

Abstract
To investigate the mechanisms by which breast cancer cells adapt and are able to grow during estrogen deprivation, human estrogen receptor-α (ERα)-positive breast cancer cells stably transfected with the aromatase gene (MCF-7Ca) were cultured in steroid-depleted medium for 6–8 months until they started proliferating. Compared with the parental MCF-7Ca cells, long-term estrogen-deprived UMB-1Ca cells exhibited increased aromatase activity (2000%), AIB1 expression (3500%) and ERα expression (100%). When MCF-7Ca cells were isolated from tumors of mice treated for 12 months with an aromatase inhibitor, letrozole, ERα was reduced (50%) whereas AIB1 levels were increased (>1000%), suggesting that the mechanism of estrogen deprivation might predetermine the signaling pathway utilized. To a lesser extent long-term estrogen-deprived MCF-7 cells (LTED) displayed an increase in AIB1, ERα and aromatase activity. Consistent with other findings, the growth of the LTED cells was inhibited by estradiol and antiestrogens, whereas the UMB-1Ca cells were slightly stimulated by estradiol and inhibited by antiestrogens and letrozole. In LTED cells treated with estradiol, levels of AIB1 and ERα (95%) were reduced. Interestingly, estradiol treatment caused no change in AIB1 and ERα expression in the UMB-1Ca cells which might explain the differential growth effect of the cells to estradiol. Together, these results demonstrate that estrogen deprivation results in the upregulation of the estrogen signaling pathway at the level of AIB1, ERα and aromatase, which might attenuate ER-mediated transcription representing one mechanism by which tumors adapt to proliferation in a low estrogenic environment.
Keywords: aromatase, breast cancer, coactivators, estrogen receptor, long-term estrogen deprivation
Introduction
In the past few years, three aromatase inhibitors have become established treatment for estrogen receptor (ER)-positive breast cancer and are proving more effective than tamoxifen. Aromatase inhibitors (letrozole, exemestane and anastrozole) act by a different mechanism from antiestrogens and reduce estrogen production. Unlike tamoxifen, aromatase inhibitors do not exert estrogenic activity but block the conversion of adrenal androgens to estrogens in the peripheral tissues of postmenopausal women. The Anastrozole, Tamoxifen Alone or in Combination (ATAC) trial compared the efficacy and tolerability of these two compounds as first-line adjuvant therapies in postmenopausal women with early breast cancer and showed the superiority of an aromatase inhibitor over tamoxifen in the adjuvant setting (1–4). Trials with other aromatase inhibitors confirm their efficacy in early-stage breast cancer after 5 years of tamoxifen treatment (5). Despite improvements in treatment, some patients remain resistant to therapy. Although aromatase inhibitors and anti-estrogens are effective in inhibiting tumor growth, tumors adapt and are able to proliferate in the presence of the drugs. The mechanisms by which tumors overcome long-term estrogen deprivation and are able to adapt from estrogen-dependent to estrogen-independent growth are complex and poorly understood. They could involve multiple factors and activation of signal transduction pathways. Gaining insight into this process could identify novel targets for new treatment modalities for postmenopausal breast cancer patients.
The p160 steroid receptor coactivator, Amplified in Breast Cancer 1 (AIB1), was shown to play a crucial role in the enhancement of breast tumor growth (6, 7) and was identified as a gene amplified in breast cancers (8). Previous reports have shown that AIB1 is amplified and overexpressed in four of five ER-positive breast and ovarian cancer cell lines and interacts with the ER in a ligand-dependent manner resulting in enhanced estrogen-dependent transcription (9). Despite data which demonstrate the involvement of AIB1 in normal growth, puberty, female reproductive function, mammary gland development and breast tumor growth (6) its role in proliferating breast tumors exposed to low estrogen is unclear. The ability of breast cancer cells to adapt to lower levels of estradiol has important implications for hormonal treatment of breast cancer.
The goal of this study was to investigate the effect of long-term estrogen deprivation on the estrogen signaling pathway utilizing aromatase-transfected and estrogen-dependent MCF-7Ca human breast cancer cells, and to determine the sensitivity of cells to antiestrogens and aromatase previously published online September 20, 2010 inhibitors. We utilized MCF-7Ca breast tumors from mice that were treated with letrozole 10 μg/day for 56 weeks until the tumors acquired the ability to proliferate in the presence of the drug. Cells were isolated from these tumors and grown in culture in the presence of letrozole (10). Results from the long-term letrozole-treated cell line (LTLT-Ca) were compared with other long-term estrogen-deprived cell lines. In an effort to understand mechanisms of resistance to estrogen deprivation, Masumara et al. reported studies of long-term estrogen-deprived MCF-7 cells (LTED) (11). After several months these cells acquired the ability to proliferate in the absence of added estrogen and developed increased sensitivity to low levels of estrogen suggesting an explanation of how cells could proliferate in conditions of estrogen deprivation. In the current study, we investigated, long-term estrogen deprived MCF-7Ca cells (UMB-1Ca) that were cultured in steroid-depleted medium for 6–8 months until they had acquired the ability to grow in the absence of added estrogen (12). These unique cells express high levels of aromatase, HER2, increased activation of AKT, and increased invasion (13). These cells were used to gain insight into the mechanism responsible for allowing cells to proliferate with estrogen deprivation. We hypothesized that long-term estrogen deprivation might be accompanied by enhanced estrogen signaling through increased aromatase activity, AIB1 and/or ER expression thereby enabling the cells to respond to low levels of estrogen. Thus, these studies highlight the importance of the interaction between the ER and AIB1 in mediating the proliferation of long-term estrogen-deprived breast cancer cells.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), 1% penicillin/streptomycin (P/S), trypsin/EDTA solution, Dulbecco’s phosphate buffered saline (DPBS) and geneticin (G418) were purchased from Life Technologies Inc. (Grand Island, NY, USA). Phenol red-free Improved Minimum Essential Medium (IMEM without phenol red) and phenol red-free trypsin/EDTA solution were obtained from Bio-fluids Inc. (Rockville, MD, USA). Fetal bovine serum (FBS) and charcoal-stripped FBS were purchased from Hyclone (Logan, UT, USA). Aromatase substrate [1β-3H]androstenedione (24.9 Ci/mmol), was purchased from New England Nuclear (Boston, MA, USA). Estradiol, androstenedione, 4-hydroxytamoxifen, RNA later™ were purchased from Sigma Chemical Company (St. Louis, MO, USA). Letrozole was kindly provided by Dr Dean Evans (Novartis Pharmaceuticals, Basel, Switzerland). Fulvestrant (anastrozole) was kindly provided by Dr Alan Wakefield (AstraZeneca Pharmaceuticals, Macclesfield, UK).
Cell culture
Human breast carcinoma MCF-7 cells were kindly provided by Dr Richard Santen (University of Virginia, VA, USA) and maintained in DMEM supplemented with 5% FBS and 1% P/S. The MCF-7 human breast cancer cell line was stably transfected with the human aromatase gene (designated MCF-7Ca cells) or they were stably transfected with the empty plasmid vector (designated MCF-7Cc cells) and both were kindly provided by Dr S. Chen (City of Hope, Duarte, CA, USA). MCF-7Ca and MCF-7Cc cells were maintained in standard medium: DMEM supplemented with 5% FBS, 1% penicillin/streptomycin and 750 μg/mL G418. The UMB-1Ca cells were developed in our laboratory as previously described (12). Briefly MCF-7Ca cells were transferred into a steroid-depleted medium, which consisted of phenol red-free IMEM supplemented with 5% charcoal-stripped FBS (CS-FBS), 1% penicillin/streptomycin solution, and 750 μg/mL G418. Cells were maintained in this medium for at least 6 months. After 6 months of estrogen deprivation, UMB-1Ca cells had the ability to proliferate in estrogen-depleted medium. The LTED (long-term estrogen-deprived) cells were kindly provided by Dr Wei Yue (University of Virginia, VA, USA) and had been developed from the MCF-7 cells by culturing in estrogen-deprived media for 3 months to 2 years until growth resumed (11). These cells were maintained in phenol red-free IMEM supplemented with 5% CS-FBS. Cells were cultured at 37°C in a humidified incubator with 5% CO2. The long-term letrozole-treated (LTLT-Ca) cells were isolated from tumors of MCF-7Ca cells grown in ovariectomized nude mice following 56 weeks of treatment with letrozole. After long-term letrozole treatment, the tumors acquired the ability to proliferate in the presence of the drug. Tumors were then removed and grown in culture in the presence of letrozole (10).
Gel electrophoresis and Western blot analysis
Equal amounts of either total protein or nuclear extract (50 μg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the Mini-PROTEAN3 electrophoresis module assembly (Bio-Rad, Hercules, CA, USA) and transferred at 4°C overnight to Hybond ECL nitrocellulose membranes (Amersham, Arlington Heights, IL, USA). Immunodetections were performed using anti-mouse monoclonal antibodies against human AIB1 (BD Transduction Laboratories, San Jose, CA, USA), human ERα (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), actin (Oncogene Research Products, Boston, MA, USA), and anti-rabbit human PR (Santa Cruz Biotechnology Inc.). Immunoreative bands were visualized using the enhanced chemiluminescence detection reagents (Amersham) according to the manufacturer’s instructions and quantitated by densitometry using ImageQuant 5.0 software.
Radiometric aromatase activity assay
The radiometric aromatase (3H2O) release assay was performed on breast cancer cell lines. Briefly, 1.5 μCi of [3H]androstenedione (Δ4A) was added to 1 mL of medium for 2 h. The medium was transferred to a glass tube and 300 μL of tricholoroacetic acid (TCA) was added to precipitate proteins. One milliliter of the mixture was extracted and mixed with 2 mL of chloroform to extract unconverted substrate and other steroids. A 0.7-mL aliquot of the aqueous phase was removed and mixed with 0.7 mL of a 2.5% activated charcoal suspension to remove residual steroids. Tritiated water (3H2O) formed during the aromatization of [3H]Δ4A to estrogen with release of tritium form C-1β and was measured by counting radioactivity in the supernatant. Nonspecific conversion was determined by performing the assays in the presence of a 100-fold excess of cold Δ4A and by performing the assay in wells that contained no cells.
3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) cell growth assay
The MTT assay is a colorimetric assay used to measure proliferation of cells. Cells were seeded at a density of 2.0×104 cells/mL into 24-well plates. The cells were allowed to attach for 24 h, fresh media was added and cells were treated for 5 days with estradiol, androstendione, letrozole, 4-hydroxytamoxifen, or Faslodex. On the day the assay was performed the medium was removed and 1 mL of a 1 mg/mL MTT stock solution was added to each well. The plates were incubated at 37°C for 3 h. The medium was then removed, 100–500 μL of DMSO was added and the plates were agitated vigorously for 5 min. The yellow tetrazolium salt (MTT) (Sigma, St. Louis, MO, USA) is reduced to a formazan product of a purple color by the mitochondrial dehydrogenase from living cells (14–16). The color can be detected by spectrophotometry at a wavelength of 560 nm.
Statistical analysis
Aromatase activity data were summarized as the mean±SEM by using the Prism version 4 software program (GraphPad Software Inc.). Western blot data and proliferation data were summarized as a percentage of the vehicle control. Student’s t-tests were used to compare relative cell proliferation or protein expression between vehicle control versus treatment groups, where a p value < 0.05 was considered significant and designated as (***, p < 0.001; **, p < 0.01; and *, p < 0.05).
Results
The effects of estradiol, androstenedione and letrozole on AIB1 and ERα expression
Western blot analyses were performed to determine if the expression of AIB1 and ERα in MCF-7 cells stably transfected with the human placental aromatase gene (MCF-7Ca cells) responded differently to hormonal treatment compared with control cells (MCF-7Cc cells). The MCF-7Cc cells maintained higher basal AIB1 compared with MCF-7Ca cells. Only when the MCF-7Ca cells were stimulated with androstenedione at 10−12 M to 10−9 M did AIB1 expression exceed that of the MCF-7Cc cells (Figure 1). Treating the MCF-7Cc cells with estradiol within the range of 10−12 M to 10−9 M, had a stimulatory effect on AIB1 at physiological levels, whereas higher estradiol (10−8 M to 10−6 M) tended to have an opposite effect on AIB1 expression (Figure 1). Similarly, this trend was observed in MCF-7Ca cells undergoing the same treatment. Thus within the physiological range (10−12 M to 10−8 M) of estradiol treatment, AIB1 levels were elevated compared with the untreated control. However, when comparing the two cell lines, AIB1 expression in MCF-7Ca cells was several-fold lower than in MCF-7Cc cells with no change in SRC-1 expression (data not shown).
Figure 1.
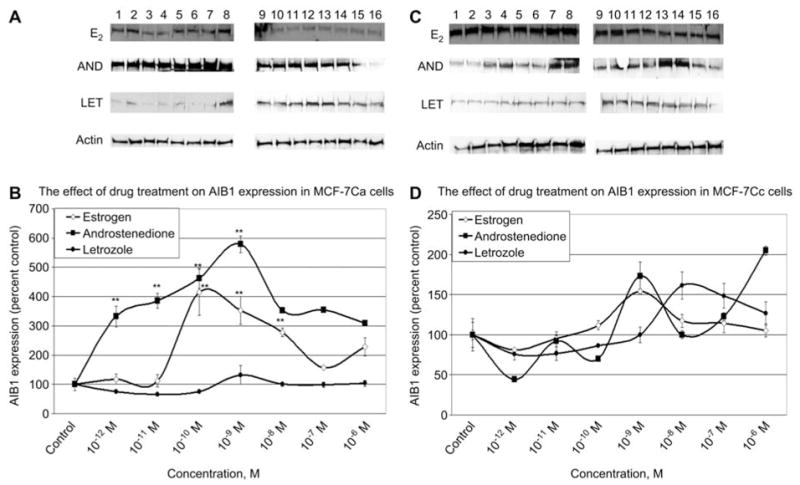
The effect of 5-day estradiol, androstenedione and letrozole treatment on AIB1 expression in MCF-7Ca and MCF-7Cc cells. Western blotting analysis of AIB1 protein expression in cell lysates. (A,C) MCF-7Ca cells and MCF-7Cc cells were treated with either vehicle control (0.1% ethanol) or various doses of estradiol, androstenedione, or letrozole for 5 days. Results of immunoblotting with anti-AIB1 are shown in duplicate. (Lanes 1–2 represent 10−12 M; lanes 3–4 represent 10−11 M; lanes 5–6 represent 10−10 M; lanes 7–8 represent 10−9 M; lanes 9–10 represent 10−8 M; lanes 11–12 represent 10−7 M; lanes 13–14 represent 10−6 M; lanes 15–16 represent vehicle control.) (B,D) Blots were striped and reprobed for β-actin and corrected for the internal loading control. Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant) and values were corrected for by normalizing to the β-actin internal control and represented as a percentage of the vehicle control±SEM (n=3).
Because androstenedione is the substrate for aromatase, MCF-7Ca cells were treated with androstenedione. The cells showed a similar pattern of AIB1 expression as MCF-7Cc cells treated with estrogen (Figure 1). The expression of AIB1 peaked at 10−9 M estradiol in MCF-7Cc cells and 10−9 M androstenedione in MCF-7Ca cells. Because estradiol upregulates AIB1 expression, it was anticipated that androstenedione would stimulate AIB1 because it is the substrate which aromatase converts to estrogen. Androstenedione treatment increased AIB1 in MCF-7Ca cells by 500% (p < 0.01) above the control. This was the highest level of expression of any hormone treatment in both cells examined, suggesting that AIB1 might be sensitive to both estrogen and androstenedione.
MCF-7Ca and MCF-7Cc cells treated with subphysiological levels of estradiol caused a modest increase in ERα, whereas higher levels did not cause any further changes or reduced the expression respectively (Figure 2). There was a dramatic induction of ERα compared with the vehicle control, when both cells were treated with androstenedione, indicating that both androgens and estrogens can affect both ERα and AIB1 in both cell lines.
Figure 2.
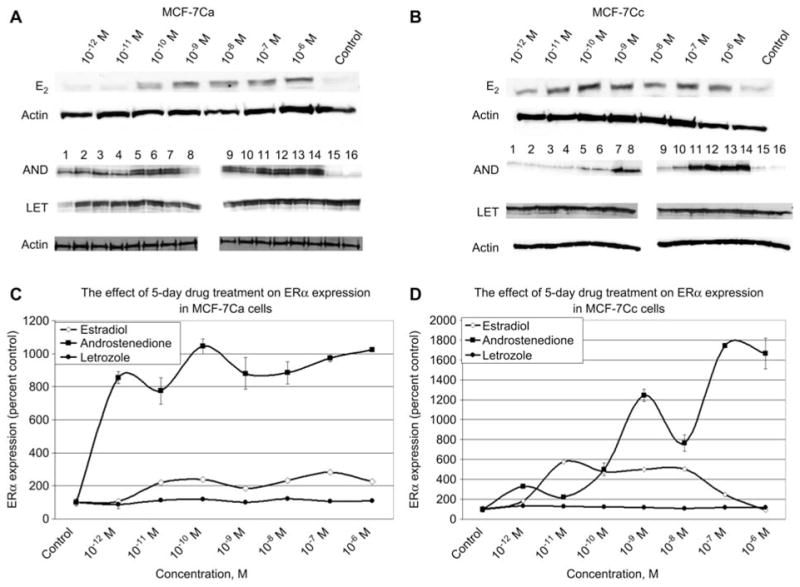
The effect of 5-day estradiol, androstenedione and letrozole treatment on ERα expression in MCF-7Ca and MCF-7Cc cells. (A,B) Western blotting analysis of ERα protein expression in nuclear extracts of MCF-7Ca and MCF-7Cc cells treated with either vehicle control (0.1% ethanol) or various doses of estradiol, androstenedione or letrozole for 5 days. Results of immunoblotting for estradiol treatment with anti-ERα is shown once whereas treatment with androstenedione and letrozole is shown in duplicate. (Lanes 1–2 represent 10−12 M; lanes 3–4 represent 10−11 M; lanes 5–6 represent 10−10 M; lanes 7–8 represent 10−9 M; lanes 9–10 represent 10−8 M; lanes 11–12 represent 10−7 M; lanes 13–14 represent 10−6 M; lanes 15–16 represent vehicle control) unless otherwise labeled. (C,D) Blots were striped and reprobed for β-actin and corrected for the internal loading control. Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant) and values are represented as a percentage of the vehicle control±SEM (n=3).
The effect of estrogen deprivation on ERα and AIB1 expression
After demonstrating the effect of estradiol on AIB1 and ERα in MCF-7Ca and MCF-7Cc cell lines, we investigated the expression profile of both proteins in cells which were long-term estrogen deprived. UMB-1Ca and LTED cells were cultured in phenol red-free, charcoal-stripped medium for 5 days in the presence of increasing concentrations of estradiol. Estradiol treatment did not cause a marked difference in AIB1 expression in both LTED and UMB-1Ca cells compared with the parental MCF-7Cc and MCF-7Ca cells, respectively (Figure 3). By contrast, there was a marked difference in ERα with estradiol treatment between parental and estrogen-deprived cells (Figure 3). Estradiol only modestly altered ERα expression in MCF-7Ca cells, with no significant change in ERα in estrogen-deprived UMB-1Ca cells (Figure 3). However, estradiol caused a dose-dependent downregulation of ERα in LTED cells. At higher concentrations of estradiol, ERα was inhibited by as much as 99% (p < 0.01). Faslodex also caused a decrease in ERα and AIB1 expression in UMB-1Ca cells (Figure 4) as well as all cell lines examined suggesting that trace levels of estrogen still remain in the media of the long-term estrogen-deprived cells. The progesterone receptor (PgR) was measured as a function of ER activity by Western blot analysis demonstrating that the ER maintained transcriptional activity in both MCF-7Ca and MCF-7Cc cells (data not shown).
Figure 3.
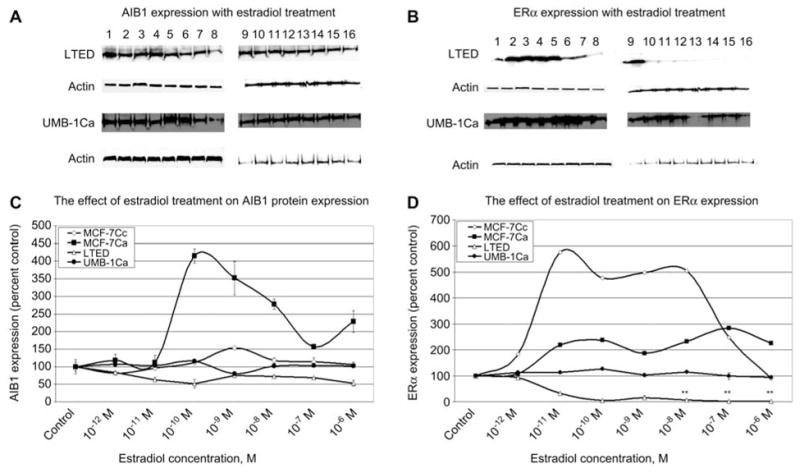
Comparison of the AIB1 and ERα expression profile in normal versus long-term estrogen deprived breast cancer cells. (A,B) Western blotting analysis of AIB1 and ERα protein expression in LTED and UMB-1Ca cells treated with either vehicle (0.1% ethanol) or various doses of estradiol for 5 days. Results of immunoblotting with anti-AIB1 and anti-ERα are shown in duplicate. (Lanes 1–2 represent 10−12 M; lanes 3–4 represent 10−11 M; lanes 5–6 represent 10−10 M; lanes 7–8 represent 10−9 M; lanes 9–10 represent 10−8 M; lanes 11–12 represent 10−7 M; lanes 13–14 represent 10−6 M; lanes 15–16 represent vehicle control.) (C,D) Blots were striped and reprobed for β-actin and corrected for the internal loading control. Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant) and values were corrected for by normalizing to the β-actin internal control and represented as a percentage of the vehicle control±SEM (n=3).
Figure 4.
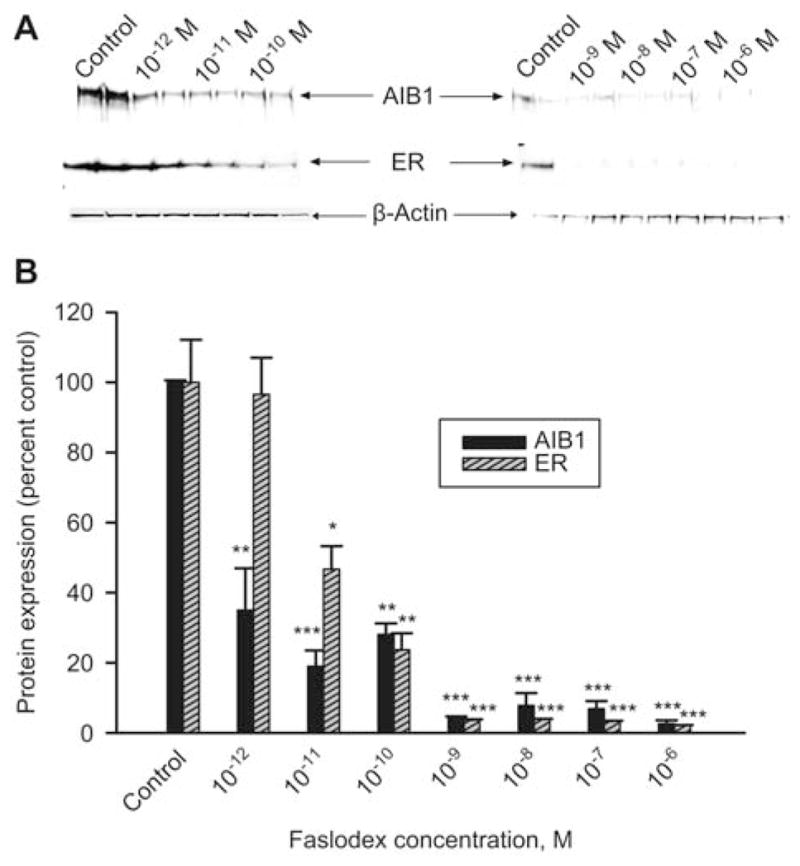
The effect of Faslodex treatment on AIB1 and ERα expression in UMB-1Ca Cells. (A) AIB1 and ERα protein expression in whole cell lysates of UMB-1Ca cells treated with either vehicle control (0.1% ethanol) or various doses of Faslodex for 5 days. Results of immunoblotting with anti-AIB1 and anti-ERα are shown in duplicate. (B) Blots were striped and reprobed for β-actin and corrected for the internal loading control. Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant) and values were corrected for by normalizing to the β-actin internal control and represented as a percentage of the vehicle control±SEM (n=3).
Long-term estrogen deprivation alters aromatase activity
Aromatase activity assays were performed in all of the MCF-7 derived cell lines to determine whether long-term estrogen deprivation was associated with changes in aromatase activity (Table 1). MCF-7 wild-type cells express low levels of aromatase (35.15±2.5 fmol/150,000 cells/h). However, LTED cells exhibited a threefold increase in aromatase activity. The MCF-7Ca cells, which were transfected with the aromatase gene, express much higher aromatase activity levels (247.04±18.7 fmol/150,000 cells/h) compared with the LTED cells. Although aromatase was a stable transfectant in MCF-7Ca cells aromatase activity was dramatically induced >eightfold (p < 0.001) in UMB-1Ca cells compared with the parental cells. The LTLT-Ca cells that were cultured in the presence of letrozole expressed levels of aromatase similar to MCF-7 cells. This further suggested the more marked reduction in available estrogen by aromatase inhibition compared with “estrogen withdrawal”. These results suggest that overall long-term estrogen deprivation via estrogen withdrawal causes an upregulation of aromatase activity, whereas long-term letrozole treatment has the opposite effect.
Table 1.
Aromatase activity in human breast cancer cells. Aromatase activity in MCF-7, MCF-7Ca, UMB1-Ca, LTED and LTLT-Ca cells were measured using the 3H2O release assay as described in materials and methods. Values represent aromatase activity (fmol/150,000 cells/h)±SEM. Cells were treated with vehicle control 24 h and then incubated with [3H]androstenedione for 18 h. The activity of the enzyme after vehicle treatment is corrected for total protein amount.
| Cell line | Aromatase activity (fmol/150,000 cells/h) |
|---|---|
| MCF-7 | 35.1477±2.5 |
| MCF-7Ca | 247.0361±18.7 |
| UMB1-Ca | 2086.7±27.6 |
| LTED | 103.9±3.9 |
| LTLT-Ca | 38.2±6.3 |
Long-term estrogen deprivation alters cell proliferation in response to estradiol
To investigate the mechanism associated with the growth advantage of long-term estrogen deprivation, the response of the LTED and MCF-7 cells in the presence of estradiol, tamoxifen and Faslodex was examined. The LTED cells displayed enhanced sensitivity to estradiol compared with the MCF-7 wild-type cells, and were stimulated (Figure 5A,B). The growth of the LTED cells were modestly stimulated in the presence of subphysiologic levels of estradiol (10−14 and 10−13 M) (Figure 6), but unaltered in the presence of 10−12 and 10−11 M estradiol (Figure 6). However, the LTED cells were inhibited in a dose-dependent manner when the concentration of estradiol was increased, supporting previous studies which reported that long-term estrogen deprivation causes estradiol hypersensitivity (11).
Figure 5.
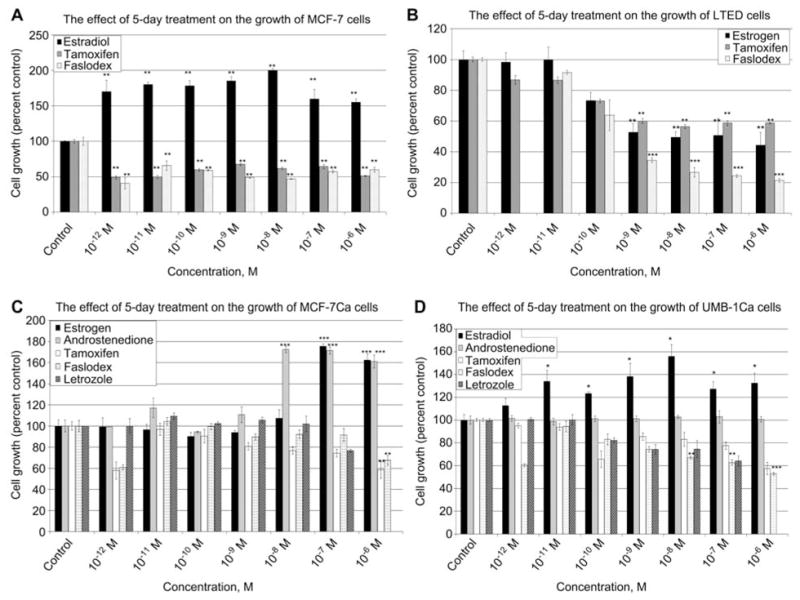
The effect of 5-day estradiol, antiestrogen and letrozole treatment on the growth of MCF-7, LTED, MCF-7Ca and UMB-1Ca cells. MTT cell proliferation assay was performed on (A) MCF-7 cells, (B) LTED cells, (C) MCF-7Ca cells and (D) UMB-1Ca cells. Cells were cultured in phenol red-free charcoal-stripped media for 2 days to deplete cells of steroids. Cells were seeded at a density of 20,000 cells/mL into 24-well plates and allowed to attach overnight. MCF-7 cells were cultured in phenol red-free IMEM for 5 days with vehicle (0.1% ethanol), estradiol, androstenedione, tamoxifen, Faslodex or letrozole. On the day the assay was performed 1 mL of a 1 mg/mL solution of MTT was added to each well and incubated for 3 h. The product was detected using a spectrophotometry at a wavelength of 560 nm. Values are represented as a percentage of the untreated control±SEM (n=3).
Figure 6.
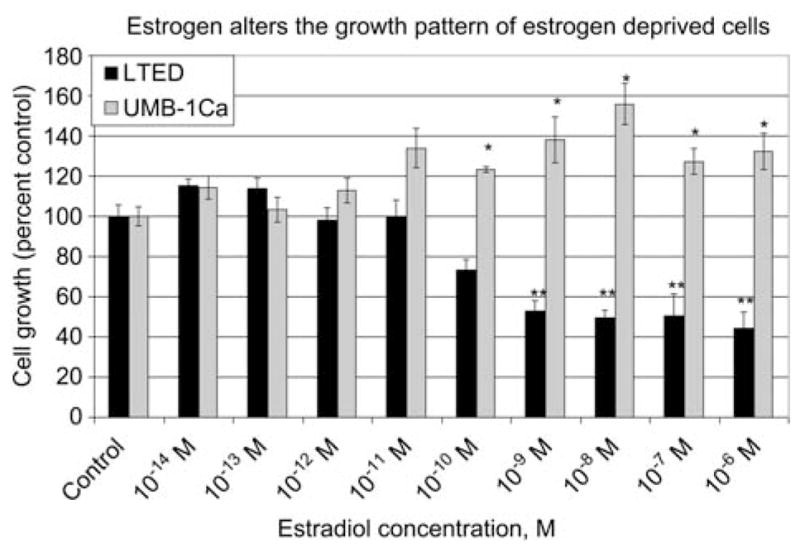
The effect of 5-day drug treatment on the growth of MCF-7Ca and UMB-1Ca Cells. MTT cell proliferation assay was performed on (A) MCF-7Ca and (B) UMB-1Ca cells. Cells were cultured in phenol red-free charcoal-stripped media for 2 days to deplete cells of steroids. Cells were seeded at a density of 20,000 cells/mL into 24-well plates and allowed to attach overnight. MCF-7Ca cells were cultured in phenol red-free IMEM for 5 days with vehicle (0.1% ethanol), estradiol, androstenedione, tamoxifen, Faslodex or letrozole. On the day the assay was performed 1 mL of a 1 mg/mL solution of MTT was added to each well and incubated for 3 h. The product was detected using spectrophotometry at a wavelength of 560 nm. Values are represented as a percentage of the untreated control±SEM (n=3).
When the LTED cells were treated with the antiestrogen tamoxifen, the maximum growth inhibition of 40% (p < 0.01) was achieved at a concentration of 10−9 M (Figure 5B) whereas higher concentrations did not cause any further inhibition. Interestingly, estradiol showed a similar dose-response effect to tamoxifen possibly reflecting the partial estrogenic action of this antiestrogen. However, when the LTED-cells were treated with Faslodex, there was a dose-dependent inhibition reaching a maximum growth inhibition of >75% (p < 0.001).
Estradiol hypersensitivity is altered by long-term estrogen-deprived cells overexpressing aromatase
Estrogen-deprived cells responded differently to estrogen-dependent cells in their response to low levels of estrogen. To determine the effect on the growth of cells overexpressing aromatase, UMB-1Ca cells were compared with cells that were not estrogen deprived (MCF-7Ca cells). Both the UMB-1Ca and the MCF-7Ca cell lines exhibited differential growth responses to estradiol treatment (Figure 5C,D). The growth response of the UMB-1Ca cells was more sensitive to the stimulatory effects of estradiol. These cells exhibited an increase in proliferation with concentrations as low as 10−11 M estradiol and continued to be stimulated with increasing doses of estradiol up to 10−6 M estradiol (Figure 5D). However, the MCF-7Ca cells required higher concentrations of estradiol (10−7 M to 10−6 M) to display an increase in proliferation (Figure 5C). Lower concentrations (10−12 M to 10−9 M) of estradiol had very little effect on the proliferation of MCF-7Ca cells. Thus, UMB-1Ca cells, which acquired the ability to grow in an estrogen-deprived environment, were more sensitive to the effects of estradiol, requiring less estradiol to stimulate growth than the MCF-7Ca cells.
Estradiol caused a growth stimulatory effect on both MCF-7Ca and UMB-1Ca cells, whereas growth was significantly inhibited by tamoxifen and Faslodex (Figure 5C,D). However, UMB-1Ca cells were slightly more sensitive to the growth inhibitory effects of antiestrogens than the MCF-7Ca cells. Faslodex (10−6 M) inhibited the growth of UMB-1Ca cells by 50% (p < 0.001), whereas MCF-7Ca cells was only inhibited by 30% (p < 0.01). Both cell lines were treated with Faslodex and tamoxifen alone and not in the presence of estradiol thereby showing a lesser inhibitory effect than would be expected in the presence of both the ligand and inhibitor.
Comparison of the differential growth effects of LTED cells and UMB-1Ca cells
We compared UMB-1Ca cells overexpressing aromatase with LTED cells that contained moderate levels of activity. To study whether the presence of aromatase could affect the level of sensitivity to estrogen, we measured the growth response of these cells to estradiol in vitro. Both cell lines were cultured in steroid-depleted medium and treated with estradiol in the range of 10−14 to 10−6 M for 5 days. The growth of the UMB-1Ca cells responded differently to estradiol than the LTED cells (Figure 6). The increase in cell number of the UMB-1Ca cells remained slightly above the control level at concentrations lower than 10−11 M estradiol, but this was not statistically different to the control. However, between the concentrations of 10−10 and 10−6 M estradiol the increase in cell number of the UMB-1Ca cells remained approximately 20% (p < 0.05) above the control. By contrast, growth of the LTED cells was inhibited by concentrations of estradiol greater than 10−11 M. Both LTED and UMB-1Ca cells contain higher levels of AIB1, ER and aromatase compared with their parental cell lines, MCF-7 and MCF-7Ca, respectively. Therefore, both the LTED and UMB-1Ca cells have an altered response to estrogen compared with the parental cells from which they were derived, MCF-7 and MCF-7Ca, respectively. These findings suggest that upregulation of the estrogen pathway has a significant role in the ability of these cells to adapt to conditions of a low estrogen environment.
Comparison of basal levels of AIB1 and ERα expression in long-term estrogen-deprived cell lines
Estradiol stimulation caused a differential growth response in the long-term estrogen-deprived cell lines. To study the mechanism responsible for these differences we measured expression of both the ERα and AIB1 in the estrogen-dependent cells and compared them with the expression of these proteins in estrogen-deprived cell lines and in cells isolated from tumors of nude mice treated with letrozole (LTLT-Ca cells). Western blot analyses were performed to investigate the endogenous AIB1 and ERα protein expression of the MCF-7Cc, LTED, MCF-7Ca as well as their variant estrogen-deprived cell lines: LTLT-Ca and UMB-1Ca cells (Figure 6). Among the cell lines which were not long-term estrogen deprived, the MCF-7Cc cells contained levels of AIB1 that were 96% (p < 0.001) higher than the MCF-7Ca cells (Figure 7). However, the levels of the ERα were similar between both the MCF-7Cc and MCF-7Ca cells (Figure 7). The LTED cells contained a slightly elevated level of AIB1 relative to the MCF-7Cc control, but the levels of the ERα was increased >200% (p < 0.001) above that of the MCF-7Cc cells (Figure 7). In the LTLT-Ca cells and UMB-1Ca cells the levels of AIB1 were dramatically elevated by as much as 900% (p < 0.001) and 3500% (p < 0.001) above the respective control levels. However, the level of the ERα was reduced in the LTLT-Ca cells by 50% (p < 0.01) but increased in the UMB-1Ca cells by more than 100% (p < 0.001). These data suggest that the marked increase in AIB1 in long-term estrogen-deprived cells expressing moderate or high levels of aromatase could increase estrogen regulated transcription under conditions of estrogen deprivation. Estrogen deprivation achieved by estrogen withdrawal also caused upregulation of aromatase expression, ERα and AIB1, in LTED and UMB-1Ca cells. Although estrogen deprivation owing to long-term letrozole treatment also caused an increase in AIB1 expression (1000-fold), by contrast, both ERα and aromatase were reduced. This provides evidence that estrogen deprivation achieved by aromatase inhibitor treatment or estrogen withdrawal can result in different cellular responses. The results suggest that the three variant cell lines represent a transition from moderate estrogen deprivation (LTED) in which upregulation of ER is sufficient for proliferation, to extreme estrogen deprivation (LTLT-Ca). These cells might no longer be able to utilize the estrogen signaling pathway for proliferation effectively, and might recruit hormone-independent pathways to activate the ER in estrogen-deprived conditions (13).
Figure 7.
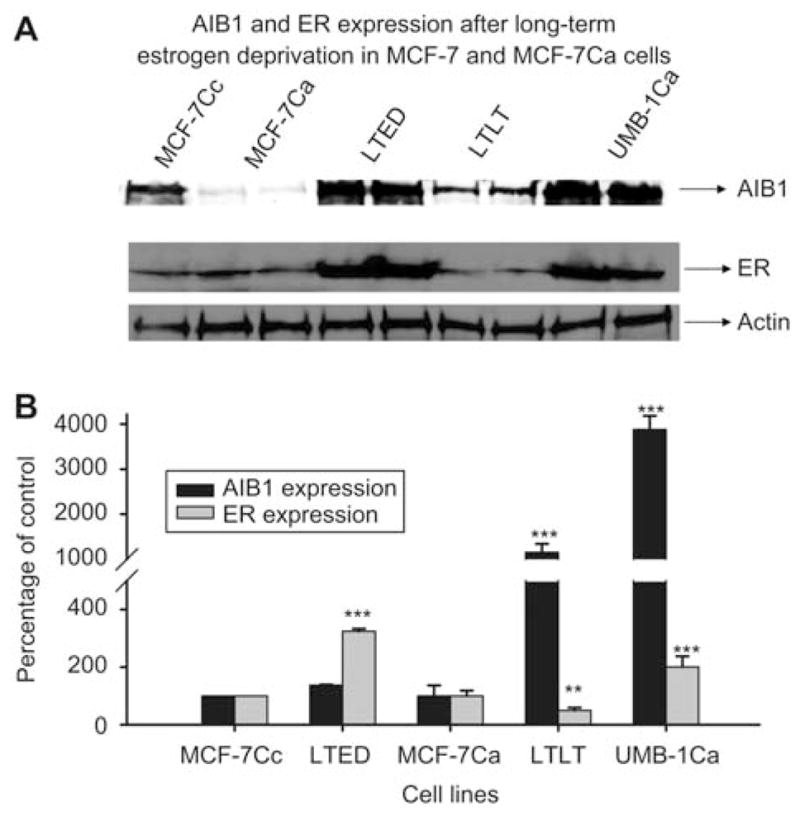
AIB1 and ERα expression after long-term estrogen deprivation in MCF-7 and MCF-7Ca cells. (A) Western immunoblotting analysis of AIB1 and ERα protein expression in whole cell lysates of MCF-7Cc, MCF-7Ca, LTED, LTLT-Ca and UMB-1Ca cells. Cells were seeded in their respective media and total cell lysates were prepared for Western blot analysis. (B) Results of immunoblotting with anti-AIB1 and anti-ERα are shown in duplicate in all lanes except MCF-7Cc lane. Blots were striped and reprobed for β-actin and corrected for the internal loading control. Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant) and values were corrected for by normalizing to the β-actin internal control and represented as a percentage of the vehicle control±SEM (n=3).
Discussion
Our results indicate that long-term estrogen deprivation in MCF-7 derived breast cancer cells is associated with alterations in the levels of aromatase, AIB1, ERα and cell growth. We hypothesize that this might be one mechanism by which these altered cells adapt and continue to proliferate in low estrogenic environments. To test this hypothesis we examined the effect of estradiol, androstenedione and letrozole treatment on the expression profile of SRC-1, AIB1, ERα and aromatase activity. No changes in SRC-1 were observed, suggesting that it might not have an important role in these cells, so we examined AIB1. Results indicated ER-positive MCF-7Ca breast cancer cells (stably transfected human aromatase) contain basal levels of AIB1 which were 25-fold lower than the MCF-7Cc control cells without androstenedione stimulation. Upon androstenedione stimulation the MCF-7Ca cells expressed similar levels of AIB1 to the MCF-7Cc cells treated with estradiol, suggesting that AIB1 expression was induced to a greater extent by an estrogenic source (aromatized androstenedione) in cells expressing aromatase (MCF-7Ca cells) than in cells with negligible levels of aromatase (MCF-7Cc cells). The change in basal AIB1 expression in the absence of hormones between both cell lines was independent of the ERα status because basal ERα levels remained relatively constant. Therefore, we can conclude that higher levels of ERα do not necessarily correlate with higher AIB1. However, once these hormone-dependent cells are stimulated with estradiol, AIB1 is dramatically elevated. This elevation, in addition to a simultaneous elevation in ER under estrogen stimulation, suggests a mechanism by which cancer cells could proliferate and result in disease progression.
The growth dependence of many breast cancers on estrogen has been exploited therapeutically by estrogen deprivation, but most patients eventually develop resistance. We hypothesize that as cells become insensitive to treatment, changes might occur in the estrogen signaling pathway whereby there are increases in the activity and level of aromatase expression, ERα and AIB1 that attenuate ER-mediated transcription. To examine this phenomenon, we chose two long-term estrogen-deprived cell lines, UMB-1Ca and LTED. Estradiol treatment of the UMB-1Ca cells caused AIB1 to remain similar to the control values. Interestingly, the expression of AIB1 was very high regardless of estradiol stimulation. By contrast, AIB1 protein levels in the LTED cells were inhibited in a dose-dependent manner by as much as 50% with estrogen treatment. Taken together with the results from the parental cells, this suggests that the estrogen-deprived cells are no longer sensitive to estrogen owing to a lack of response of AIB1 with estradiol treatment.
Increased concentrations of estradiol led to both decreased AIB1 and proliferation of LTED cells. A similar study previously reported that a reduction in AIB1 reduces the estrogen-dependent proliferation of human MCF-7 breast cancer cells (17) suggesting that AIB1 exerts a rate-limiting role for hormone-dependent human breast tumor growth. The decrease in expression of AIB1 could result from the marked reduction of ERα. However, because ERα is significantly downregulated by estradiol treatment, AIB1 might still be present to compensate for the lack of ERα to maintain transcriptional activity. This is probably not the mechanism involved in the UMB-1Ca cells, because they respond to estrogens differently.
Understanding mechanisms of resistance to long-term estrogen deprivation could provide insight into the limitations of existing therapies suggesting ways to improve treatment strategies. Our data show that AIB1 and ERα of both long-term estrogen-deprived cells (UMB-1Ca or LTED) are upregulated compared with the parental cells. Estrogen withdrawal by culturing cells in the absence of estradiol was not the only mechanism by which upregulation of AIB1 was achieved. Cells deprived of estrogen by long-term letrozole treatment (LTLT-Ca cells) also demonstrated upregulation of AIB1 but not ER or aromatase activity. From this, it can be extrapolated that breast tumors deprived of estrogen via an antiestrogen or aromatase inhibitor, can result in increased AIB1 resulting in enhanced transcription and ultimately conferring a growth advantage to breast cancer cells. An explanation for the upregulation of AIB1, ER and aromatase with estrogen deprivation could be a negative regulatory mechanism. As breast cancer cells are deprived of estrogen, this might trigger a cell survival response allowing for an increase in AIB1 and ERα, enabling the cells to continue to proliferate.
The data also indicated that long-term estrogen deprivation was associated with an increase in aromatase expression as compared with the parental cell line as demonstrated by the UMB-1Ca and LTED cells. Upregulation of aromatase as a result of estrogen deprivation could be the response of the cells to the new environment and an attempt to compensate for the loss of estrogen required for growth. In addition to enhanced aromatase activity, AIB1 and ERα signaling, we also observed that estrogen deprivation results in the upregulation of growth factor signaling pathways leading to a more aggressive and hormone refractory phenotype (13). Ample biological evidence supports the idea that crosstalk between growth factor receptor and ER pathways is important in the regulation of breast cancer cells (18). It appears that during estrogen deprivation, several components of the estrogen pathway are increased ultimately leading to enhanced proliferation.
Our data indicate that the growth of the estrogen-deprived cell lines (UMB-1Ca and LTED cells) respond differently to estradiol. Both cell lines adapted to estrogen deprivation by developing increased sensitivity to estradiol. The LTED cells showed increased sensitivity to low doses of estradiol and were inhibited by higher concentrations of estradiol, whereas the UMB-1Ca cells continued to proliferate with increasing levels of estradiol with no differences in the basal levels of AIB1 and ERα. This suggests that although both cell lines initially express comparable levels of AIB1 and ER, their responses to estradiol are different and might utilize AIB1 and the ER through separate mechanisms.
Until now the mechanism which allows the growth of LTED cells to be inhibited in the presence of estradiol while not inhibiting the growth of the UMB-1Ca cells has not been elucidated. This is the first report to identify the mechanism responsible for the differences in the response of both cell lines to estradiol treatment. In LTED cells, ERα dramatically decreased to <80% of the control with estradiol treatment, whereas ERα in UMB-1Ca cells remained within ±20% of the control. Although both cell lines are not dependent on estrogen for growth, the lack of sufficient ER renders the LTED cells unable to proliferate in the presence of estradiol regardless of AIB1 expression. UMB-1Ca cells maintain sufficient levels of both AIB1 and ER which are required for proliferation in response to estradiol, explaining why the growth of these cells is not inhibited by higher doses of estradiol. Recently it was suggested that estrogen deprivation might trigger activation of a non-genomic, estrogen-regulated MAPK pathway (19). However, if this pathway was activated in the LTED cells, in the presence of estradiol, the MAPK signaling cascade would also be downregulated owing to insufficient ER compared with UMB-1Ca cells. In summary, the LTED cells have a lower capacity to utilize estradiol than UMB-1Ca cells owing to decreased ER in the presence of estrogen.
Overall, our studies indicate that estrogen-deprived UMB-1Ca cells contain multiple factors which contribute to their more aggressive and altered phenotype. LTED and UMB-1Ca cells proliferating in the presence of low levels of estradiol have adapted by increasing AIB1, ER and aromatase expression, suggesting that these cells have an altered response to estrogens yet remain responsive to low concentrations. Studies with LTLT-Ca cells derived from letrozole-treated mice demonstrated that exposure to letrozole has a different effect on estrogen signaling than cells deprived of estradiol. Letrozole caused downregulation in AIB1 and ER compared with UMB-1Ca cells, possibly owing extreme estrogen deprivation. Recently, we have shown that LTLT-Ca cells have activated MAPK signaling in response to estrogen deprivation. These data are clinically relevant, because long-term letrozole therapy not only reduces ER levels compared with estrogen deprivation, but also causes a reduction in AIB1 levels. Therefore, targeting this pathway might be useful for treating patients whose tumors have developed resistance and failed hormonal treatment.
Acknowledgments
This work was supported by an NIH grant number RO1CA062483-22 (A.M.H.) and F31CA083172-03S1 (S.L.T.).
Footnotes
Disclosure of potential conflicts of interest
Authors S.L.T., G.S. and A.H.B. have nothing to declare.
References
- 1.Litherland S, Jackson IM. Antiestrogens in the management of hormone-dependent breast cancer. Cancer Treat Rev. 1988;15:183–94. doi: 10.1016/0305-7372(88)90002-3. [DOI] [PubMed] [Google Scholar]
- 2.Early Breast Cancer Trialists’ Collaborative Group. Systemic treatment of early breast cancer by hormonal, cytotoxic or immune therapy: 133 randomized trials among 31,000 recurrences and 24,000 deaths among 75,000 women. Lancet. 1992;339:71–85. [PubMed] [Google Scholar]
- 3.ATAC Trialists’ Group. The ATAC (Arimidex, Tamoxifen, Alone or in Combination) adjuvant breast cancer trial in post-menopausal women. Breast Cancer Res Treat. 2001;69:210. [Google Scholar]
- 4.Baum M, Buzdar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T for the ATAC (Arimidex, Tamoxifen Alone or in Combination) Trialists’ Group. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomized trial. Lancet. 2002;359:2131–9. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 5.Coombes RC, Hall E, Gibson LJ, Paridaens R, Jassem J, Delozier T, Jones SE, Alvarez I, Bertelli G, Ortmann O, Coates AS, Bajetta E, Dodwell D, Coleman RE, Fallowfield LJ, Mickiewicz E, Andersen J, Lonning PE, Cocconi G, Stewart A, Stuart N, Snowdon CF, Carpentieri M, Massimini G, Bliss JM Intergroup Exemestane Study. A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. N Engl J Med. 2004;350:1081–92. doi: 10.1056/NEJMoa040331. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, O’Malley BW. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc Natl Acad Sci USA. 2000;97:6379–84. doi: 10.1073/pnas.120166297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hudelist G, Czerwenka K, Kubista E, Marton E, Pischinger K, Singer CF. Expression of sex steroid receptors and their cofactors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res Treat. 2003;78:193–204. doi: 10.1023/a:1022930710850. [DOI] [PubMed] [Google Scholar]
- 8.Oñate SA, Tsai SY, Tsai MJ, O’Malley BW. Sequence and characterization of a coactivator for the steroid receptor superfamily. Science. 1995;270:1354–7. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 9.Anzick S, Kononen J, Walker RL, Azorosa DO, Tanner MM, Guan X, Sauter G, Kallioniemi O, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–7. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 10.Jelovac D, Sabnis G, Long BJ, Macedo L, Goloubeva OG, Brodie AMH. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 11.Masumara S, Santner SJ, Heitjan DF, Santen RJ. Estrogen deprivation causes estradiol hypersensitivity in human cancer cells. J Clin Endocrinol Metab. 1995;80:2918–25. doi: 10.1210/jcem.80.10.7559875. [DOI] [PubMed] [Google Scholar]
- 12.Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:5092–5. [PubMed] [Google Scholar]
- 13.Sabnis G, Jelovac D, Long BJ, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005;65:3903–10. doi: 10.1158/0008-5472.CAN-04-4092. [DOI] [PubMed] [Google Scholar]
- 14.Barile FA. Cellular methods of general toxicity. In: Barile FA, editor. Introduction to in vitro cytotoxicology mechanisms and methods. Florida: CRC Press; 1994. pp. 54–5. [Google Scholar]
- 15.Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods. 1986;89:271–7. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- 16.Mossmann T. Rapid colorimetric assay for cellular growth and survival: application and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 17.List H-J, Reiter R, Singh B, Wellstein A, Riegel AT. Expression of the nuclear coactivator AIB1 in normal and malignant breast tissue. Breast Cancer Res Treat. 2001;68:21–8. doi: 10.1023/a:1017910924390. [DOI] [PubMed] [Google Scholar]
- 18.Osborne C, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, Wong J, Allred DC, Clark GM, Schiff R. Role of the ER coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95:353–61. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 19.Song R-X, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, Santen RJ. Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol. 2002;16:116–27. doi: 10.1210/mend.16.1.0748. [DOI] [PubMed] [Google Scholar]