

Abstract
Factors underlying vulnerability to alcoholism are largely unknown. We identified an innate endophenotype in rodents that predicted individual risk for alcohol-related behaviors that was associated with decreased expression of the neuroplasticity-related cell-adhesion molecule PSA-NCAM. Depletion of PSA-NCAM within the ventromedial prefrontal cortex was sufficient to render mice unable to extinguish alcohol seeking, indicating a causal role of naturally occurring variation. These data demonstrate a novel mechanism of aberrant prefrontal neuroplasticity that underlies enhanced propensity for inflexible addiction-related behavior.
Alcoholism is associated with executive dysfunction and impaired decision-making1 that results in maladaptive behaviors, including the inability to restrict alcohol intake and high rates of relapse. Due to the effects of drugs of abuse on mesocorticolimbic circuitry, it has remained difficult to elucidate the neurobiological differences that exist prior to alcohol exposure that confer risk for addiction from the adaptations caused by alcohol itself. Identifying behaviorally relevant variations in this circuit is expected to be crucial to the prevention and treatment of alcohol use disorders.
To investigate these preexisting differences, it was critical to identify an endophenotype that was predictive of alcoholism-related behaviors prior to any alcohol exposure. After repeated pairings, cues associated with reward are thought to be able to motivate and energize instrumental behavior e.g., 2, 3, and in human populations it has been shown that risk for alcoholism is associated with enhanced cue reactivity e.g., 4. To investigate how individual differences in cue-related behaviors relate to alcohol-seeking behaviors, we trained an outbred strain of mice on a Pavlovian-to-instrumental transfer (PIT) task. Animals first learned that a cue was predictive of delivery of a food reinforcer, and then they were trained to make an instrumental response (lever press) for the same reinforcer. The ability of the food-paired cue to invigorate responding was assessed in the absence of food reinforcement, and animals were assigned to High or Low PIT groups based on their lever-press performance in this session (Fig. 1a). Mice were then trained over several days to make a different response (nosepoke) for an unsweetened alcohol reinforcer that was paired with a distinct cue (see Supp. Methods; Supp. Fig. 1). This response was extinguished (i.e., not reinforced), and, after extinction, responding was reinstated by presentation of the alcohol-paired cue. Our data indicated that high levels of cue-motivated behavior (high PIT) was predictive of resistance to extinction (Fig. 1b) and enhanced cue-induced reinstatement of alcohol seeking (Fig. 1c). No differences in alcohol consumption were seen during training (See Supp. Fig. 2). Additionally, the predictive relationship between food PIT and extinction and reinstatement was specific to alcohol reinforcement as PIT status was not related to inflexible sucrose-seeking behavior (See Supp. Methods; Supp. Fig. 3, 4).
Figure 1.
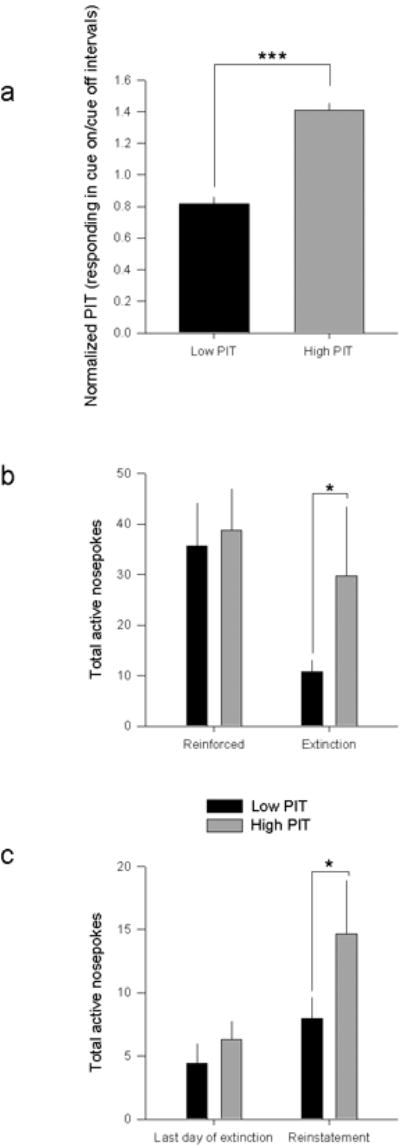
a. Mice were separated by median split into High and Low PIT groups that are significantly different from one another (F1,62= 41.82; p<.001) on normalized responding (normalized lever presses during Cue On to Cue Off periods), but no effect of Pavlovian approach (F1,62= .462, p=.5). b. High PIT mice show resistance to extinction on the first session A repeated measures ANOVA revealed an interaction between PIT and session on responding (F1, 29=4.7572, p=.03). Post hoc analyses indicate that there is an effect of Extinction in Low PIT animals (p=.013) but no effect in High PIT animals (p>.7). Responding on subsequent extinction days was equivalent between groups by a repeated measures ANOVA (no effect of PIT x day; p>.2). c. High PIT mice show greater cue-induced reinstatement than Low PIT mice. Repeated measures ANOVA revealed an interaction between PIT and session on active responding (F1,29= 6.3778, p=.017), as well as a main effect of reinstatement (F1,29= 8.3147, p<.01). Post-hoc analyses indicate that High PIT mice make a greater number of nosepokes during the reinstatement session than during the last extinction session (p=.016), while Low PIT mice respond equivalently (p=.5), and further, that High PIT mice make a greater number of nosepokes during the reinstatement session than Low PIT mice (p=.0372), indicating that High PIT mice show a greater effect of reinstatement than Low PIT mice. *p<.05, **p<.01, ***p<.001. Error bars ± SEM.
By identifying which individuals were at risk for resistance to extinction of alcohol seeking prior to any alcohol exposure, we may be able to determine the precise neurobiological factors that produce susceptibility for alcoholism-related behaviors. Alterations in neural plasticity have recently been identified as a risk factor for addictive behavior in animal models and alcohol consumption itself produces neuronal adaptations that may promote the transition from casual drinking to dependencee.g., 5. Based on the established role of plasticity in the ventromedial PFC (vmPFC) in the extinction of learned fear6, we hypothesized that preexisting differences in plasticity-related molecules in the vmPFC prior to any alcohol exposure would underlie innate differences in extinction behavior.
Neural cell adhesion molecule (NCAM) and its modified form, polysialylated NCAM (PSA-NCAM), are critically involved in learning and memorye.g., 7,8. In its unmodified form, NCAM molecules on pre and postsynaptic membranes form heteromers that serve to stabilize the synapse. The addition of PSA to NCAM confers anti-adhesive properties that may be necessary to promote activity-dependent synaptic plasticity and dendritic remodelinge.g, 9,18. Additionally, polymorphisms in the NCAM gene have been shown to relate to risk for alcoholism in human populations10, though a direct functional relationship to vulnerability cannot be proven and a causal role for plasticity-associated molecules in brain has not yet been demonstrated. Based on the established role of vmPFC plasticity in extinction6 and the putative involvement of NCAM and its modifications in alcoholism-related behaviors, we hypothesized that low PIT animals that rapidly extinguish and resist cue-induced reinstatement of alcohol-seeking would show higher PSA-NCAM in the vmPFC than high risk, thereby reflecting greater potential for synaptic plasticity. Accordingly, we demonstrated that prior to any alcohol exposure, low PIT animals show higher PSA-NCAM in the vmPFC (Fig. 2a), but not in other brain regions, including the dorsomedial PFC (See Supp. Fig. 6-7). As no alterations were seen in total NCAM levels (See Supp. Fig. 5), it is as yet unclear exactly what underlies these innate individual differences in PSA-NCAM. It is possible that differential PSA-NCAM expression is the result of differences in the neuronal activity that impact the addition of PSA to NCAM by the polysialyltransferases that mediate this and/or differences in the endocytosis of NCAM or PSA11. If PSA-NCAM mediated plasticity in the vmPFC is causally related to extinction of alcohol seeking, we predicted that manipulating levels of PSA-NCAM directly would impact extinction ability. To demonstrate this, in a separate cohort of mice we first infused a bacterial enzyme (endo-neuraminidase; endo-N) to cleave PSA from NCAM into the vmPFC prior to extinction (Fig. 2b; Supp. Fig. 8). Our data show that loss of polysialylated NCAM in the vmPFC resulted in an inability to extinguish alcohol seeking (Fig. 2c), confirming that infralimbic PSA-NCAM is critical for successful extinction of alcohol seeking. Given that promotion of prelimbic plasticity can result in a loss of flexible behavior12, we confirmed that endo-N infusion in the prelimbic PFC had no effect on extinction or reinstatement (Supp. Fig. 9,10). Notably, loss of vmPFC PSA-NCAM did not alter cue-induced reinstatement of alcohol seeking (Fig. 2d). Therefore, innate differences in cue-induced reinstatement may be the result of separate, as yet unidentified, neurobiological differences between high and low PIT mice that may act in concert with or independent of the identified region-specific differences in vmPFC PSA-NCAM plasticity to produce a resiliency to extinction of alcohol seeking.
Figure 2.
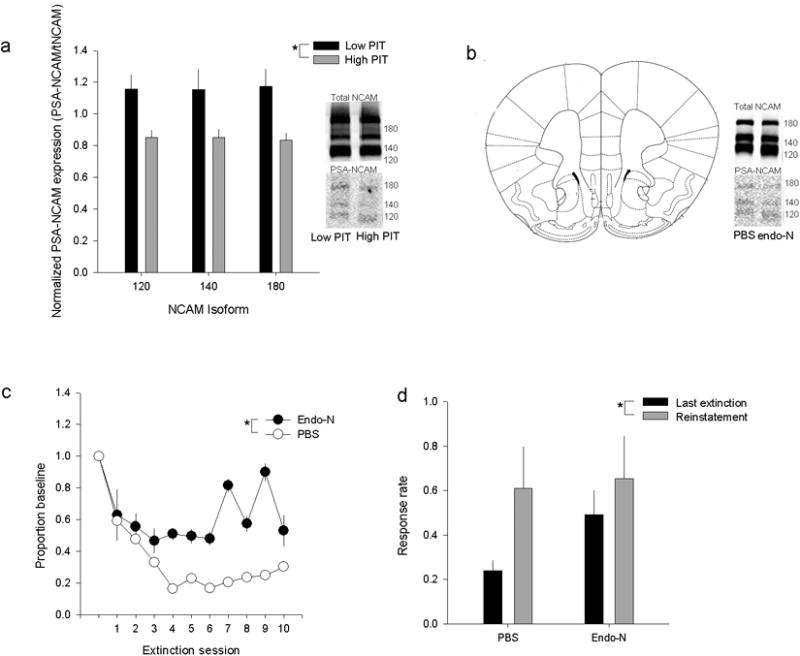
a. High PIT status predicted lower levels of PSA-NCAM in the vmPFC. A repeated measures ANOVA showed a main effect of PIT status on normalized PSA-NCAM expression in the vmPFC (PSA-NCAM/total NCAM; F1,34=6.0155, p=.015). No interaction was seen, indicating that this effect was consistent across isoforms. Representative bands are cropped from complete blots (Supp. Fig 10). b. Site of endo-N infusion and representative bands showing decreased PSA-NCAM expression at the time of reinstatement. c. Loss of PSA-NCAM in the vmPFC resulted in resistance to extinction across multiple sessions. A main effect of infralimbic endo-N treatment on responding during extinction by repeated measures ANOVA (F1,13= 5.65, p=.03), but no main effect of PIT status, consistent with a lack of effect on multiple days of extinction in outbred mice, or of day of extinction. d. Treatment with endo-N did not influence cue-induced reinstatement of alcohol seeking during the between sessions reinstatement session (p>.2). Nor was there an interaction between endo-N and session (p>.25) or an effect during a ‘within-session’ test performed to confirm that baseline differences in responding during extinction were not responsible for the lack of effect of prefrontal endo-N treatment on cue-induced reinstatement or that impaired consolidation of extinction learning was responsible for any effects on reinstatement (F1,16=.89, p>.35). A main effect of session was seen (F1,16=9.63, p<.01), indicating that all mice significantly increased responding during the reinstatement session.
*p<.05, **p<.01, ***p<.001. Error bars ± SEM.
Together, these data indicate that genes and environment interact to produce differences in neuroplasticity that underlie resistance to extinction and relapse of alcohol-seeking behavior. Specifically, we found that an endophenotype indicative of resistance to extinction of alcohol seeking and increased risk for relapse was associated with decreased expression of PSA-NCAM in the vmPFC. Moreover, we demonstrated that loss of vmPFC PSA-NCAM more robustly impaired the extinction of alcohol seeking independent of risk status. Given that altering the potential for synaptic plasticity in the vmPFC by explicitly ablating PSA-NCAM impedes the ability to extinguish alcohol-seeking behavior, we suggest that individual differences in synaptic stability may be responsible for this behavioral phenotype.
Our work indicates that normal plasticity in the vmPFC is necessary for the extinction of alcohol-related behavior. In accordance with this finding, previous work has demonstrated that infusion of BDNF, a pro-plasticity neurotrophin molecule, into the vmPFC facilitates the extinction of fear memories6. Our data show a novel role for a plasticity promoting post-translational modification of a cell-adhesion molecule in the extinction of drug memories. This growing body of evidence suggests that individual differences in multiple prefrontal plasticity-associated processes may be indicative of risk for several neuropsychological disorders, including addiction. Additionally, the transition to addiction may also involve drug/alcohol-induced alterations in cell adhesion molecules13,14. Further research using this model in mice is expected to enable us to detect additional preexisting neurobiological differences between high and low risk populations, as well as differences in response to alcohol consumption, that will aid in the development of pharmacological interventions. As PIT is measurable in human populations15 this paradigm may also enable behavioral screening to indicate which individuals are more likely to develop alcohol use disorders and to tailor prevention strategies accordingly.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health Grants P50 AA012870 (JRT), R01 DA011717 (JRT), F31 AA020135 (JMB), K01 DA031745 (MMT) and the Connecticut Department of Mental Health and Addiction Services. The PSA-NCAM antibody developed by TM Jessell, J Dodd & S Brenner-Morton was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Iowa City, IA 52242.
Footnotes
Contributions: JMB, MMT and JRT designed the study and wrote the experiments. JMB performed experiments and analyzed data.
References
- 1.Stavro K, Pelletier J, Potvin S. Addiction Biology. 2012 doi: 10.1111/j.1369-1600.2011.00418.x. [DOI] [PubMed] [Google Scholar]
- 2.Berridge KC, Robinson TE. Trends Neurosci. 2003;26(9):507–13. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- 3.Balleine BW, Dickinson A. Neuropharmacology. 1998;37(4-5):407–19. doi: 10.1016/s0028-3908(98)00033-1. [DOI] [PubMed] [Google Scholar]
- 4.Karaken DA, Bragulat V, Dzemidzic M, Cox C, Talavage T, Davidson D, O'Connor SJ. NeuroImage. 2010;50:267–276. doi: 10.1016/j.neuroimage.2009.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCool BA. Neuropharmacology. 2011;61(7):1097–108. doi: 10.1016/j.neuropharm.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Science. 2010;328(5983):1288–90. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conboy L, Bisaz R, Markram K, Sandi C. Adv Exp Med Biol. 2010;663:271–96. doi: 10.1007/978-1-4419-1170-4_18. [DOI] [PubMed] [Google Scholar]
- 8.Senkov O, Sun M, Weinhold B, Gerardy-Schahn R, Schachner M, Dityatev A. J Neurosci. 2006;26(42):10888–109898. doi: 10.1523/JNEUROSCI.0878-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kochlamazashvili G, Bukalo O, Senkov O, Salmen B, Gerardy-Schahn R, Engel AK, Schachner M, Dityatev A. J Neurosci. 2012;32(7):2263–2275. doi: 10.1523/JNEUROSCI.5103-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang BZ, Kranzler HR, Zhao H, Gruen JR, Luo X, Gelernter J. Hum Mol Genet. 2007;16(23):2844–53. doi: 10.1093/hmg/ddm240. [DOI] [PubMed] [Google Scholar]
- 11.Bonfanti L. Progress in Neurobiology. 2006;80(3):129–64. doi: 10.1016/j.pneurobio.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Graybeal C, Feyder M, Schulman E, Saksida LM, Bussey TJ, Brigman JL, Holmes A. Nat Neurosci. 2011;14(12):1507–9. doi: 10.1038/nn.2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gourley SL, Taylor JR, Koleske AJ. Commun Integr Biol. 2011;4(1):30–3. doi: 10.4161/cib.4.1.14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiggins A, Smith RJ, Shen HW, Kalivas PW. J Neurosci. 2011;31(45):16177–84. doi: 10.1523/JNEUROSCI.3816-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talmi D, Seymour B, Dayan P, Dolan RJ. J Neurosci. 2008;28(2):360–8. doi: 10.1523/JNEUROSCI.4028-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.