

Abstract
EMMPRIN, a transmembrane glycoprotein known to pro-mote survival, invasion and metastasis of tumor cells through multiple pathways and mechanisms, has been found to be overexpressed in various types of cancer cells. Here we report that loss of the function of p53, a tumor suppressor protein that is mutated in approximately 50% of human cancers, contributes to the upregulation of EMMPRIN protein. We observed an inverse association between the activity of p53 and the level of EMMPRIN protein in several cancer cell lines. We further demonstrated that p53 is able to negatively regulate EMMPRIN protein, but downregulation of EMMPRIN by p53 is independent of repression of the EMMPRIN transcription. Furthermore, downregulation of EMMPRIN by p53 can be rescued by chloroquine, a lysosome inhibitor, but not by MG132, a proteasome inhibitor, suggesting an involvement of the lysosomal pathway in the p53-regulated degradation of EMMPRIN. Downregulation of EMMPRIN by p53 leads to a decrease in the activity of MMP-9 and an inhibition of tumor cell invasion. Our study suggests that the upregulation of EMMPRIN seen in many cancers can be attributed to, at least in part, the dysfunction of p53 and thus provides new evidence for the roles of p53 in tumor development and progression.
Keywords: p53, extracellular matrix metalloproteinase inducer, matrix metalloproteinase, tumor progression
Introduction
Extracellular matrix metalloproteinase inducer (EMMPRIN or CD147) is a member of the immunoglobulin family and a glycoprotein enriched on the surface of various types of tumor cells. The roles of EMMPRIN in tumor progression have become increasingly appreciated. For example, it is known that expression of EMMPRIN promotes invasion and metastasis of cancer cells by increasing the production of several matrix metalloproteinases (MMPs)1-5 and by upregulating the expression vascular endothelial growth factor and angiogenesis.6 We previously reported that overexpression of EMMPRIN in multidrug resistant cancer cells not only contributes to the enhanced invasive ability, but also to the induction of MDR1 gene whose product is P-glycoprotein, a multidrug transporter.7-9 We have also demonstrated that expression of EMMPRIN confers tumor cells resistance to anoikis through inhibition of Bim, a pro-apoptotic BH3-only protein.10 Additionally, EMMPRIN has been reported to play an important role in regulating the efflux of lactate and membrane localization of monocarboxylate transporters11 and the metabolism of glucose by trafficking with monocarboxylate transporters12 in human breast cancer cells. Despite the progresses in understanding the functions of EMMPRIN and its importance in cancer biology, little is known about the regulation of expression of this protein, except a recent report implicating the ERK1/2 and p38 signaling pathways in activating the expression of EMMPRIN.13
Tumor suppressor protein p53 is known to regulate the expression of numerous genes14 and play critical roles in important cellular events such as cell cycle regulation, DNA damage repair, apoptosis, autophagy, etc. For instance, p21 can be transcriptionally activated by p53 under stress conditions such as DNA damage, thereby causing cell cycle arrest through p21 binding and inhibiting of cyclin-dependent kinase complex. Recent studies have also shown that p53 is involved in the control of motility, invasion and metastasis of cancer cells through regulating several molecular signaling pathways including RhoA-ROCK pathway,15 SDF-1/CXCL12,16 CXCR4.17 Although p53 mutation is known to occur in approximately 50% of human cancers, and the roles of p53 in cancer development and progression have been extensively studied and well appreciated, how loss of p53 function contributes to cancer invasion and metastasis has not been fully understood. In the current study, we demonstrated that wild-type p53 negatively modulates the protein level of EMMPRIN through the lysosomal degradation pathway, and downregulation of EMMPRIN by p53 suppresses invasive potential of cancer cells. Our finding of the role of p53 in regulating EMMPRIN expression provides additional evidence and insights into the importance of this tumor suppressor protein in modulation of malignant phenotype.
Results
Effects of p53 status on EMMPRIN protein expression
We observed that the human prostate cancer cell lines, LNCaP, DU-145 and PC-3, which differ in their status of p53,18 expressed different levels of EMMPRIN (Fig. 1A). Among these three cell lines, the p53-null PC-3 and p53 mutant DU-145 lines expressed higher levels of EMMPRIN, as compared with LNCaP cells that harbor wild-type p53 (Fig. 1A). Expression of EMMPRIN in these cell lines correlated with their respective invasive ability, as PC-3 and DU-145 cells showed significantly greater invasiveness than LNCaP cells (Fig. 1B). Silencing of EMMPRIN expression with siRNA in PC-3 cells (Fig. 1C) significantly reduced the number of invading tumor cells (Fig. 1D). These observations suggest that p53 may play a role in suppressing tumor cell invasion through controlling EMMPRIN expression. To further study the role of p53 in regulating EMMPRIN expression, we utilized the LNCaP cells transfected with a human temperature-sensitive p53 vector, tsp Val138. At 39°C the transfectants (LVCaP cells) express mutant p53, whereas at 32°C these cells express a functionally wild-type p53 protein.19 As shown in Figure 2A, LVCaP cells cultured at 32°C expressed lower levels of EMMPRIN as compared with the cells cultured at 39°C. To verify the effect of p53 on EMMPRIN expression, we transfected the p53-null cells, PC-3, with a wild-type p53 expression vector and then measured the expression of EMMPRIN protein. Introduction of p53 resulted in a “dose-dependent” activation of p21 transcription (Fig. 3B), an indicator of p53 activity and downregulation of EMMPRIN protein (Fig. 2B). p21 protein level did not appear to change in those cells (Fig. 2B), probably due to that p21 protein also undergoes p53-independent posttranscriptional regulation such as protein or mRNA turnover. Similar results were previously shown by others.20,21 The effect of p53 on EMMPRIN expression was also observed in the mutant p53-harboring T98G cells transfected with the wild-type p53 expression vector (Fig. 2C). Additionally, in MCF-7 cells that express endogenous wild-type p53, γ-irradiation (5 Gy) caused an activation of p53, as indicated by an increase in p21 protein level, and a noticeable reduction of EMMPRIN protein (Fig. 2D). By contrast, treatment with γ-irradiation did not decrease EMMPRIN protein in NCI/ADR-RES cells whose p53 is dysfunctional (Fig. 2D).

Figure 1. Expressions of EMMPRIN in human prostate cancer cell lines with different p53 status correlate with their respective invasive potential. (A) Cell lysates were prepared from LNCaP, DU-145 and PC-3 cells and equal amounts (50 μg) of proteins were resolved by 15% SDS-PAGE. Proteins were transferred to nitrocellulose membrane, and EMMPRIN was detected by immunoblotting with a monoclonal anti-EMMPRIN antibody. Ran was used as a loading control. (B) Cells suspended in media supplemente with 0.1% BSA were plated on Matrigel-covered filters in a Boyden chamber (3–5 x 105 cells/well). Following incubation at 37°C for 48 h, the cells that reached the underside of the Matrigel-coated membrane were stained with hematoxylin and eosin and counted under a microscope at a magnification of x200. Each bar represents the mean ± SD of quadruplicate determinations from one of three independent experiments. (C) PC-3 cells in exponential phase of growth were transfected with an EMMPRIN-targeted siRNA (100 nM) or a non-targeting RNA. Forty-eight hours later, cell lysates were prepared from the transfected cells and EMMPRIN was detected as described above. (D) PC-3 cells in exponential phase of growth were transfected with an EMMPRIN-targeted siRNA (100 nM) or a non-targeting RNA. Forty-eight hours later, cells were plated on Matrigel-covered filters in a Boyden chamber (3–5 x 105 cells/well). After incubation at 37°C for 48 h, invasive ability of the cells was determined as described above. Each bar represents the mean ± SD of quadruplicate determinations from one of three independent experiments.

Figure 2. Increases in wild-type p53 downregulate EMMPRIN protein. (A) LVCaP cells, which harbored a temperature-sensitive p53 construct, were cultured at 32°C (functional wt p53) and 39°C (mutant p53) for 5 d. EMMPRIN, p21 (as a positive control) and Ran (as a loading control) were detected by western blot. (B and C) PC-3 and T98G cells were transfected with different amounts of p53 plasmid. Twenty-four hours later, EMMPRIN was measured by western blot. (D) western blot analysis of EMMPRIN in MCF-7 (wt p53) and NCI/ADR-RES (mutant p53) cells treated with γ-irradiation (5 Gy) for different periods of time (0, 24, 48 and 72 h). Protein expression was quantified using the ImageJ software (NIH, Bethesda, MD). Results shown are representative of three similar experiments.
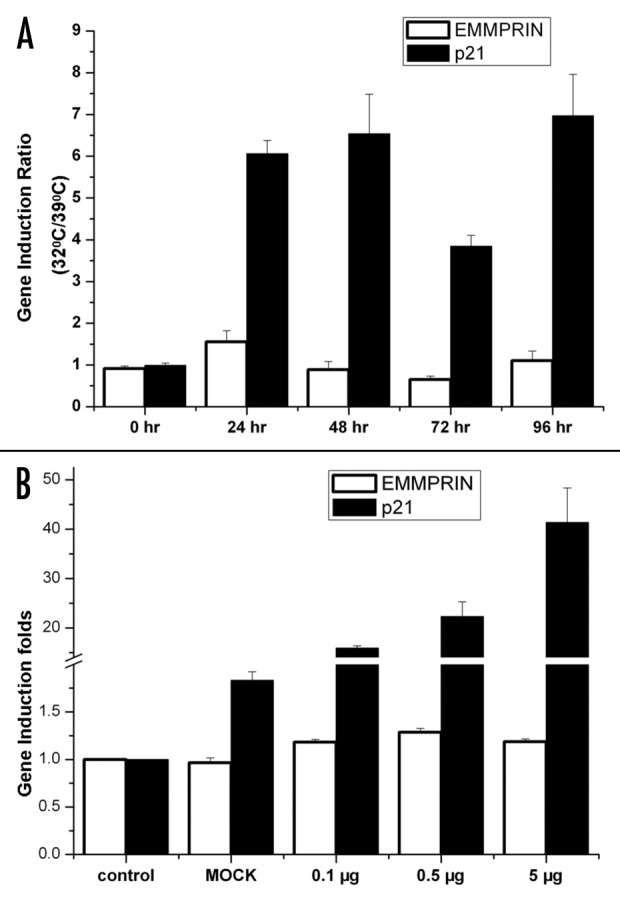
Figure 3. EMMPRIN transcription is not affected by p53. (A) LVCaP cells were cultured at 32°C and 39°C for indicated times. Total RNA was extracted by TRIzol® reagent, and EMMPRIN mRNA was determined by qRT-PCR. The mRNA level ratios between 32 and 39°C in different time points were shown. p21 was used as a positive control. (B) PC-3 cells were transfected with different amounts of p53 plasmid. Twenty-four hours later, total RNA was extracted from each sample and EMMPRIN mRNA was determined by qRT-PCR. p21 was used as a positive control. EMMPRIN or p21 mRNA levels of the control samples were arbitrarily set at one, and the mRNA levels of EMMPRIN or p21 in the treated samples were normalized to the control. Results are the mean ± SD of triplicate determinations from one of three identical experiments.
p53 does not affect EMMPRIN transcription
p53 is known to participate in transcriptional regulation of numerous genes. To determine whether the effect of p53 on EMMPRIN expression was exerted at transcriptional level, we measured the expression of EMMPRIN mRNA in cells with different status of p53 using quantitative real-time RT-PCR. Figure 3A shows that LVCaP cells cultured at 32°C (wild-type p53) had high ratios of p21 mRNA induction over the cells cultured at 39°C (mutant p53). In contrast, the ratios of expression of EMMPRIN mRNA at 32°C vs. 39°C were barely changed (Fig. 3A). In PC-3 cells transfected with a wild-type p53 expression vector, the expression of p21 mRNA was induced in a p53 “dose-dependent” manner (Fig. 3B); by contrast, introduction of p53 into PC-3 cells had no effect on the expression of EMMPRIN mRNA (Fig. 3B). To verify these results, we performed the real-time RT-PCR analyses using three different sets of EMMPRIN and β-actin primers, which bind to different sequences of these two genes, and obtained similar results (data not shown). These experiments indicate that p53 does not affect the transcription of EMMPRIN.
Lysosomal pathway is involved in the downregulation of EMMPRIN by p53
It has been reported that p53 is able to modulate degradation of certain proteins via either proteasomal22 or lysosomal23 pathway. Therefore, we next sought to determine whether the downregulation of EMMPRIN by p53 occurred at protein level. To explore this possibility, we treated cells with either MG132, an inhibitor of proteasome, or chloroquine, an inhibitor of the late endosome-lysosome transport. As shown in Figure 4A, treatment of LVCaP cells with MG132 barely caused an increase in the accumulation of EMMPRIN protein either at 32 or 39°C; notably, treatment with chloroquine rescued the downregulation of EMMPRIN protein in LVCaP cells cultured at 32°C (expressing a functionally wild-type p53 protein). Furthermore, chloroquine also rescued the downregulation of EMMPRIN protein in PC-3 cells transfected with a wild-type p53 expression vector (Fig. 4B); in contrast, MG132 did not show the rescuing effect on the downregulation of EMMPRIN protein in these p53-expressing cells (Fig. 4B). These results suggest that the lysosomal pathway is involved in the downregulation of EMMPRIN by p53.

Figure 4. Downregulation of EMMPRIN by p53 is not affected by proteasomal inhibitor, but is sensitive to lysosomal inhibitor. (A) LVCaP cells were cultured at 32°C or 39°C in the presence or absence of MG132 (10 μM) or chloroquine (50 μM). Twenty-four hours later, EMMPRIN, p21 and Ran were determined by western blot. Ran was used as a loading control. Protein expressions were quantified using the ImageJ software (NIH, Bethesda, MD). (B) PC-3 cells transfected with different amounts of p53 plasmid were treated with MG132 (10 μM) or choloroquine (50 μM) for 24 h. At the end of the treatment, EMMPRIN, p21 and Ran were determined by western blot. Results shown are representative of three similar experiments.
p53-mediated downregulation of EMMPRIN inhibits MMP-9 activity and tumor cell invasion
To evaluate the functional significance of the p53-mediated downregulation of EMMPRIN, we assayed the activity of MMP-9, one of the downstream effectors of EMMPRIN, in the media from the cultures of PC-3 and T98G cells with or without expressing a functional p53. Figure 5A shows that in PC-3 and T98G cells transfected with a wild-type p53 expression vector, the level of pro-MMP-9 was significantly decreased as compared with that in the control cells. Treatment with Ilomastat, an MMP-9 inhibitor, further lowered the level of pro-MMP-9 in these tumor cells (Fig. 5A). The active MMP-9 present in the conditioned media from the cultures of PC-3 and T98G cells transfected with the wild-type p53 was also reduced as compare with that of the control cells, as analyzed by zymography (Fig. 5B). Using the conditioned media from the cultures of PC-3 and T98G cells with or without expressing the wild-type p53, we also measured the invasive ability of LNCaP cells, which showed relative low EMMPRIN expression and low invasive ability (Fig. 1A and B). As shown in Figure 5C, the invasive ability of LNCaP cells were significantly increased when cultured in the conditioned media from the cultures of PC-3 or T98G cells that lack a functional p53, in comparison to that of the cells cultured in unconditioned media. By contrast, when cultured in the conditioned media from the cultures of the p53-transfected cells the invasive ability of LNCaP cells were significantly reduced (Fig. 5C). In the presence of Ilomastat, the invasive potential of LNCaP cells was further decreased (Fig. 5C). These results indicate that a functional p53 can suppress invasive potential of tumor cells through modulating the EMMPRIN-MMP-9 pathway.

Figure 5. Effects of p53 on MMP-9 activity and tumor cell invasion. (A) PC-3 and T98G cells were transfected with different amounts of p53 plasmid, and then cultured at 37°C and 5% CO2 for 48 h in the absence or presence of 25 μM of Ilomastat. Pro-MMP-9 in the conditioned media was assayed using the MMP-9 ELISA Kit (Calbiochem). Each bar represents the mean ± SD of quadruplicate determinations from one of three independent experiments. (B) Conditioned media from cell cultures were collected and concentrated; active MMP-9 in the conditioned media was measured by gelatin zymography. MMP-9 activity was visualized as clear bands. (C) PC-3 and T98G cells were transfected with 25 μg of p53 plasmid and then cultured at 37°C and 5% CO2 for 24 h. The conditioned media (with or without MMP-9 inhibitor, Ilomastat, 25 μM) from the above cell culture were then used for culturing LNCaP cells in the upper chamber of the inserts. After a 48 h incubation, the inserts were transferred into a second 24-well plate containing 500 μL/well of 4 μg/mL calcein AM (Invitrogen, Carlsbad, CA) in HBSS. Invading cells were quantified by measuring fluorescence using a Victor3 Multi Label plate reader. Each bar represents the mean ± SD of quadruplicate determinations from one of three independent experiments. Results are representative of three similar experiments.
Discussion
Based on our observation that cancer cell lines with different status of p53 function expressed different levels of EMMPRIN protein, i.e., the p53-null (PC-3) and -mutant (DU-145) tumor cells expressed higher level of EMMPRIN than LNCaP cells harboring wild-type p53 (Fig. 1A), and the expression levels of EMMPRIN in these three cell lines correlated with their respective invasive capacity (Fig. 1B–D), we hypothesized that the tumor suppressor protein, p53, plays a role in regulating the expression EMMPRIN and invasive potential of tumor cells. Because these three cell lines harbor other disparities between them in addition to the difference in their p53 status, such as their androgen receptor status,24 we performed further experiments to test the effect of p53 on EMMPRIN expression. Our hypothesis is supported by our experiments showing that restoration of p53 function by genetic approaches resulted in a reduction of the protein level of EMMPRIN (Fig. 2A–C), and that induction of p53 in MCF-7 cells by γ-irradiation lead to a decrease in EMMPRIN expression (Fig. 2D). By contrast, γ-irradiation did not cause a downregulation of EMMPRIN in NCI/ADR-RES cells whose p53 is inactivated (Fig. 2D), another evidence for a role for p53 in controlling the level of EMMPRIN.
Many of the p53-regulated events such as apoptosis and cell cycle are mediated through its effects on gene transcription.25,26 Nevertheless, inhibition of EMMPRIN expression by p53 appears to be independent of transcriptional regulation, as we did not observe any effect of p53 on the expression of EMMPRIN mRNA (Fig. 3). Interestingly, we found that downregulation of EMMPRIN by p53 was sensitive to inhibition of endosome-lysosome traffic by chloroquine, but not to inhibition of proteasome by MG132 (Fig. 4), suggesting that the p53-mediated regulation of EMMPRIN occurs in the lysosomal pathway, one of the two alternative systems for protein degradation in cells. Indeed, using a web-based algorithm PEST find, we did not find in EMMPRIN any valid PEST sequences, a region that usually renders proteins susceptible to ubiquitination and proteasomal degradation.27,28 Recently, the p53-regulated lysosomal degradation of other protein has been reported by Valbuena et al.23 In addition, the role of p53 in the regulation of the lysosomal function has also been reported.29,30 However, the precise mechanism by which how p53 regulates protein degradation remains unclear.
The roles of p53 in regulating cell proliferation and survival have been extensively studied and well defined.31 Evidence for implication of p53 deficiency in tumor invasion and metastasis also exist. For instance, it was reported that loss of p53 function enhances invasion and metastasis in a mouse model of hepatocellular carcinoma.32 Also, p53 mutation was frequently observed in invasive tumors in a genetic model of colorectal tumorigenesis.33 The pathways and molecular mechanisms underlying the effects of p53 on tumor cell migration, invasion and metastasis are also being revealed. For example, Guo et al. reported that Rho family GTPases and phosphoinositide 3-kinase are involved in cell -migration and invasion promoted by loss of p53 function. The results of the current study demonstrate that inactivation of p53 may promote tumor cell invasion via upregulating EMMPRIN, a cell surface glycoprotein that stimulates the productions of MMPs and hyaluronan, and activates several survival pathways. We also showed that restoration of p53 function could decrease the activity of MMPs such as MMP9 (Fig. 5A and B) and inhibit invasion of tumor cells (Fig. 5C). As EMMPRIN has been shown to play a promotive role in tumorigenesis, invasion and metastasis.34,35 The regulation of EMMPRIN by p53 may therefore represent one of the mechanisms by which this tumor suppressor protein controls development and progression of tumors. The study reported here further underscores the importance of restoring p53 function as an anti-cancer treatment, and suggests the potential of targeting EMMPRIN as a therapeutic strategy, in particular, for p53-deficient tumors.
Materials and Methods
Cell cultures
Human prostate cancer cell lines LNCaP, DU-145 and PC-3 were maintained in RPMI 1640 media with L-glutamine. Temperature-sensitive p53 transfectant, LVCaP line, was established and cultured as previously described.19 Human glioblastoma T98G cells were maintained in F-10 media; human breast cancer MCF-7 cells and ovarian carcinoma NCI/ADR-RES cells were cultured in RPMI-1640 media. All of the media contain 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin. Cell cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2, 95% air and were free of contamination by mycoplasma or fungi. All of the cell lines were discarded after 3 mo, and new cell cultures were propagated from frozen stocks.
Antibodies and reagents
The antibodies used in this study were purchased from the following sources: monoclonal anti-EMMPRIN antibody, Research Diagnostics, Inc., (Flanders, NJ); monoclonal anti-p53 antibody, Santa Cruz Biotechnology, Inc., (Santa Cruz, CA); monoclonal anti-p21 antibody, Calbiochem; monoclonal anti-ran and anti-β-actin antibody, Sigma (St. Louis, MO). Halt Protease Inhibitor Cocktail was purchased from Pierce Biotechnology, Inc.; MMP-9 inhibitor, Ilomastat, was purchased from Chemicon International Inc., (Billerica, MA); MG132 and chloroquine were purchased from Sigma; EMMPRIN-siRNA was purchased from Dharmacon RNA Technologies (Lafayette, CO).
siRNA, plasmid, and transfection
siRNA against EMMPRIN was transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. EMMPRIN siRNAs sequences were: 5′-GUA CAA GAU CAC UGA CUC UUU-3′.10 pRC/CMV-wt p53 expression plasmid was a generous gift from Dr. Arnold J. Levine (The Cancer Institute of New Jersey). Transfection of plasmid was also performed using the Lipofectamine 2000.
DNA damage treatment
Cells were cultured in 100-mm dishes for 24 h, and then were treated with γ-irradiation at 5 Gy (CIS BioInternational IBL 437C 137Cs γ-irradiation source; dose rate, 0.49 Gy/min).
Western blot analysis
Cell were lysed with TNT buffer (20 mM TRIS-HCl, pH 7.4, 200 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail) and cell lysates were collected. Proteins were resolved by 15% SDS-PAGE. Following transfer of proteins to nitrocellulose, blots were incubated in blocking solution consisting of 5% milk in 10 mM TRIS-HCl, pH 8.0, 150 mM NaCl and 0.1% Tween 20 at room temperature for 1 h, and then immunoblotted with the respective antibodies. Detection of protein by enzyme-linked chemiluminescence (ECL) was performed according to the manufacturer’s protocol (Pierce Biotechnology, Inc., Rockford, IL). Actin or Ran were used as a loading control.
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted with TRIzol® reagent (Invitrogen, Carlsbad, CA). Two μg of mRNA were reverse transcribed into cDNA using an Omniscript RT kit (Qiagen GmbH, Hilden, Germany) in 20 μl reaction mixture, according to the manufacturer’s instructions. Primer sets of EMMPRIN gene were designed using the MacVector software (MacVector, Inc., Cary, NC). The primer sequences were as follows: EMMPRIN: forward, 5′-CCT CAC CTG CTC CTT GAA TGA C-3′; reverse, 5′-CAC CTT GAA CTC CGT TTT CTG G-3′; p21: forward, 5′-ACA GCA GAG GAA GAC CAT GTG-3′; reverse, 5′-GGG CTT CCT CTT GGA GAA GAT-3′;36 β-actin: forward, 5′-CCA CAC TGT GCC CAT CTA CG-3′; reverse, 5′-CAG CGG AAC CGC TCA TTG CCA ATG G-3′. qRT-PCR analysis was performed on a Stratagene MX 3005 amplification system (Stratagene, La Jolla, CA). For amplification, 10 μl of SYBR Green PCR master mix, 4 μl of diluted cDNA, and 200 nM primers were used in a 20 μl reaction mixture, and all samples were amplified in triplicates. The cycling conditions included a 3 min polymerase activation at 95°C followed by 40 cycles at 95°C for 15 sec, 67°C for 30 sec, and 72°C for 30 sec. The annealing temperature of p21 and β-actin was 55°C. β-actin was used as internal control. Calculations were performed as described by Livak and Schmittgen.37
Assays of pro-MMP-9 and active MMP-9
Pro-MMP-9 in conditioned media from cell culture was measured using an MMP-9 ELISA kit (Calbiochem, Cambridge, MA) according to the product manual.
MMP activity (active MMP-9) was assayed using a zymographic method as described previously.7 Briefly, conditioned media from cell culture were collected and concentrated. Concentrated conditioned media (10 μg of protein) were mixed with Laemmli loading buffer (without reducing agent) and subjected to gelatin (DIFCO Laboratories, Detroit, MI) zymography. MMP-9 activity was visualized as clear bands with Coomassie brilliant blue R-250 staining.
Invasion assay
Invasive ability of tumor cells was measured as previous described, using the 24-well BD Biocoat Matrigel invasion chambers with 8-μm polycarbonated filters (Becton Dickinson, Bedford, MA). Briefly, cells (2.5 x 104 cells) were seeded in the Matrigel-coated inserts. Inserts were then placed into lower chambers filled with complete media with FBS or conditioned media. After incubation at 37°C for 48 h, the inserts were stained with hematoxylin and eosin and counted under a microscope at a magnification of x200. Alternatively, the inserts were transferred into a second 24-well plate containing 500 μL/well of 4 μg/mL calcein AM (Invitrogen, Carlsbad, CA) in HBSS. Following a 1 h incubation at 37°C and 5% CO2, invading cells were quantified by measuring fluorescence using a Victor3 Multi Label plate reader (PerkinElmer, Wellesley, MA).
Acknowledgments
Supported by grants from the US Public Health Service CA66077.
Glossary
Abbreviations:
- EMMPRIN
extracellular matrix metalloproteinase inducer
- MMP
matrix metalloproteinases
- RT-PCR
reverse transcriptase-polymerase chain reaction
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/9207
References
- 1.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 2.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caudroy S, Polette M, Nawrocki-Raby B, Cao J, Toole BP, Zucker S, et al. EMMPRIN-mediated MMP regulation in tumor and endothelial cells. Clin Exp Metastasis. 2002;19:697–702. doi: 10.1023/A:1021350718226. [DOI] [PubMed] [Google Scholar]
- 4.Sun J, Hemler ME. Regulation of MMP-1 and MMP-2 production through CD147/extracellular matrix metalloproteinase inducer interactions. Cancer Res. 2001;61:2276–81. [PubMed] [Google Scholar]
- 5.Zhang W, Erkan M, Abiatari I, Giese NA, Felix K, Kayed H, et al. Expression of extracellular matrix metalloproteinase inducer (EMMPRIN/CD147) in pancreatic neoplasm and pancreatic stellate cells. Cancer Biol Ther. 2007;6:218–27. doi: 10.4161/cbt.6.2.3623. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Nakada MT, Rafferty P, Laraio J, McCabe FL, Millar H, et al. Regulation of vascular endothelial growth factor expression by EMMPRIN via the PI3K-Akt signaling pathway. Mol Cancer Res. 2006;4:371–7. doi: 10.1158/1541-7786.MCR-06-0042. [DOI] [PubMed] [Google Scholar]
- 7.Yang JM, Xu Z, Wu H, Zhu H, Wu X, Hait WN. Overexpression of extracellular matrix metalloproteinase inducer in multidrug resistant cancer cells. Mol Cancer Res. 2003;1:420–7. [PubMed] [Google Scholar]
- 8.Li QQ, Wang WJ, Xu JD, Cao XX, Chen Q, Yang JM, et al. Up-regulation of CD147 and matrix metalloproteinase-2, -9 induced by P-glycoprotein substrates in multidrug resistant breast cancer cells. Cancer Sci. 2007;98:1767–74. doi: 10.1111/j.1349-7006.2007.00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li QQ, Wang WJ, Xu JD, Cao XX, Chen Q, Yang JM, et al. Involvement of CD147 in regulation of multidrug resistance to P-gp substrate drugs and in vitro invasion in breast cancer cells. Cancer Sci. 2007;98:1064–9. doi: 10.1111/j.1349-7006.2007.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang JM, O’Neill P, Jin W, Foty R, Medina DJ, Xu Z, et al. Extracellular matrix metalloproteinase inducer (CD147) confers resistance of breast cancer cells to Anoikis through inhibition of Bim. J Biol Chem. 2006;281:9719–27. doi: 10.1074/jbc.M508421200. [DOI] [PubMed] [Google Scholar]
- 11.Slomiany MG, Grass GD, Robertson AD, Yang XY, Maria BL, Beeson C, et al. Hyaluronan, CD44, and emmprin regulate lactate efflux and membrane localization of monocarboxylate transporters in human breast carcinoma cells. Cancer Res. 2009;69:1293–301. doi: 10.1158/0008-5472.CAN-08-2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res. 2007;67:4182–9. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]
- 13.Huang Z, Wang C, Wei L, Wang J, Fan Y, Wang L, et al. Resveratrol inhibits EMMPRIN expression via P38 and ERK1/2 pathways in PMA-induced THP-1 cells. Biochem Biophys Res Commun. 2008;374:517–21. doi: 10.1016/j.bbrc.2008.07.058. [DOI] [PubMed] [Google Scholar]
- 14.Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–93. [PMC free article] [PubMed] [Google Scholar]
- 15.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moskovits N, Kalinkovich A, Bar J, Lapidot T, Oren M. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66:10671–6. doi: 10.1158/0008-5472.CAN-06-2323. [DOI] [PubMed] [Google Scholar]
- 17.Mehta SA, Christopherson KW, Bhat-Nakshatri P, Goulet RJ, Jr., Broxmeyer HE, Kopelovich L, et al. Negative regulation of chemokine receptor CXCR4 by tumor suppressor p53 in breast cancer cells: implications of p53 mutation or isoform expression on breast cancer cell invasion. Oncogene. 2007;26:3329–37. doi: 10.1038/sj.onc.1210120. [DOI] [PubMed] [Google Scholar]
- 18.Rubin SJ, Hallahan DE, Ashman CR, Brachman DG, Beckett MA, Virudachalam S, et al. Two prostate carcinoma cell lines demonstrate abnormalities in tumor suppressor genes. J Surg Oncol. 1991;46:31–6. doi: 10.1002/jso.2930460108. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan GF, Yang JM, Vassil A, Yang J, Bash-Babula J, Hait WN. Regulation of expression of the multidrug resistance protein MRP1 by p53 in human prostate cancer cells. J Clin Invest. 2000;105:1261–7. doi: 10.1172/JCI9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoneda K, Yokoyama T, Yamamoto T, Hatabe T, Osaki T. p53 gene mutations and p21 protein expression induced independently of p53, by TGF-beta and gamma-rays in squamous cell carcinoma cells. Eur J Cancer. 1999;35:278–83. doi: 10.1016/S0959-8049(98)00291-3. [DOI] [PubMed] [Google Scholar]
- 21.Nakanishi Y, Pei XH, Takayama K, Bai F, Izumi M, Kimotsuki K, et al. Polycyclic aromatic hydrocarbon carcinogens increase ubiquitination of p21 protein after the stabilization of p53 and the expression of p21. Am J Respir Cell Mol Biol. 2000;22:747–54. doi: 10.1165/ajrcmb.22.6.3877. [DOI] [PubMed] [Google Scholar]
- 22.Rizos H, Scurr LL, Irvine M, Alling NJ, Kefford RF. p14ARF regulates E2F-1 ubiquitination and degradation via a p53-dependent mechanism. Cell Cycle. 2007;6:1741–7. doi: 10.4161/cc.6.14.4428. [DOI] [PubMed] [Google Scholar]
- 23.Valbuena A, Vega FM, Blanco S, Lazo PA. p53 downregulates its activating vaccinia-related kinase 1, forming a new autoregulatory loop. Mol Cell Biol. 2006;26:4782–93. doi: 10.1128/MCB.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, et al. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–25. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 25.Mirza A, Wu Q, Wang L, McClanahan T, Bishop WR, Gheyas F, et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–54. doi: 10.1038/sj.onc.1206477. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 27.Marchal C, Haguenauer-Tsapis R, Urban-Grimal D. A PEST-like sequence mediates phosphorylation and efficient ubiquitination of yeast uracil permease. Mol Cell Biol. 1998;18:314–21. doi: 10.1128/mcb.18.1.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornitzer D, Raboy B, Kulka RG, Fink GR. Regulated degradation of the transcription factor Gcn4. EMBO J. 1994;13:6021–30. doi: 10.1002/j.1460-2075.1994.tb06948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fogarty MP, McCormack RM, Noonan J, Murphy D, Gowran A, Campbell VA. A role for p53 in the beta-amyloid-mediated regulation of the lysosomal system. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.09.018. In press. [DOI] [PubMed] [Google Scholar]
- 30.Gowran A, Campbell VA. A role for p53 in the regulation of lysosomal permeability by delta 9-tetrahydrocannabinol in rat cortical neurones: implications for neurodegeneration. J Neurochem. 2008;105:1513–24. doi: 10.1111/j.1471-4159.2008.05278.x. [DOI] [PubMed] [Google Scholar]
- 31.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 32.Lewis BC, Klimstra DS, Socci ND, Xu S, Koutcher JA, Varmus HE. The absence of p53 promotes metastasis in a novel somatic mouse model for hepatocellular carcinoma. Mol Cell Biol. 2005;25:1228–37. doi: 10.1128/MCB.25.4.1228-1237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 34.Zucker S, Hymowitz M, Rollo EE, Mann R, Conner CE, Cao J, et al. Tumorigenic potential of extracellular matrix metalloproteinase inducer. Am J Pathol. 2001;158:1921–8. doi: 10.1016/S0002-9440(10)64660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reimers N, Zafrakas K, Assmann V, Egen C, Riethdorf L, Riethdorf S, et al. Expression of extracellular matrix metalloproteases inducer on micrometastatic and primary mammary carcinoma cells. Clin Cancer Res. 2004;10:3422–8. doi: 10.1158/1078-0432.CCR-03-0610. [DOI] [PubMed] [Google Scholar]
- 36.Roth MJ, Hu N, Johnson LL, Quon-Hang W, Ahnen DJ, Iwamoto M, et al. beta-Catenin splice variants and downstream targets as markers for neoplastic progression of esophageal cancer. Genes Chromosomes Cancer. 2005;44:423–8. doi: 10.1002/gcc.20251. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]