

Abstract
Immunoglobulin (Ig) G4-related disease (also known as ‘IgG4-related sclerosing disease’, ‘IgG4-related systemic disease’ or ‘hyper-IgG4-disease’) is a recently recognized systemic fibroinflammatory disease associated with IgG4-positive plasma cells in tissue lesions. IgG4-related disease was initially described as autoimmune pancreatitis, but it is now known to affect virtually any organ. The authors describe a patient presenting with multi-organ manifestations, including airway inflammation mimicking asthma, pulmonary parenchymal infiltrates, intrathoracic lymphadenopathy, submandibular gland swelling and a kidney mass.
Keywords: Airflow obstruction, Asthma, IgG4-related disease
Abstract
La maladie liée aux immunoglobulines (Ig) G4 (aussi appelée maladie sclérosante liée aux IgG4 ou syndrome d’hyper-IgG4) est une maladie fibro-inflammatoire systémique récemment reconnue qui s’associe à des cellules plasmatiques positives aux IgG4 contenues dans les lésions tissulaires. La maladie liée aux IgG4 a d’abord été décrite comme une pancréatite auto-immune, mais on sait maintenant qu’elle peut toucher à peu près n’importe quel organe. Les auteurs décrivent le cas d’un patient atteint de manifestations multiorganiques, y compris une inflammation des voies aériennes imitant l’asthme, des infiltrats pulmonaires parenchymateux, une lymphadénopathie intrathoracique, un œdème des glandes sous-maxillaires et une masse rénale.
Learning objectives
Learn multiorgan manifestations of immunoglobulin G4-related disease (IgG4-RD) and its responsiveness to corticosteroids.
Recognize IgG4-RD as a possible etiology of refractory asthma-like symptoms.
Pre-test
What is IgG4-RD?
What laboratory tests are of value in making a diagnosis of IgG4-RD?
What are the typical pathological findings seen in IgG4-RD?
What is the first-line treatment for IgG4-RD?
CASE PRESENTATION
A 44-year-old male nonsmoker presented with a two-year history of sinus congestion, wheezing, dyspnea and cough. Pharmacological treatment, including antibiotics, inhaled corticosteroids and antihistamines, did not improve his symptoms. Submandibular gland enlargement was apparent one year after the onset of symptoms. Left submandibular gland biopsy performed at another hospital revealed chronic sialadenitis. Sialadenitis and other symptoms improved after several weeks of oral prednisone; however, these manifestations recurred three months after discontinuation of prednisone therapy. A computed tomography (CT) scan of the neck, chest and abdomen demonstrated enlarged submandibular glands, as well as hilar and mediastinal lymphadenopathy, a 2.5 cm spiculated infiltrate in the left upper lobe of the lung and a 2.5 cm low-attenuation mass in the left kidney (Figures 1A, 1B, 1C and 1D), with no evidence of pancreatic abnormalities. The patient was referred to the Mayo Clinic (Minnesota, USA) for further evaluation.
Figure 1).
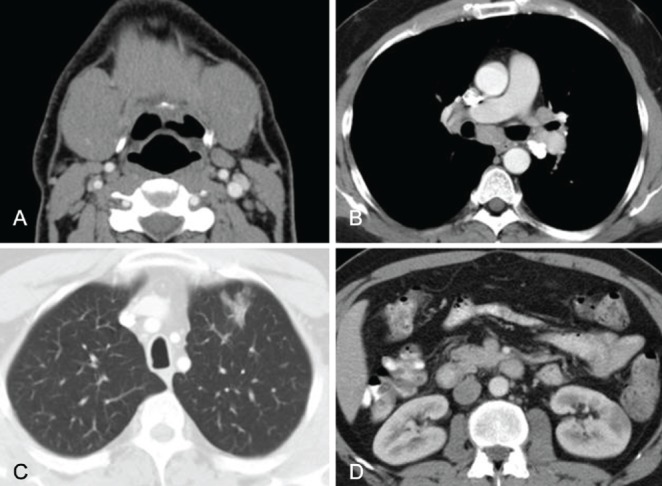
Computed tomography (CT). A Neck CT demonstrating enlarged submandibular glands. B Chest CT demonstrating hilar and mediastinal lymphadenopathy. C Chest CT demonstrating a 2.5 cm spiculated infiltrate in the left upper lobe. D Abdominal CT demonstrating a 2.5 cm low-attenuation mass in the left kidney
Physical examination was unremarkable except for bilateral submandibular gland enlargement. Pulmonary function testing (PFT) demonstrated borderline airflow obstruction with a forced expiratory volume in 1 s (FEV1)/forced vital capacity ratio of 0.6, FEV1 of 85% of predicted normal value and positive methacholine challenge. Serum protein electrophoresis showed polyclonal hypergammaglobulinemia with serum immunoglobulin (Ig) level of 25 g/L (normal range 0.6 g/L mg/dL to 15 g/L) and IgG4 level of 20.2 g/L (normal range 0.08 g/L to 1.4 g/L). Other laboratory tests were normal including complete blood count with differentials, chemistries, angiotensin converting enzyme level and vasculitis/connective tissue serological panel. Bronchoscopy demonstrated inflammatory changes along the tracheobronchial tree. Mucosal biopsy of the right carina 1 revealed dense chronic inflammation in the bronchial wall. Immunostaining for IgG4 highlighted 15 to 20 IgG4-positive plasma cells per high-power field (Figure 2A and 2B). Needle aspiration of hilar and mediastinal lymphadenopathy was negative for malignancy. Outside submandibular gland pathology slides were retrieved and examined using immunostaining, which demonstrated numerous IgG4-positive plasma cells with an IgG4+/IgG+ cell ratio of 0.6 (Figure 2C and 2D). Diagnosis of IgG4-RD was made and the patient was started on prednisone 40 mg daily. One month later, his symptoms had resolved and the submandibular glands had returned to normal size. Repeat CT showed near-complete resolution of the focal lung infiltrate, intrathoracic lymphadenopathy and left kidney mass. PFT results had returned to normal. Serum IgG4 level decreased to 0.55 g/L. Corticosteroid therapy was slowly tapered over the following two months.
Figure 2).
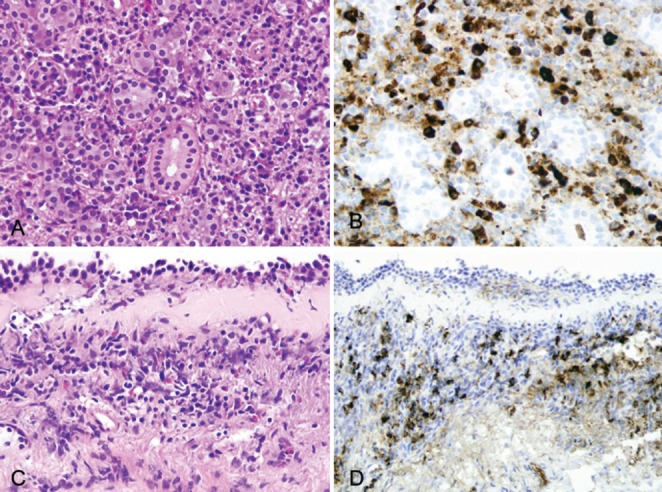
Microscopic examination. A Submandibular gland biopsy demonstrating the features of chronic sialadenitis (hematoxylin and eosin stain, original magnification ×400). B Increased immunoglobulin (Ig) G4-positive plasma cells within the chronic inflammatory infiltrate in submandibular gland biopsy (IgG4 immunostain, original magnification ×400). C Bronchoscopic biopsy demonstrating chronic inflammatory infiltrate and thickened basement membrane in bronchial mucosa (hematoxylin and eosin stain, original magnification ×200). D Increased IgG4-positive plasma cells within the inflammatory infiltrate in bronchial mucosa (IgG4 immunostain, original magnification ×200)
Three months after discontinuation of corticosteroid therapy, his sinopulmonary symptoms recurred without evidence of pulmonary parenchymal infiltrate or left kidney mass. His serum IgG4 level was noted to be elevated to 1.96 g/L. He was restarted on prednisone 20 mg daily for two months, followed by 12 months of maintenance therapy with prednisone 7.5 mg daily. The maintenance therapy was tapered over a one-month period and he has not experienced a recurrence of symptoms for one year since discontinuation of the maintenance therapy.
DISCUSSION
IgG4-RD is a recently recognized fibroinflammatory systemic disease that often presents as autoimmune pancreatitis (AIP) but can affect virtually any organ. There have been reports describing a broad spectrum of organ involvement including biliary system, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid gland, pericardium and skin (1–5). Thus, the clinical presentation varies widely and depends on the organs affected. The pancreas is one of the most commonly affected organs, and patients with AIP often present with abdominal pain and obstructive jaundice. However, an inflammatory pulmonary infiltrate or a kidney lesion may not cause symptoms. Patients with IgG4-RD typically demonstrate high serum IgG4 levels and polyclonal hypergammaglobulinemia; however, approximately 20% of patients with biopsy-proven IgG4-RD may have normal serum IgG4 values at diagnosis (6,7). Characteristic histopathological features include abundant IgG4-positive lymphoplasmacytic infiltration, fibrosis, and obliterative phlebitis or arteritis (1,4,8). In extrapancreatic sites of IgG4-RD, all three pathological features may not be present (5,9). Immunohistochemical staining for IgG4 and IgG are often used to quantify the amount of IgG4-positive cells in the affected tissues. Several different cut-offs have been proposed for diagnosis of IgG4-RD, which include the absolute number of IgG4-positive cells per high-power field >10 to 50, and the ratio of IgG4-positive cells to IgG-positive cells >0.3 to 0.4 (5,10–12). Eosinophilic infiltration is apparent in the affected tissues; however, the role of eosinophils in the pathophysiology of IgG4-RD has not been determined (13,14). Thus, the diagnosis of IgG4-RD requires integration of three components: clinico-radiological features (including serum IgG4 level), histopathological findings and immunostaining results.
Men are three to four times more frequently affected than women, and the median age at diagnosis is approximately 60 years (2,5). The mainstay of treatment is oral corticosteroids, with most patients demonstrating a good initial response (1,15).
Our case was unique in that the patient presented with clinical features mimicking asthma associated with other organ involvement. PFT demonstrated borderline airflow obstruction with positive methacholine challenge, and bronchoscopy revealed inflammatory changes in the tracheobronchial tree. Immunostaining of the endobronchial mucosal biopsy showed an increased number of IgG4-positive plasma cells. One case of IgG4-RD has previously been reported manifesting central airway stenosis due to lymphadenopathy in a patient with AIP; however, there has been no previous description of diffuse mucosal inflammation in the tracheobronchial tree as seen in our patient (16).
IgG4-RD is an under-recognized disorder and it appears likely that some patients with central airway inflammation related to IgG4-RD may have been misdiagnosed with asthma. Along this line, a recent cross-sectional study demonstrated that 19% of IgG4-RD patients had been previously diagnosed with allergic disorders, such as bronchial asthma, sinusitis or allergic rhinitis (5). The measurement of serum IgG4 level may facilitate the diagnosis of IgG4-RD if the clinical presentation seems atypical for asthma or the patient exhibits multiple organ involvement.
Although multiple organs (salivary glands, intrathoracic lymph nodes, lung and kidney) were affected, our patient did not demonstrate evidence of pancreatobiliary disease, which is the most common presentation of IgG4-RD (5). Salivary gland involvement is the second most common manifestation and it generally causes firm, nodular swelling that is associated with pain, tenderness and decreased saliva production (4,5). Intrathoracic lymphadenopathy is also a relatively common manifestation of IgG4-RD. A previous study showed that 78% of AIP patients had intrathoracic lymphadenopathy (17). Other pulmonary manifestations involve parenchymal infiltrates, pleural effusion and pleuritis (1). The most common CT or ultrasonographic findings of renal involvement are single or multiple low-attenuation lesions followed by bilateral diffuse kidney enlargement (3,18). Tubulointerstitial nephritis is a dominant pathological feature characterized by abundant IgG4-positive plasma cell infiltration into the renal interstitium with fibrosis (3,18). Overall, radiological changes in any affected organs can mimic infectious or malignant infiltrates and its differentiation can be difficult via radiological findings alone. Serum IgG4 level can be within the normal range in 20% of patients with IgG4-RD and elevated in other diseases (6,7,19). Thus, an elevated serum IgG4 level alone is insufficient to confirm the diagnosis of IgG4-RD. No correlation has been observed between serum levels of other IgG subclasses and IgG4-RD. In addition, IgG4-RD patients are at higher risk of developing malignancy than the general population (20,21). Therefore, biopsy is often required to make a definitive diagnosis of IgG4-RD or to differentiate another organ manifestation from malignancy.
IgG4-RD is a corticosteroid-responsive disorder. Although the optimal regimen of corticosteroid therapy has not been determined, most patients respond to a prednisone dose of 0.6 mg/kg per day (22). Favourable response is usually observed in two to four weeks of treatment. After the initial two to four weeks of therapy, the prednisone dose is gradually decreased over the following few months, with continued monitoring for complete resolution or possible recurrence. Maintenance therapy with lower doses of prednisone (5 mg to 10 mg daily) has been shown to reduce the relapse rate in patients with pancreatic manifestation of IgG4-RD (22).
Although many patients demonstrate an initial favourable response to corticosteroids, a substantial number of patients experience a relapse after or during the corticosteroid tapering process. A previous study involving AIP patients demonstrated relapse rates of 32% within six months of discontinuing treatment, 56% within one year and 92% within three years (22). There have been a limited number of reports regarding treatment with other immunosuppressive agents in patients with recurrent or refractory IgG4-RD. Azathioprine, mycophenolate mofetil and rituximab have been used; however, the data regarding their efficacy are limited to small retrospective case series and case reports (23–25).
CONCLUSIONS
IgG4-RD is a recently recognized systemic fibroinflammatory disease characterized by a high serum IgG4 level, abundant IgG4-positive lymphoplasmacytic infiltration in various organs and steroid responsiveness. Clinical presentations can mimic various diseases such as infectious, malignant and other inflammatory disorders. Increased awareness of IgG4-RD is important to avoid diagnostic delay and to facilitate the appropriate treatment to prevent irreversible organ damage.
Post-test.
-
What is IgG4-RD?
IgG4-RD is a recently recognized fibroinflammatory systemic disease. It was initially described as AIP; however, it is now known to affect virtually every organ system including biliary system, salivary glands, periorbital tissues, kidneys, lungs, lymph nodes, meninges, aorta, breast, prostate, thyroid gland, pericardium and skin. Patients with IgG4-RD generally manifest high serum IgG4 levels and polyclonal hypergammaglobulinemia.
-
What laboratory tests are of value in making diagnosis of IgG4-RD?
Elevated serum IgG4 level and polyclonal hypergammaglobu-linemia in serum protein electrophoresis can be useful in making a diagnosis of IgG4-RD. However, isolated abnormalities in these tests are not highly sensitive or specific. Clinicoradiological correlation and pathological confirmation (histopathological findings and immunostaining results) are warranted.
-
What are the typical pathological findings seen in IgG4-RD?
Typical pathological findings include abundant IgG4-positive lymphoplasmacytic infiltration, fibrosis, and obliterative phlebitis or arteritis.
-
What is the first-line treatment for IgG4-RD?
IgG4-RD is a corticosteroid-responsive disorder. Although the optimal regimen of corticosteroid therapy has not been determined, most patients respond to prednisone therapy at 0.6 mg/kg per day. Favourable response is usually observed within two to four weeks of treatment. Thereafter, the prednisone dose is gradually decreased over the following few months with continued monitoring for complete resolution or possible recurrence.
Footnotes
AUTHOR CONTRIBUTIONS: HS, TRA and JHR identified and managed the case. ESY evaluated the pathology of the case. HS and RH composed the manuscript, and TRA, ESY and JHR reviewed and edited it.
REFERENCES
- 1.Ryu JH, Sekiguchi H, Yi ES. Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J. 2012;39:180–6. doi: 10.1183/09031936.00025211. [DOI] [PubMed] [Google Scholar]
- 2.Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:57–66. doi: 10.1097/BOR.0b013e3283418057. [DOI] [PubMed] [Google Scholar]
- 3.Raissian Y, Nasr SH, Larsen CP, et al. Diagnosis of IgG4-related tubulointerstitial nephritis. J Am Soc Nephrol. 2011;22:1343–52. doi: 10.1681/ASN.2011010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khosroshahi A, Deshpande V, Stone JH. The clinical and pathological features of IgG(4)-related disease. Curr Rheumatol Rep. 2011;13:473–81. doi: 10.1007/s11926-011-0213-7. [DOI] [PubMed] [Google Scholar]
- 5.Zen Y, Nakanuma Y. IgG4-related disease: A cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812–9. doi: 10.1097/PAS.0b013e3181f7266b. [DOI] [PubMed] [Google Scholar]
- 6.Kamisawa T, Takuma K, Tabata T, et al. Serum IgG4-negative autoimmune pancreatitis. J Gastroenterol. 2011;46:108–16. doi: 10.1007/s00535-010-0317-2. [DOI] [PubMed] [Google Scholar]
- 7.Ebbo M, Daniel L, Pavic M, et al. IgG4-related systemic disease: Features and treatment response in a French cohort: Results of a multicenter registry. Medicine (Baltimore) 2012;91:49–56. doi: 10.1097/MD.0b013e3182433d77. [DOI] [PubMed] [Google Scholar]
- 8.Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: A clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33:1886–93. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 9.Shrestha B, Sekiguchi H, Colby TV, et al. Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with autoimmune pancreatitis: Report of 6 and 12 cases with similar histopathology. Am J Surg Pathol. 2009;33:1450–62. doi: 10.1097/PAS.0b013e3181ac43b6. [DOI] [PubMed] [Google Scholar]
- 10.Cheuk W, Chan JK. IgG4-related sclerosing disease: A critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol United States. 2010:303–32. doi: 10.1097/PAP.0b013e3181ee63ce. [DOI] [PubMed] [Google Scholar]
- 11.Smyrk TC. Pathological features of IgG4-related sclerosing disease. Curr Opin Rheumatol. 2011;23:74–9. doi: 10.1097/BOR.0b013e328341347b. [DOI] [PubMed] [Google Scholar]
- 12.Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD) Mod Rheumatol. 2012;25:1181–92. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 13.Sato Y, Kojima M, Takata K, et al. Systemic IgG4-related lymphadenopathy: A clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol. 2009;22:589–99. doi: 10.1038/modpathol.2009.17. [DOI] [PubMed] [Google Scholar]
- 14.Deshpande V, Khosroshahi A, Nielsen GP, et al. Eosinophilic angiocentric fibrosis is a form of IgG4-related systemic disease. Am J Surg Pathol. 2011;35:701–6. doi: 10.1097/PAS.0b013e318213889e. [DOI] [PubMed] [Google Scholar]
- 15.Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:67–71. doi: 10.1097/BOR.0b013e328341a240. [DOI] [PubMed] [Google Scholar]
- 16.Ito M, Yasuo M, Yamamoto H, et al. Central airway stenosis in a patient with autoimmune pancreatitis. Eur Respir J. 2009;33:680–3. doi: 10.1183/09031936.00051408. [DOI] [PubMed] [Google Scholar]
- 17.Fujinaga Y, Kadoya M, Kawa S, et al. Characteristic findings in images of extra-pancreatic lesions associated with autoimmune pancreatitis. Eur J Radiol. 2010;76:228–38. doi: 10.1016/j.ejrad.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 18.Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int. 2010;78:1016–23. doi: 10.1038/ki.2010.271. [DOI] [PubMed] [Google Scholar]
- 19.Ryu JH, Horie R, Sekiguchi H, et al. Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. Int J Rheumatol. 2012;2012:6. doi: 10.1155/2012/232960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi N, Ghazale AH, Smyrk TC, et al. Possible association between IgG4-associated systemic disease with or without autoimmune pancreatitis and non-Hodgkin lymphoma. Pancreas. 2009;38:523–6. doi: 10.1097/MPA.0b013e31819d73ca. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto M, Takahashi H, Tabeya T, et al. Risk of malignancies in IgG4-related disease. Mod Rheumatol. 2011 doi: 10.1007/s10165-011-0520-x. [DOI] [PubMed] [Google Scholar]
- 22.Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–7. doi: 10.1136/gut.2008.172908. [DOI] [PubMed] [Google Scholar]
- 23.Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: Clinical profile and response to therapy. Gastroenterology. 2008;134:706–15. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Khosroshahi A, Bloch DB, Deshpande V, Stone JH. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62:1755–62. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 25.Topazian M, Witzig TE, Smyrk TC, et al. Rituximab therapy for refractory biliary strictures in immunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol. 2008;6:364–6. doi: 10.1016/j.cgh.2007.12.020. [DOI] [PubMed] [Google Scholar]