/ajp.156.7.1046. [DOI] [PubMed] [Google Scholar]
- Kumari V, Gray JA, Geyer MA, ffytche D, Soni W, Mitterschiffthaler MT, Vythelingum GN, Simmons A, Williams SC, Sharma T. Neural correlates of tactile prepulse inhibition: a functional MRI study in normal and schizophrenic subjects. Psychiatry Res. 2003;122:99–113. doi: 10.1016/s0925-4927(02)00123-3. [DOI] [PubMed] [Google Scholar]
- Kumari V, Peters ER, Fannon D, Antonova E, Premkumar P, Anilkumar AP, Williams SC, Kuipers E. Dorsolateral prefrontal cortex activity predicts responsiveness to cognitive-behavioral therapy in schizophrenia. Biol Psychiatry. 2009;66:594–602. doi: 10.1016/j.biopsych.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari V, Premkumar P, Fannon D, Aasen I, Raghuvanshi S, Anilkumar AP, Antonova E, Peters ER, Kuipers E. Sensorimotor gating and clinical outcome following cognitive behaviour therapy for psychosis. Schizophr Res. 2012;134:232–238. doi: 10.1016/j.schres.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunugi H, Tanaka M, Hori H, Hashimoto R, Saitoh O, Hironaka N. Prepulse inhibition of acoustic startle in Japanese patients with chronic schizophrenia. Neurosci Res. 2007;59:23–28. doi: 10.1016/j.neures.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Kurtz MM, Moberg PJ, Gur RC, Gur RE. Approaches to cognitive remediation of neuropsychological deficits in schizophrenia: a review and meta-analysis. Neuropsychol Rev. 2001;11:197–210. doi: 10.1023/a:1012953108158. [DOI] [PubMed] [Google Scholar]
- Kurtz MM, Wexler BE, Fujimoto M, Shagan DS, Seltzer JC. Symptoms versus neurocognition as predictors of change in life skills in schizophrenia after outpatient rehabilitation. Schizophr Res. 2008;102:303–311. doi: 10.1016/j.schres.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz MM, Seltzer JC, Fujimoto M, Shagan DS, Wexler BE. Predictors of change in life skills in schizophrenia after cognitive remediation. Schizophr Res. 2009;107:267–274. doi: 10.1016/j.schres.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvajo M, McKellar H, Joseph A, Gogos JA. Molecules, signaling and schizophrenia. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 629–656. [DOI] [PubMed] [Google Scholar]
- Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q J Nucl Med. 1998;42:211–221. [PubMed] [Google Scholar]
- Lazarewicz MT, Ehrlichman RS, Maxwell CR, Gandal MJ, Finkel LH, Siegel SJ. Ketamine modulates theta and gamma oscillations. J Cogn Neurosci. 2010;22:1452–1464. doi: 10.1162/jocn.2009.21305. [DOI] [PubMed] [Google Scholar]
- Lee T, Seeman P. Elevation of brain neuroleptic/dopamine receptors in schizophrenia. Am J Psychiatry. 1980;137:191–197. doi: 10.1176/ajp.137.2.191. [DOI] [PubMed] [Google Scholar]
- Leucht S, Arbter D, Engel RR, Kissling W, Davis JM. How effective are second-generation antipsychotic drugs? A meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14:429–447. doi: 10.1038/sj.mp.4002136. [DOI] [PubMed] [Google Scholar]
- Levitt JJ, Bobrow L, Lucia D, Srinivasan P. A selective review of volumetric and morphometric imaging in schizophrenia. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 243–282. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Morris HM. Cell and receptor type-specific alterations in markers of GABA neurotransmission in the prefrontal cortex of subjects with schizophrenia. Neurotox Res. 2008;14:237–248. doi: 10.1007/BF03033813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Papadakis K, Csernansky J, Litman R, Volavka J, Jia XD, Gage A MEM-MD-29 Study Group. A randomized, placebo-controlled study of memantine as adjunctive treatment in patients with schizophrenia. Neuropsychopharmacology. 2009;34:1322–1329. doi: 10.1038/npp.2008.200. [DOI] [PubMed] [Google Scholar]
- Light GA, Braff DL. Stability of mismatch negativity deficits and their relationship to functional impairments in chronic schizophrenia. Am J Psychiatry. 2005a;162:1741–1743. doi: 10.1176/appi.ajp.162.9.1741. [DOI] [PubMed] [Google Scholar]
- Light GA, Braff DL. Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch Gen Psychiatry. 2005b;62:127–136. doi: 10.1001/archpsyc.62.2.127. [DOI] [PubMed] [Google Scholar]
- Light GA, Hsu JL, Hsieh MH, Meyer-Gomes K, Sprock J, Swerdlow NR, Braff DL. Gamma band oscillations reveal neural network cortical coherence dysfunction in schizophrenia patients. Biol Psychiatry. 2006;60:1231–1240. doi: 10.1016/j.biopsych.2006.03.055. [DOI] [PubMed] [Google Scholar]
- Light GA, Braff DL, Sprock J, Cadenhead KS, Swerdlow NR. Prepulse inhibition of startle is positively associated with higher order cognition in women. Abstr Soc Neuroscience. 2007a;806:17. [Google Scholar]
- Light GA, Swerdlow NR, Braff DL. Preattentive sensory processing is associated with cognitive and psychosocial functioning in healthy adults. J Cog Neurosci. 2007b;19:1624–1632. doi: 10.1162/jocn.2007.19.10.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn GS, Javitt DC. Phencyclidine (PCP)-induced deficits of prepulse inhibition in monkeys. Neuroreport. 2001;12:117–120. doi: 10.1097/00001756-200101220-00031. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Luciana M, Collins PF. Dopamine modulates working memory for spatial but not object cues in normal humans. J Cogn Neurosci. 1997;4:58–68. doi: 10.1162/jocn.1997.9.3.330. [DOI] [PubMed] [Google Scholar]
- Luciana M, Depue RA, Arbisi P, Leon A. Facilitation of working memory in humans by a D2 dopamine receptor agonist. J Cogn Neurosci. 1992;4:58–68. doi: 10.1162/jocn.1992.4.1.58. [DOI] [PubMed] [Google Scholar]
- Luciana M, Collins PF, Depue RA. Opposing roles for dopamine and serotonin in the modulation of human spatial working memory functions. Cereb Cortex. 1998;8:218–226. doi: 10.1093/cercor/8.3.218. [DOI] [PubMed] [Google Scholar]
- Lynch D, Laws KR, McKenna PJ. Cognitive behavioural therapy for major psychiatric disorder: does it really work? A meta-analytical review of well-controlled trials. Psychol Med. 2010;40:9–24. doi: 10.1017/S003329170900590X. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:229–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- Marder SR, Buchanan RW. Pharmacological strategies to improve cognition in schizophrenia: results from TURNS and MATRICS-CT. American College of Neuropsychopharmacology (ACNP); Hollywood, FL: 2009. [Google Scholar]
- Mattay VS, Callicott JH, Bertolino A, Heaton I, Frank JA, Coppola R, Berman KF, Goldberg TE, Weinberger DR. Effects of dextroamphetamine on cognitive performance and cortical activation. Neuroimage. 2000;12:268–275. doi: 10.1006/nimg.2000.0610. [DOI] [PubMed] [Google Scholar]
- Mattay VS, Goldberg TE, Fera F, Hariri AR, Tessitore A, Egan MF, Kolachana B, Callicott JH, Weinberger DR. Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc Natl Acad Sci U S A. 2003;100:6186–6191. doi: 10.1073/pnas.0931309100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk SR, Meltzer HY. The role of cognition in vocational functioning in schizophrenia. Schizophr Res. 2000;45:175–184. doi: 10.1016/s0920-9964(99)00198-x. [DOI] [PubMed] [Google Scholar]
- McGurk SR, Mueser KT. Cognitive functioning, symptoms, and work in supported employment: a review and heuristic model. Schizophr Res. 2004;70:147–173. doi: 10.1016/j.schres.2004.01.009. [DOI] [PubMed] [Google Scholar]
- McGurk SR, Mueser KT, Feldman K, Wolfe R, Pascaris A. Cognitive training for supported employment: 2–3 year outcomes of a randomized controlled trial. Am J Psychiatry. 2007a;164:437–441. doi: 10.1176/appi.ajp. 164.3.437. [DOI] [PubMed] [Google Scholar]
- McGurk SR, Twamley EW, Sitzer DI, McHugo GJ, Mueser KT. A meta-analysis of cognitive remediation in schizophrenia. Am J Psychiatry. 2007b;164:1791–1802. doi: 10.1176/appi.ajp.2007.07060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk SR, Mueser KT, DeRosa TJ, Wolfe R. Work, recovery, and comorbidity in schizophrenia: a randomized controlled trial of cognitive remediation. Schizophr Bull. 2009;35:319–335. doi: 10.1093/schbul/sbn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medalia A, Choi J. Cognitive remediation in schizophrenia. Neuropsychol Rev. 2009;19:353–364. doi: 10.1007/s11065-009-9097-y. [DOI] [PubMed] [Google Scholar]
- Medalia A, Richardson R. What predicts a good response to cognitive remediation interventions? Schizophr Bull. 2005;31:942–953. doi: 10.1093/schbul/sbi045. [DOI] [PubMed] [Google Scholar]
- Mehta MA, Owen AM, Sahakian BJ, Mavaddat N, Pickard JD, Robbins TW. Methylphenidate enhances working memory by modulating discrete frontal and parietal lobe regions in the human brain. J Neurosci. 2000;20:1–6. doi: 10.1523/JNEUROSCI.20-06-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta MA, Swainson R, Gogilvie AD, Sahakian BJ, Robbins TW. Improved short-term spatial memory but impaired reversal learning following the dopamine D2 agonist bromocriptine in human volunteers. Psychopharmacology. 2001;159:10–20. doi: 10.1007/s002130100851. [DOI] [PubMed] [Google Scholar]
- Mueser KT, Bellack AS, Douglas MS, Wade JH. Prediction of social skill acquisition in schizophrenic and major affective disorder patients from memory and symptomatology. Psychiatry Res. 1991;37:281–296. doi: 10.1016/0165-1781(91)90064-v. [DOI] [PubMed] [Google Scholar]
- Müller U, von Cramon Y, Pollmann S. D1- versus D2-receptor modulation of visuospatial working memory in humans. J Neurosci. 1998;18:2720–2728. doi: 10.1523/JNEUROSCI.18-07-02720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuechterlein KH, Green MF, Kern RS, Baade LE, Barch DM, Cohen JD, Essock S, Fenton WS, Frese FJ, 3rd, Gold JM, Goldberg T, Heaton RK, Keefe RS, Kraemer H, Mesholam-Gately R, Seidman LJ, Stover E, Weinberger DR, Young AS, Zalcman S, Marder SR. The MATRICS consensus cognitive battery: part 1. Test selection, reliability, and validity. Am J Psychiatry. 2008;165:203–213. doi: 10.1176/appi.ajp.2007.07010042. [DOI] [PubMed] [Google Scholar]
- Parnas AS, Weber M, Richardson R. Effects of multiple exposures to D-cycloserine on extinction of conditioned fear in rats. Neurobiol Learn Mem. 2005;83:224–231. doi: 10.1016/j.nlm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- Penn DL, Meyer PS, Evans E, Wirth RJ, Cai K, Burchinal M. A randomized controlled trial of group cognitive-behavioral therapy vs. enhanced supportive therapy for auditory hallucinations. Schizophr Res. 2009;109:52–59. doi: 10.1016/j.schres.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Pilling S, Bebbington P, Kuipers E, Garety P, Geddes J, Martindale B, Orbach G, Morgan C. Psychological treatments in schizophrenia: II. Meta-analyses of randomized controlled trials of social skills training and cognitive remediation. Psychol Med. 2002;32:783–791. doi: 10.1017/s0033291702005640. [DOI] [PubMed] [Google Scholar]
- Porto PR, Oliveira L, Mari J, Volchan E, Figueira I, Ventura P. Does cognitive behavioral therapy change the brain? A systematic review of neuroimaging in anxiety disorders. J Neuropsychiatry Clin Neurosci. 2009;21:114–125. doi: 10.1176/appi.neuropsych.21.2.114. [DOI] [PubMed] [Google Scholar]
- Quartermain D, Mower J, Rafferty MF, Herting RL, Lanthorn TH. Acute but not chronic activation of the NMDA-coupled glycine receptor with D-cycloserine facilitates learning and retention. Eur J Pharmacol. 1994;257:7–12. doi: 10.1016/0014-2999(94)90687-4. [DOI] [PubMed] [Google Scholar]
- Quednow BB, Wagner M, Mössner R, Maier W, Kühn KU. Sensorimotor gating of schizophrenia patients depends on catechol O-Methyltransferase Val158Met polymorphism. Schizophr Bull. 2010;36:341–346. doi: 10.1093/schbul/sbn088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberg A, Harvey PD. Neuropsychological impairments in schizophrenia: Integration of performance-based and brain imaging findings. Psychol Bull. 2007;133:833–858. doi: 10.1037/0033-2909.133.5.833. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- Rice DP. The economic impact of schizophrenia. J Clin Psychiatry. 2009;60(Suppl 1):4–6. discussion 28–30. [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR, Lahti AC. Dopaminergic synapses in the caudate of subjects with schizophrenia: relationship to treatment response. Synapse. 2009;63:520–530. doi: 10.1002/syn.20623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev. 2003;9:275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussos P, Giakoumaki SG, Bitsios P. Tolcapone effects on gating, working memory, and mood interact with the synonymous catechol-O-methyltransferase rs4818c/g polymorphism. Biol Psychiatry. 2009;66:997–1004. doi: 10.1016/j.biopsych.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Saxena S, Gorbis E, O’Neill J, Baker SK, Mandelkern MA, Maidment KM, Chang S, Salamon N, Brody AL, Schwartz JM, London ED. Rapid effects of brief intensive cognitive-behavioral therapy on brain glucose metabolism in obsessive-compulsive disorder. Mol Psychiatry. 2009;14:197–205. doi: 10.1038/sj.mp. 4002134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JM, Stoessel PW, Baxter LR, Martin KM, Phelps ME. Systematic changes in cerebral glucose metabolic rate after successful behavior modification treatment of obsessive-compulsive disorder. Arch Gen Psychiatry. 1996;53:109–113. doi: 10.1001/archpsyc.1996.01830020023004. [DOI] [PubMed] [Google Scholar]
- Sellwood W, Wittkowski A, Tarrier N, Barrowclough C. Needs-based cognitive-behavioural family intervention for patients suffering from schizophrenia: 5-year follow-up of a randomized controlled effectiveness trial. Acta Psychiatr Scand. 2007;116:447–452. doi: 10.1111/j.1600-0447.2007.01097.x. [DOI] [PubMed] [Google Scholar]
- Siegel BV, Trestman RL, O’Faithbheartaigh S, Mitropoulou V, Amin F, Kirrane R, Silverman J, Schmeidler J, Keefe RS, Siever LJ. D-Amphetamine challenge effects on Wisconsin Card Sort Test. Performance in schizotypal personality disorder. Schizophr Res. 1996;20:29–32. doi: 10.1016/0920-9964(95)00002-x. [DOI] [PubMed] [Google Scholar]
- Silbersweig DA, Stern E, Frith C, Cahill C, Holmes A, Grootoonk S, Seaward J, McKenna P, Chua SE, Schnorr L, Jones T, Frackowiak RSJ. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378:176–179. doi: 10.1038/378176a0. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Bergen SE, Sun Z, Marcsisin MJ, Sampson AR, Lewis DA. Anatomical evidence of impaired feedforward auditory processing in schizophrenia. Biol Psychiatry. 2007;61:854–864. doi: 10.1016/j.biopsych.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR. Cortico-striatal substrates of cognitive, motor and sensory gating: Speculations and implications for psychological function and dysfunction. In: Panksepp J, editor. Advances in biological psychiatry. Vol. 2. JAI; Greenwich, CT: 1996. pp. 179–208. [Google Scholar]
- Swerdlow NR. Are we studying and treating schizophrenia correctly? Schizophr Res. 2011;130:1–10. doi: 10.1016/j.schres.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Shoemaker JM, Ross L, Wasserman LC, Talledo J, Auerbach PP. Effects of amantadine and bromocriptine on startle and sensorimotor gating: Parametric studies and cross-species comparisons. Psychopharmacology. 2002;164:82–92. doi: 10.1007/s00213-002-1172-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Light GA, Cadenhead KS, Sprock J, Hsieh MH, Braff DL. Startle gating deficits in a large cohort of patients with schizophrenia: Relationship to medications, symptoms, neurocognition, and level of function. Arch Gen Psychiatry. 2006a;63:1325–1335. doi: 10.1001/archpsyc.63.12.1325. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Talledo J, Sutherland AN, Nagy D, Shoemaker JM. Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology. 2006b;31:2011–2021. doi: 10.1038/sj.npp.1301043. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008;199:331–388. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Lelham SA, Sutherland Owens AN, Chang WL, Sassen SDT, Talledo JA. Pramipexole effects on startle gating in rats and normal men. Psychopharmacology. 2009a;205:689–698. doi: 10.1007/s00213-009-1577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J. The effects of memantine on prepulse inhibition. Neuropsychopharmacology. 2009b;34:1854–1864. doi: 10.1038/npp.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Light GA, Thanam S, Slater A, Talledo JA. Abstract, American College of Neuropsychopharmacology (ACNP) 2010. Dec 5–9, Memantine and amphetamine effects on neurocognition and sensorimotor gating in healthy subjects. [Google Scholar]
- Swerdlow NR, Talledo JA, Kei J, Thanam S, Lamb SN, Light GA. Abstract, American College of Neuropsychopharmacology (ACNP) 2011. Dec 4–8, Amphetamine effects on MATRICS performance in healthy adults: Prelude to a genetic analysis. [Google Scholar]
- Tai S, Turkington D. The evolution of cognitive behavior therapy for schizophrenia: current practice and recent developments. Schizophr Bull. 2009;35:865–873. doi: 10.1093/schbul/sbp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talledo JA, Sutherland Owens AN, Schortinghuis T, Swerdlow NR. Amphetamine effects on startle gating in normal women and female rats. Psychopharmacology. 2009;204:165–175. doi: 10.1007/s00213-008-1446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub E, Uswatte G, Elbert T. New treatments in neurorehabilitation founded on basic research. Nat Rev Neurosci. 2002;3:228–236. doi: 10.1038/nrn754. [DOI] [PubMed] [Google Scholar]
- Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull. 2007;33:69–94. doi: 10.1093/schbul/sbl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twamley EW, Narvaez JM, Becker DR, Bartels SJ, Jeste DV. Supported employment for middle-aged and older people with schizophrenia. Am J Psychiatr Rehabil. 2008a;11:76–89. doi: 10.1080/15487760701853326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twamley EW, Savla GN, Zurhellen CH, Heaton RK, Jeste DV. Development and pilot testing of a novel compensatory cognitive training intervention for people with psychosis. Am J Psychiatr Rehabil. 2008b;11:144–163. doi: 10.1080/15487760801963678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twamley EW, Burton CZ, Vella L. Compensatory cognitive training for psychosis: who benefits? Who stays in treatment? Schizophr Bull. 2011;37(Suppl 2):S55–S62. doi: 10.1093/schbul/sbr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006;52:155–168. doi: 10.1016/j.neuron.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Urban N, Abi-Dargham A. Neurochemical imaging in schizophrenia. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 215–242. [DOI] [PubMed] [Google Scholar]
- van der Linden D, Massar SA, Schellekens AF, Ellenbroek BA, Verkes RJ. Disrupted sensorimotor gating due to mental fatigue: preliminary evidence. Int J Psychophysiol. 2006;62:168–174. doi: 10.1016/j.ijpsycho.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Volk DW, Lewis DA. Prefrontal cortical circuits in schizophrenia. In: Swerdlow NR, editor. Behavioral neurobiology of schizophrenia and its treatment. Current topics in behavioral neuroscience. Springer; Heidelberg: 2010. pp. 485–508. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Barro M, Csomor PA, Feldon J. Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry. 2006;60:597–603. doi: 10.1016/j.biopsych.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Volz H, Gaser C, Hager F, Rzanny R, Ponisch J, Mentzel H, Kaiser WA, Sauer H. Decreased frontal activation in schizophrenics during stimulation with the Continuous Performance Test—a functional magnetic resonance imaging study. Eur Psychiatry. 1999;14:17–24. doi: 10.1016/s0924-9338(99)80711-1. [DOI] [PubMed] [Google Scholar]
- Weike AI, Bauer U, Hamm AO. Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biol Psychiatry. 2000;47:61–70. doi: 10.1016/s0006-3223(99)00229-2. [DOI] [PubMed] [Google Scholar]
- Willenborg B, Schmoller A, Caspary J, Melchert UH, Scholand-Engler HG, Jauch-Chara K, Hohagen F, Schweiger U, Oltmanns KM. Memantine prevents hypoglycemia-induced decrements of the cerebral energy status in healthy subjects. J Clin Endocrinol Metab. 2011;96:E384–E388. doi: 10.1210/jc.2010-1348. [DOI] [PubMed] [Google Scholar]
- Wong D, Wagner H, Tune L, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, Toung JK, Malat J, Williams JA, O’Tuama LA, Snyder SH, Kuhar MJ, Gjedde A. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science. 1986;234:1558–1563. doi: 10.1126/science.2878495. [DOI] [PubMed] [Google Scholar]
- Wonodi I, Cassady SL, Adami H, Avila M, Thaker GK. Effects of repeated amphetamine administration on antisaccade in schizophrenia spectrum personality. Psychiatry Res. 2006;141:237–245. doi: 10.1016/j.psychres.2005.07.028. [DOI] [PubMed] [Google Scholar]
- Wykes T, Sturt E, Katz R. The prediction of rehabilitative success after three years. The use of social, symptom and cognitive variables. Br J Psychiatry. 1990;157:865–870. doi: 10.1192/bjp.157.6.865. [DOI] [PubMed] [Google Scholar]
- Wykes T, Huddy V, Cellard C, McGurk SR, Czobor P. A meta-analysis of cognitive remediation for schizophrenia: methodology and effect sizes. Am J Psychiatry. 2011;168:472–485. doi: 10.1176/appi.ajp. 2010.10060855. [DOI] [PubMed] [Google Scholar]
- Wynn JK, Sugar C, Horan WP, Kern R, Green MF. Mismatch negativity, social cognition, and functioning in schizophrenia patients. Biol Psychiatry. 2010;67:940–947. doi: 10.1016/j.biopsych.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdanys K, Tampi RR. A systematic review of off-label uses of memantine for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1362–1374. doi: 10.1016/j.pnpbp.2008.01.008. [DOI] [PubMed] [Google Scholar]