

Abstract
We previously demonstrated that treatment of human androgen-responsive prostate cancer cell lines LNCaP and CWR22-Rv1 with 12-O-tetradecanoylphorbol 13-acetate (TPA), a known protein kinase C (PKC) activator, decreases ATM protein levels, thus de-repressing the enzyme ceramide synthase (CS) and promoting apoptosis as well as radio-sensitizing these cells.1 Here we show that PKCα mediates the TPA effect on ATM expression, since ATM suppression and apoptosis induced by either TPA or diacylglycerol-lactone (DAG-lactone), both inducing PKCα activation,2 are abrogated in LNCaP cells following transfection of a kinase-dead PKCα mutant (KD-PKCα). Similarly, KD-PKCα blocks the apoptotic response elicited by combination of TPA and radiation, whereas expression of constitutively active PKCα is sufficient to sensitize cells to radiation alone, without a need to pre-treat the cells with TPA. These findings identify CS activation as a downstream event of PKCα activity in LNCaP cells. Similar results were obtained in CWR22-Rv1 cells with DAG-lactone treatment. Using the LNCaP orthotopic prostate model it is shown that treatment with TPA or DAG-lactone induces significant reduction in tumor ATM levels coupled with tumor growth delay. Furthermore, while fractionated radiation alone produces significant tumor growth delay, pretreatment with TPA or DAG-lactone significantly potentiates tumor cure. These findings support a model in which activation of PKCα downregulates ATM, thus relieving CS repression by ATM and enhancing apoptosis via ceramide generation. This model may provide a basis for the design of new therapies in prostate cancer.
Keywords: ATM, PKCα, apoptosis, ionizing radiation, prostate
Introduction
The PKC family of serine/threonine kinases consists of multiple isozymes involved in a myriad of signal transduction pathways. PKC isozymes are classified into calcium-dependent or conventional/classical cPKCs (α, βI, βII and γ), calcium-independent or novel nPKCs (δ, ε, η and θ) and atypical aPKCs (ζ and μ).3 TPA activates classical and novel isoforms by mimicking DAG function,3 thereby inducing cellular transformation, proliferation, differentiation and apoptosis resistance.4,5 PKC activated by TPA inhibits apoptotic responses to TNFα,6,7 chemotherapy,8 growth factor withdrawal9 and radiation.10 However, in some cell types TPA-activated PKC induces, rather than attenuates apoptosis, such as in EBV-infected Burkitt Lymphoma cells,11 immature thymocytes,12 salivary gland acinar cells,13 human MCF-7 breast cancer cells14 and LNCaP cells.1,15 Using genetic and pharmacologic inhibition of specific PKC isozymes Garcia-Bermejo et al.2 showed that both PKCα and PKCδ mediate TPA-induced apoptosis in LNCaP cells, although the mechanism of action remains undefined.
We have recently reported that TPA treatment reversed apoptosis resistance of LNCaP human prostate cancer cells to radiation.16 This response was mediated by ceramide formed by de novo synthesis through activation of the biosynthetic enzyme CS. Our studies also showed that pre-treatment of LNCaP cells with TPA reduced ATM protein levels, which in turn abrogated CS repression by ATM kinase, leading to increased sensitivity to radiation-induced apoptosis.1
ATM, the gene mutated in ataxia telangiectasia (AT), is a member of the PI-3 kinase family and a component of the DNA damage surveillance/repair system.17 Previously, we showed that EBV-immortalized B cells from AT patients exhibited markedly increased radiation-induced CS activation, ceramide generation and apoptosis.18 While stable transfection of wild type ATM cDNA reversed these events, anti-sense inactivation of ATM in normal B cells conferred the AT phenotype. Collectively, these findings indicated that ATM inactivation leads to CS de-repression, allowing DNA damage-mediated apoptosis. Consistent with this notion, treatment of LNCaP, CWR22-Rv1 (TPA-sensitive), PC3 and DU-145 (TPA-insensitive) human prostate cells with antisense-ATM oligonucleotides (ODNs), led to markedly reduced cellular ATM levels, enhanced radiation-induced CS activation and apoptosis with radiation doses as low as 1 Gy.1 Thus the ATM-regulated CS pathway appears as a central regulator of radiation-induced apoptosis in human prostate cancer cells.
A new class of conformationally-constrained DAG analogs (DAG-lactones) was recently described with enhanced selectivity for specific PKC isoforms. In particular, a branched DAG-lactone (HK654) selectively binds and activates PKCα in LNCaP cells.2 In view of the current literature implicating PKCα in TPA-mediated prostate cancer cell death,2 we pursued the mechanistic significance of PKCα in this process by both pharmacologic and genetic-based approaches. Using dominant negative and constitutively active PKCα mutants, we found that this isoform participates in regulation of ATM expression and apoptosis induction in LNCaP cells. Furthermore, we show that PKCα activation leads to downregulation of ATM protein in prostate tumors in vivo and consequently enhanced radiation-induced tumor response.
Results
Prolonged activation of PKCα in TPA-treated LNCaP cells
PKCα activity was initially measured in membrane and cytosolic fractions of TPA-stimulated LNCaP cells at multiple time points up to 24 h (Fig. 1). PKCα activity rises in membrane to over 600% of baseline within 15 min and remains at that level for four hours. Membrane PKCα activity drops between 4 and 20 h, but remains 2-fold higher as compared with the control level. These results suggest that in LNCaP cells there is persistent activation of PKCα in response to TPA treatment, confirming previous findings by others of persistent translocation of PKCα2,30 to plasma membrane.
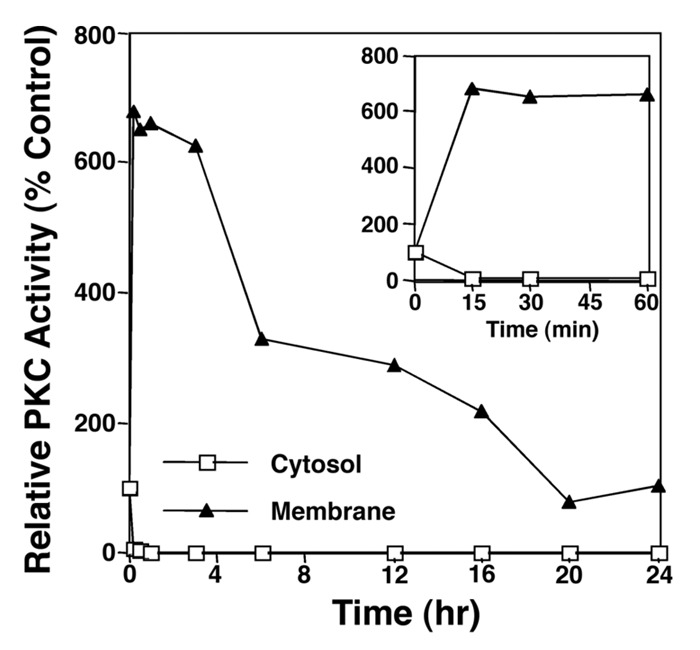
Figure 1. PKCα activation is prolonged in LNCaP cells. Monolayers of LNCaP cells were stimulated for different times with TPA (10 ng/ml) and phosphotransferase activity of partially purified PKC activity was measured as described in the ‘Methods’ section. The inset box shows in more detail the PKC activity up to one hour after stimulation. The results are representative of three experiments.
TPA-induced apoptosis and ATM downregulation are mediated by PKCα
To assess the role of PKCα in TPA-induced apoptosis, LNCaP cells were transiently transfected with either a kinase-dead PKCα mutant (KD-PKCα) or control vector (empty vector) 24 h prior to addition of TPA (10 ng/ml). Figure 2A shows, that while ATM protein levels decreased in control transfectants at 16 h after TPA treatment, KD-PKCα expression blocked ATM downregulation. In contrast, transfection with KD-PKCδ did not affect TPA-induced ATM downregulation.
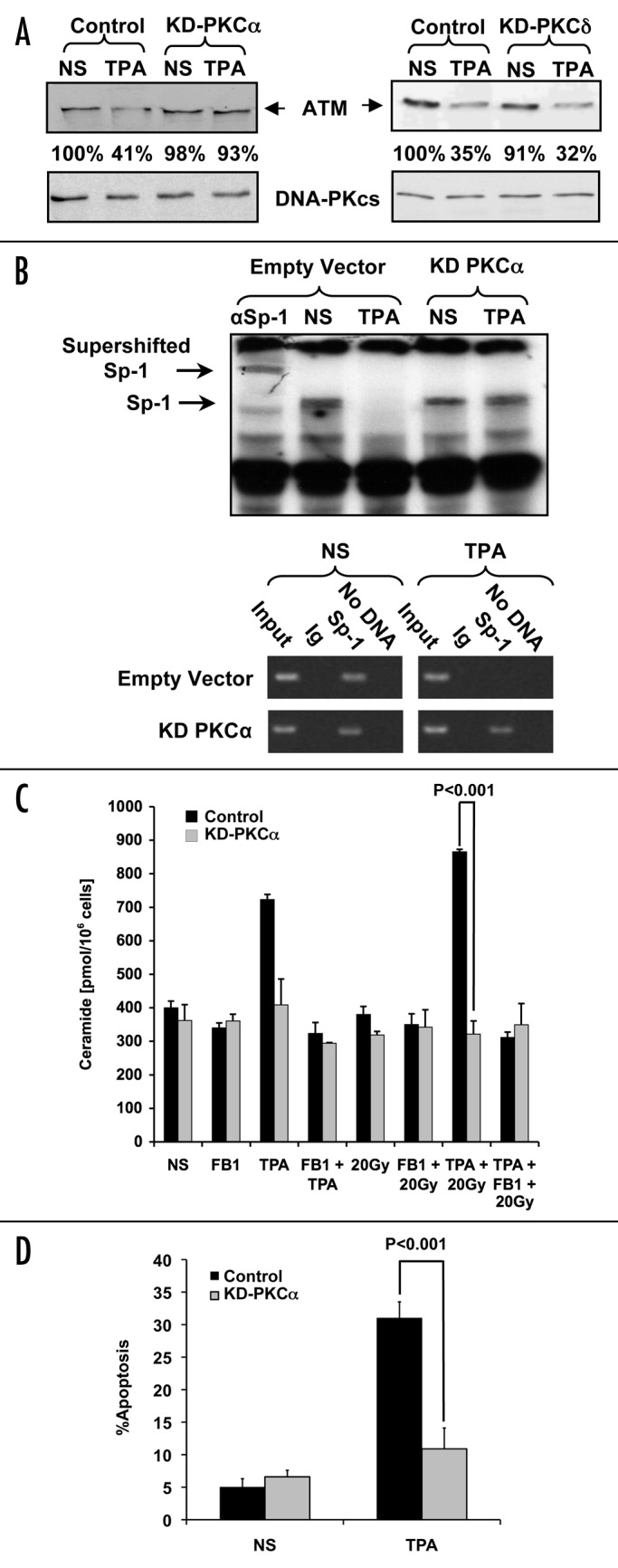
Figure 2. TPA-mediated apoptosis and ATM downregulation are regulated by PKCα. (A) LNCaP cells were transfected with 4 μg of either empty vector, KD-PKCα or KD-PKCδ 24 h before stimulation for 16 h with TPA (10 ng/ml). Samples (30 μg/lane) were analyzed by western blot for ATM expression with a monoclonal anti-ATM antibody (1:1000). The relative abundance of ATM protein is shown as a percentage of the non-stimulated control (NS). In parallel, DNA-PKcs expression was measured to establish equivalent loading. The results are representative of three experiments. (B) Top: Gel-shift assay of Sp-1 binding to the ATM promoter was performed with LNCaP cells that had been transfected with either KD-PKCα or the control vector. Transfectants were incubated with TPA (10 ng/ml) for two h and compared with NS samples treated with the vehicle. The first lane shows super-shifted Sp-1 (αSp1) caused by incubating the sample with anti-Sp-1 antibody. The results are representative of three experiments. Bottom: ChIP analysis of Sp-1 binding to the ATM promoter. The cells were treated as for the gel-shift before ChIP was performed. The Input lane shows sheared DNA before ChIP and serves as a positive control. Ig is irrelevant isotype-matched Ab. Sp-1 has been immunoprecipitated with anti-Sp-1 Ab, and the No DNA lane has substituted ddH2O instead of DNA and shows that the band amplified comes from the purified DNA. (C) Measurement of ceramide levels in LNCaP cells transfected with 3 μg of KD-PKCα or empty vector. The results represent the mean average ± s.d. of triplicate measurements from two separate experiments. (D) Apoptosis of LNCaP cells was measured in cells co-transfected with 0.4 μg of GFP vector and either 4 μg of KD-PKCα or empty vector 24 h before TPA addition. At 24 h post-TPA treatment apoptosis was scored using bis-benzimide staining. Only transfected cells displaying green fluorescence were scored. Values representing apoptotic cells are the mean average ± s.d. of duplicate measurements from three experiments.
We previously demonstrated that ATM downregulation by TPA was due to inhibition of Sp-1 binding to the ATM promoter.1 Therefore, Sp-l DNA-binding to the ATM promoter was measured in response to TPA in KD-PKCα transfected LNCaP cells. In extracts from TPA-treated LNCaP cells transfected with control vector, there was virtually no Sp-l binding to the ATM promoter. However, cells transfected with KD-PKCα showed no inhibition of Sp-1 binding to ATM promoter in response to TPA (Fig. 2B top). These findings indicate that PKCα activity is most likely involved in loss of Sp-1 binding upon phorbol ester treatment. Pre-treatment of cell extracts with anti-Sp-l antibodies produced a super-shift of the Sp-1 band (Fig. 2B top, first lane), confirming specificity of this effect. To ensure that Sp-1 binding was specific to ATM, ChIP was also performed (Fig. 2B bottom). Cells treated with TPA and either transfected with KD-PKCα or control-transfected (empty vector) were analyzed. Again, treatment of control-transfected cells with TPA yielded no Sp-1 binding to the ATM promoter as measured by the lack of amplified ATM promoter sequence. However after transfection with KD-PKCα there was significant Sp-1 binding to the ATM promoter with TPA addition (as evidenced by amplified ATM promoter DNA present), validating the crucial role of PKCα in this event. These results support the hypothesis that endogenous PKCα plays a specific role in mediating TPA-induced suppression of ATM protein levels.
Since ATM downregulation is necessary but not sufficient for elevating ceramide levels, we next determined whether PKCα plays a role in ceramide generation in response to TPA and/or radiation. LNCaP cells transfected with KD-PKCα or empty vector were treated with TPA and/or 20 Gy and ceramide levels measured 12 h later. Control transfectants treated with TPA, or the combination of TPA + 20 Gy, exhibited dramatic increases in ceramide levels (Fig. 2C), whereas KD-PKCα-expressing cells were resistant to stimulated ceramide elevation (Fig. 2C). These results suggest that PKCα catalytic activity is necessary for TPA-induced ceramide generation in these cells. Additionally, treatment with FB1, a specific competitive inhibitor of CS, suppressed ceramide generation in TPA-treated vector-transfected control cells, confirming that ceramide generation occurs via CS activation. Figure 2D shows that TPA-mediated apoptosis is similarly inhibited by KD-PKCα, from a level of 31% +/− 2.5% to a near basal level of 11.5% +/− 3.5% (p < 0.001), while there were no significant effects of KD-PKCα on the non-stimulated cells (Control = 6.6% +/− 1.0%, KD-PKCα = 10.9% +/− 3.2% p > 0.7). Collectively, these data suggest that PKCα mediates TPA-induced apoptosis via suppression of ATM, consequently conferring CS activation that leads to ceramide generation and apoptosis.
Constitutively-activated (CA) PKCα suppresses ATM and confers apoptosis
To confirm that PKCα mediates TPA-induced apoptosis, LNCaP cells were transiently transfected with CA-PKCα or empty vector and ATM protein levels were examined 16 h after TPA and/or radiation treatment. Figure 3A shows that CA-PKCα expression substantially reduced ATM levels compared with vector transfected controls (to 64% of control). Treatment of CA-PKCα-transfected cells with TPA yielded additional ATM reduction (to 36% of control), a level equivalent to that found for control transfectants treated with TPA alone. Although radiation alone caused a minimal effect, CA-PKCα expression enhanced radiation-induced ATM downregulation with TPA. The lack of additivity of CA-PKCα and maximal TPA on ATM levels is consistent with their acting via the same mechanism, whereas the enhanced radiation effect implicates an additional mechanism.
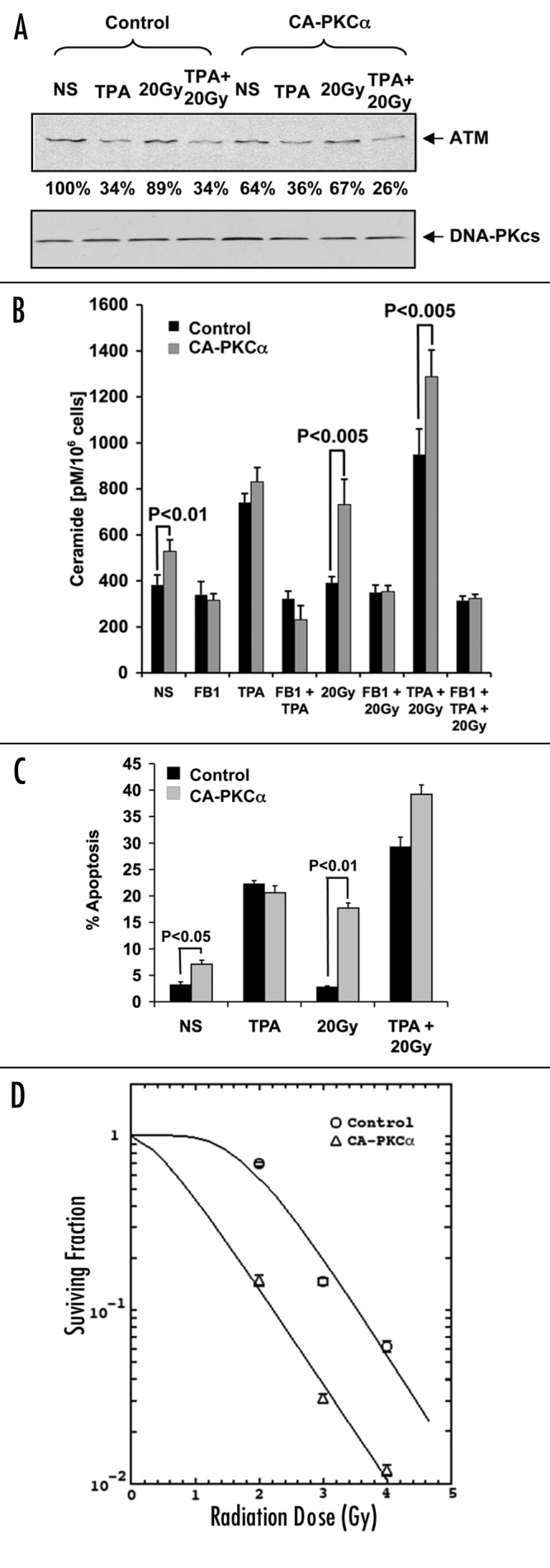
Figure 3. Effects of CA-PKCα on TPA-mediated apoptosis and ATM levels. LNCaP cells were transfected (t = 0) with 4 μg of CA-PKCα or control vector (and co-transfected with 0.4 μg GFP for apoptosis experiments), followed after 24 h with 20 Gy irradiation. (A) ATM expression was measured by western blot in transfected cells treated with 10 ng/ml TPA and/or 20 Gy of radiation. Sixteen hours following irradiation (t = 40 h), cell lysates were prepared. The results are representative of three experiments. (B) Measurement of ceramide levels in LNCaP cells (irradiated or non-irradiated) was performed 12 h post-irradiation (t = 36 h), as described in ‘Materials and Methods’. Values represent the mean average ± s.d. of triplicate measurements from two experiments. (C) Apoptosis of transfected cells treated with TPA and/or radiation (20 Gy). These experiments were performed as described in the legend of Figure 2. Results represent the mean average ± s.d. of triplicate measurements from three experiments. (D) LNCaP cells were transfected with either CA-PKCα or empty vector, incubated for 48 h and irradiated at various doses. The cells were subsequently replated at known concentration before culture for two weeks. Colonies of cells representing at least six divisions were scored. The curves were plotted using the single hit, multiple target (SHMT) model of radiation sensitivity. Each data point represents the mean average of six separate determinations, and show standard error.
These effects on ATM were reflected in alterations in total cellular ceramide. CA-PKCα transfectants exhibited increased ceramide levels compared with vector cells, from a baseline of 383 ± 42 pmol/106 cells to 538 ± 50 (p < 0.01, Figure 3B). Ceramide levels were further increased in CA-PKCα transfectants by TPA addition to 831 ± 62 pmol/106, a level similar to that induced by TPA alone in vector controls (Fig. 3B). Thus TPA and CA-PKCα are non-additive with respect to ceramide elevation. Alternately, 20 Gy alone did not affect ceramide levels, while CA-PKCα expression plus 20 Gy produced a moderate yet significant increase in ceramide levels to 732 ± 110 pmol/106 (p < 0.005 vs. CA-PKCα), which was further enhanced by TPA to 1287 ± 116 pmol/106 cells (p < 0.005 vs. CA-PKCα + 20 Gy). In all instances this increase was inhibited by FB1, implicating CS as the source or ceramide elevation. These results are most consistent with PKCα and TPA acting via the same pathway to increase ceramide levels, by reducing ATM protein that de-represses CS activity, a sequence of events that also leads radiation sensitization.
The additional benefit provided by CA-PKCα with regard to TPA + radiation-induced ceramide generation, was similarly reflected in apoptosis. CA-PKCα transfected LNCaP cells, which display a moderate increase in ceramide also display a modest increase in apoptosis from a baseline of 3.2 ± 0.5% to 7.1 ± 0.7% (p < 0.05). Apoptosis levels were further increased in CA-PKCα transfectants by TPA addition to 20.6 ± 1.32%, a level similar to that induced by TPA alone in vector controls (Fig. 3C), mimicking the effects on ceramide generation (Fig. 3B). Thus TPA and CA-PKCα are non-additive also with respect to apoptosis.
CA-PKCα expression however sensitized LNCaP cells to radiation-induced apoptosis (Fig. 3C), increasing apoptosis at 48 h to 17.7 ± 1.0% (p < 0.01) and in combination with TPA to 39.2 ± 1.8%. Collectively, these data indicate that CA-PKCα acts via the same mechanism as TPA to suppress ATM levels, enhance CS-mediated ceramide generation and confer apoptosis, events that also regulate radiation sensitization.
In addition we assessed the radiosensitizing effect of CA-PKCα using the colony-forming assay. Figure 3D shows that CA-PKCα transfected LNCaP cells acquired significant radiosensitization as reflected in a decrease in Dq (0.434 Gy for CA-PKCα and 1.845 Gy for the control p = 0.05), shifting the survival curve to a lower dose range. There was, however, no significant change in the Do (0.783 for CA-PKCα vs. 0.743 for LNCaP transfected with empty vector). At the clinically relevant radiation dose of 2 Gy, CA-PKCα transfected LNCaP cells exhibited significantly fewer surviving cells (a survival fraction of 0.73 for the control and 0.15 for CA-PKCα; p = 0.01). These data indicate a correlation between the apoptotic and the clonogenic assays in demonstrating the radiosensitizing effect of CA-PKCα in LNCaP cells.
Effects of DAG-lactone on LNCaP cells
Recently the PKCα activator HK-654, a branched diacylglycerol (DAG)-lactone, was shown to mimic the action of TPA with respect to apoptosis in LNCaP cells.2 While HK-654 consisted of a racemic mixture, the active R-enantiomer showed a 2-fold higher affinity for PKCα.19 To determine whether the active R-enantiomer (JH-131E-153; Figure 4A) acts like TPA to decrease ATM expression, cells were treated with JH-131E-153 or the inactive analog JH-131A-109. Figure 4B demonstrates that JH-131E-153 treatment reduced ATM protein levels to as low as 27% of those found after treatment with JH-131A-109. This effect is equivalent to that observed upon TPA treatment in LNCaP cells (Fig. 2A).1 Further, like TPA, JH-131E-153 significantly increased ceramide levels in LNCaP (Fig. 4C), from baseline of 363.5 ± 30.8 pmol/106 cells to 1120 ± 3.3 pmol/106 cells (p < 0.001). Radiation (20 Gy) further increased ceramide at 12 h to 1420 ± 31.1 pmol/106 cells (p < 0.001). Importantly, the inactive analog JH131A-109 did not allow for ceramide generation in either setting, while FB1 (40 μM) decreased active DAG-lactone-induced ceramide elevation to basal levels, further implicating CS as the source of ceramide.

Figure 4. Effect of specific PKCα activator DAG-lactone on LNCaP cells. (A) Chemical structures are shown for the racemic mixture (HK654) used previously,2 its active R-enantomer (JH-131E-153), and an inactive analog (JH-131A-109). (B) western blot analysis of ATM levels following 16 h incubation with either DAG-lactone or control. The relative abundance of ATM is shown as percent of NS controls. DNA-PKcs levels were used as a loading control. The results are representative of three experiments. (C) LNCaP cells were treated for 16 h with 10 μM of active DAG-lactone JH-131E-153, or inactive analog JH-131A-109 prior to irradiation with 20 Gy. Where indicated, FB1 (40 μM) treatment was applied three h before irradiation. Ceramide levels were measured 12 h post-irradiation and the bars represent s.d. values. These experiments were repeated three times. (D) Each DAG-lactone was added to the cells as described in (C). Following an additional 24 h, apoptotic cells were determined as in Figure 2D. The results are mean average ± s.d. of duplicate determinations from three experiments. (E) Active and inactive DAG-lactones (10 μM) were added to either wild-type stably-transfected LNCaP cells (WT8.4) or to empty-vector transfected cells (Control) and apoptosis was quantified 48 h later. The results are the mean average ± s.d. of duplicate determinations from three experiments.
Apoptotic death measurements in LNCaP cells incubated with 10 μM of JH-131E-153 for 48 h (Fig. 4D) showed that while the inactive homolog JH-131A-109 did not induce significant apoptosis, JH-131E-153 generated 25% ± 0.9% apoptosis in these cells, partially inhibited by FB1 (15.2% ± 0.6% Figure 4D; p < 0.01 vs. control). Although 20 Gy alone did not induce apoptosis, in combination with JH-131E-153 the number of apoptotic cells rose to 33.5% ± 1.6% (p = 0.005 vs. JH-131E-153 alone), similar to the synergy observed with TPA and radiation (Fig. 3C). This effect was also partially inhibited by FB1 (20.8% +/− 0.1% Figure 4D; p < 0.01 vs. 20 Gy. This finding implies that, like TPA, JH-131E-153 signals apoptosis through the CS-mediated pathway and thereby confers radiosensitivity.
To confirm that ATM reduction was required for DAG-lactone induced apoptosis, full-length ATM was stably transfected into LNCaP cells under control of cytomegalovirus (CMV) promoter, WT8.4. Figure 4E shows that LNCaP cells overexpressing wild type ATM are more resistant to DAG-lactone-induced cell death compared with the LNCaP cells transfected with the empty vector (24.1% ± 2.2% for vector control and 8.8% ± 1.8% for WT8.4, p < 0.01), a response similar to that observed with TPA.26 The inactive DAG-lactone JH131E-109 showed no significant apoptosis in both cell lines.
Effect of DAG-lactone on CWR22-Rv1 cells
To determine whether the DAG-lactone effect is a LNCaP cell-specific phenomenon, the androgen-responsive CWR22-Rv1 prostate cancer cell line, was tested. We previously showed that TPA lowered ATM protein levels, generated a significant increase in ceramide levels and induced apoptosis in this cell line.1 Ceramide levels were examined in response to DAG-lactone in these cells (Fig. 5A). Similar to results obtained with LNCaP cells, the active lactone JH131E-153 significantly raised ceramide levels from a baseline of 140 ± 16.5 pmol/106 cells to 496.1 ± 18.5 pmol/106 cells (p < 0.005), while the inactive analog JH131A-109 yielded no effect on ceramide levels. Consistent with our previous results, the ceramide baseline level in CWR22-Rv1 cells was lower than the baseline in LNCaP cells.26 Also consistent with the previous data, radiation alone increased ceramide levels generation in untreated and JH131A-109 treated CWR22-Rv1 cells (to 304.5 ± 37.6 pmol/106 cells for untreated and to 299.2 ± 40.6 pmol/106 cells for JH131A-109 treated cells). Radiation (20 Gy) further increased ceramide levels to 750.9 ± 39.3 pmol/106 cells (p < 0.01 vs. untreated or JH131A-109 treated cells) in cells pretreated with the active lactone, JH131E-153. This increase in ceramide was inhibited almost completely by FB1 (40 μM) to 380.7 ± 40.3 pmol/106 cells (p < 0.01 vs. radiation alone) indicating that CS is the source of ceramide generation.
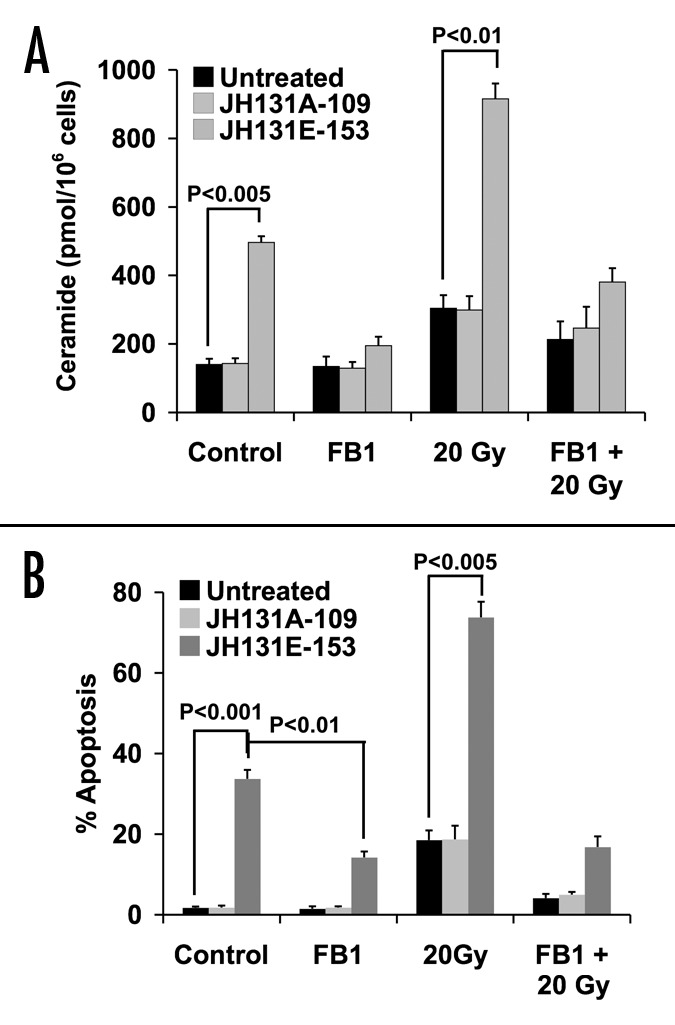
Figure 5. Effect of specific PKCα activator DAG-lactone on CWR22-Rv1 cells. (A) CWR22-Rv1 cells were treated for 16 h with 10 μM of active DAG-lactone JH-131E-153, or inactive analog JH-131A-109 prior to irradiation with 20 Gy. Where indicated, FB1 (40 μM) treatment was added three h before irradiation. Ceramide levels were measured 24 h post-irradiation and the bars represent s.d. values. These experiments were repeated three times. (B) Each DAG-lactone was added to the cells as described in (Fig. 4B). Following an additional 72 h, apoptotic cells were determined as in Figure 2D. The results are the mean average ± s.d. of duplicate determinations from three experiments.
Similar to TPA, the active DAG-lactone JH131E-153 displayed maximal apoptotic (33.7% ± 2.2%, p < 0.001) effect in CWR22-Rv1 cells at 72 h (Fig. 5B). While CWR22-Rv1 displayed modest radiation-induced apoptosis (18.5% ± 2.4% for untreated, 18.7% ± 3.4% with treatment with inactive DAG-lactone control JH131A-109), pretreatment with JH131E-153 resulted in significantly more apoptosis (73.8% ± 3.8%, p < 0.005 vs. irradiated control). Pretreatment with FB1 at 40 μM concentration markedly attenuated the apoptotic responses to DAG-lactone (14.2% ± 1.5% for JH131E-153 alone, vs. 1.5% ± 0.6% and 1.8% ± 0.5% for untreated and JH131A-109 treated respectively, p < 0.01) and DAG-lactone and radiation treatments (16.8% ± 2.7% for JH131E-153 and irradiation, vs. 4.1% ± 1 and 5% ± 0.7% for untreated and JH131A-109 treated, respectively, p < 0.001). Taken together, these data indicate that DAG-lactone induced-apoptosis is not a LNCaP cell-specific phenomenon.
Effect of PKC activation on LNCaP tumor response in vivo
The main goal of these studies was to demonstrate the ability to translate these results in an animal model. In contrast to the known tumor-promoting effects of TPA, JH-131E-153 was not transforming in tumor promotion sensitive JB6 cells in vitro (Young M and Marquez VE, unpublished data). To explore a potential for tumor radiosensitization, the orthotopic LNCaP model was employed using Swiss nude mice. LNCaP tumors reaching a size of ~100 mm3 were injected i.p. with either TPA (40 μg/kg), JH-131E-153 (12 mg/kg) or vehicle control (DMSO). Two injections were delivered at 24 h intervals with prostate resection at 48 h after the first injection. Figure 6A shows that ATM levels in tumor extracts were significantly decreased following DAG-lactone treatment, dropping to a level between 7% and 3% of vehicle control. These results compare favorably with TPA treatment that reduced ATM levels to 12–27% of baseline.

Figure 6. Effect of PKCα stimulation on orthotopic prostate cancer model. (A) DAG-lactone was injected twice i.p. into orthotopically-implanted nude mice at 24 h intervals at a concentration of 12 mg/kg/day. Prostate tumors were removed 48 h after the first injection, homogenized and prepared for western blot as described in the ‘Materials and Methods’. The density of the bands was measured and is shown in comparison to the control. The results shown are representative of three separate experiments. (B) western blots for prostate tumor samples were probed with either phospho-serine specific Abs to PKCα or PKCδ, and compared with total levels of PKCα or PKCδ as a control. The results are representative of two separate experiments. (C) PSA levels in TPA-treated mice. Mice were injected i.p. with 40 μg/kg TPA or DMSO solvent control for five days. Radiation-treated mice were locally irradiated with 4 Gy 16 h after each TPA injection for a total of 20 Gy. Control non-irradiated mice n = 20, radiation-treated control mice n = 12, TPA-non-irradiated mice n = 17, and TPA-treated irradiated mice n = 10. (D) PSA levels in DAG-lactone treated mice. Mice were injected i.p. with 12 mg/kg/day of DAG-lactone or with DMSO solvent control for ten days. Radiation-treated mice were locally irradiated at 1 Gy 16 h after each injection to a total of 10 Gy. Control non-irradiated mice n = 14, radiation-treated control mice n = 14, DAG-lactone treated mice n = 13, radiation-treated DAG-lactone mice n = 12. The values represent the mean ± standard errors.
To determine whether DAG-lactone treatment was activating PKCα in vivo, the same samples were probed for an increase in PKCα serine phosphorylation at position 657, while for PKCδ serine-643 phosphorylation was examined. Figure 6B shows that while DAG-lactone treatment induced a significant serine-657 phosphorylation in PKCα, no corresponding increase in serine-643 phosphorylation was detected for PKCδ. Total levels of PKCα and PKCδ remained similar throughout the experiment. These results indicate, similar to the in vitro data, that DAG-lactone also specifically activated PKCα in the in vivo LNCaP tumor model.
We next tested whether the reduction in ATM after TPA treatment correlated with radiation response using fractionated radiation scheme employing 4 Gy delivered daily five times per week, Monday-Friday, to a total dose of 20 Gy. Mice received daily i.p. injections of TPA (40 μg/kg) 16 h prior to each radiation treatment. Tumor response was assessed indirectly by measuring serum PSA levels, shown to correlate with tumor growth.30 PSA doubling time in our orthotopically-implanted tumors was approximately ten days (data not shown). Figure 6C shows that fractionated radiotherapy alone resulted in an initial tumor growth response. However by week four PSA started to rise and by week seven when mice were sacrificed the tumor had increased to 1 cm3. In contrast, the combination of TPA and fractionated radiation completely inhibited tumor growth for the duration of the experiment (eight weeks) and no elevation of serum PSA was detected during this period (p < 0.001 vs. control). These data indicate that TPA significantly enhances the LNCaP tumor response to radiotherapy.
JH-131E-153 similarly improved the outcome of fractionated radiotherapy (Fig. 6D). For these studies, 1 Gy radiation was delivered daily five times per week for two weeks, Monday-Friday, to a total dose of 10 Gy. Mice received daily i.p. injections of JH-131E-153 (12 mg/kg) 16 h prior to each radiation treatment during the first week, whereas during the second week only two additional injections were delivered at a 48 h interval, with the first injection delivered on Monday. Serum PSA continued to increase in control mice treated with either vehicle or JH-131E-153 alone, while tumors treated with radiation plus vehicle displayed a delay of almost four weeks before PSA levels increased (p < 0.01 vs. control). In contrast, JH-131E-153 combined with 10 Gy fractionated radiotherapy completely suppressed tumor growth and PSA elevation for at least 8 weeks (p < 0.001 vs. control). Taken together, these results strongly suggest that activation of PKCα with either TPA or JH-131E-153 significantly improves the radiation response of LNCaP prostate tumors, indicating JH-131E-153 might serve as a potent radiosensitizer in vivo.
Discussion
The present studies provide further evidence that TPA-induced apoptosis in androgen-sensitive human prostate cells is mediated in vitro and in vivo by ATM downregulation. This downregulation appears to be driven by catalytically active PKCα (Fig. 2), which inhibits Sp-1 binding to the ATM promoter (Fig. 2B), leading to de-repression of CS activity (Fig. 2C) and apoptosis induction (Fig. 2D). Previous studies by our and other laboratories showed that prolonged activation/translocation of PKCα to the plasma membrane mediated TPA-induced apoptosis in LNCaP cells (Fig. 1).31 TPA activation of MAPK, a potential downstream target of PKC pathway, had no effect on ATM downregulation, thus ruling out a possible role for MAPK-mediated post-translational regulation (data not shown). Here we show that catalytically competent PKCα is necessary for transcriptionally-regulated TPA-induced ATM downregulation and apoptosis, since engineered expression of kinase-dead PKCα blocked all effects attributed to TPA. Consistent with these findings, overexpression of constitutively active PKCα also recapitulated the TPA effect, although its ability to reduce ATM levels was more modest than that of TPA (Fig. 3A). While it is possible that other PKC isoforms might be involved in the TPA effect, overexpressing constitutively active PKCα simply might deliver a less powerful signal than TPA, accounting for the differences. In this regard, we have excluded a role for PKCδ in ATM downregulation, an isoform known to be involved in TPA-induced apoptosis, as dominant-negative kinase-dead PKCδ was without effect (Fig. 2A).
The present studies also indicate that like TPA, CA-PKCα can synergize with ionizing radiation to yield increased ceramide levels and apoptosis (Fig. 3). Expression of kinase-dead PKCα or inhibition of CS by FB1 was sufficient to block effects by TPA alone or TPA plus radiation. In view of our earlier findings that ionizing radiation activates CS,16 the present results are consistent with a model (Fig. 7) in which the CS-activating effects of radiation and TPA collaborate in raising the level of ceramide that in turn initiates apoptosis. As shown in our previous work, downregulation of ATM is necessary, but not sufficient, to produce apoptosis in TPA-treated human prostate tumor cells. A second event elicited by either TPA or radiation must occur in order to activate CS and generate ceramide. Although the present work showed that PKCδ is not involved in ATM downregulation, a functional role for this isoform in the “second event” in TPA-induced apoptosis in LNCaP cells, is currently under investigation in our laboratory.
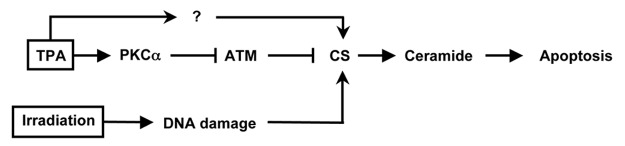
Figure 7. Schematic model: The present results are consistent with a model in which downregulation of ATM is mediated by activation of PKCα. This event is necessary, but not sufficient to produce apoptosis in human prostate tumor cells. A second event elicited by either TPA or radiation must occur in order to activate CS and generate ceramide to initiate in apoptosis.
TPA has been shown to act as a tumor promoter in skin of mice “initiated” with aromatic hydrocarbons, although topical application of TPA alone has no tumorigenic effects.32-34 Intravenous TPA therapy administered at similar doses as used in our study has recently been used safely in patients with myelocytic leukemia and other malignancies.35,36 However, long-term effects of such therapy are as yet unknown. As an alternative to TPA, we tested a branched DAG-lactone, shown by Garcia-Bermejo et al. to mimic TPA effects in LNCaP cells.2 Although DAG-lactone activates PKCα and induces apoptosis like TPA, it does not produce the same transforming effects as TPA (Young M and Marquez VE, unpublished data). Here we demonstrated that this branched DAG-lactone lowered ATM levels in LNCaP cells in culture and in orthotopic tumors resulting in both contexts in significantly increased ceramide levels and apoptosis, via activation of PKCα. These findings signify involvement of the same ATM-CS pathway as used by TPA. This is not a LNCaP cell-specific phenomenon, since DAG-lactone activated this pathway and induced apoptosis in another human androgen-sensitive prostate cancer cell line CWR22-Rv1 (Fig. 5). Furthermore, the dramatic anti-tumor effect produced by DAG-lactone treatment of mice was greatly potentiated when combined with a fractionated protocol of radiation consisting of doses as low as 10 x 1 Gy.
Our studies strongly suggest that activation of PKCα through either TPA or DAG-lactone can significantly improve radiosensitivity and responsiveness of prostate tumors. We suggest that the DAG-lactones represent a novel class of compounds worthy of further consideration as a treatment for human prostate cancer.
Materials and Methods
Materials
Cell culture products were obtained through Mediatech (800-Cellgro, VA), except for fetal calf serum, which was purchased from Gemini Bio-products (CA). TPA and the DNA-binding fluorochrome bis-benzimide trihydrochloride were from Sigma-Aldrich (St. Louis, MO). The branched DAG-lactone (JH-131E-153)19 utilized in the present investigation is the pure R-enantiomer of the racemic mixture (HK654) used by Garcia-Bermejo.2 An inactive DAG-lactone (JH-131A-109) was also employed as negative control.20 Phosphoserine-specific and non-phospho-specific antibodies to PKCα and PKCδ were purchased from Santa Cruz Biotechnology Inc., (CA).
Cell lines
The human prostate cancer cell line LNCaP were acquired from the ATCC. CWR22-Rv1 was kindly provided by Dr. Jacobberger from Case Western Reserve University (Cleveland, OH).21 Cells were cultured at 37°C in a humidified 5% CO2 atmosphere in RPMI-1640 supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin and 10 mM HEPES (pH 7.2). For experiments, cells were cultured in the same medium except 0.2% human albumin (HA) was substituted for FCS. TPA was diluted to a concentration of 1 mg/ml in DMSO and stored at -20°C in the dark. LNCaP cells were typically treated with TPA (10 ng/ml) for 16 h prior to irradiation.
Measurement of PKC activity
PKC activity was measured as described by Haimovitz-Friedman et al.22 by a method modified from Makowske et al. and Niles and Loewy.23,24 Briefly, following stimulation with either TPA or DAG-lactone, cells were harvested by scraping into ice-cold buffer A (20 mM TRIS-HCl, pH 7.5; 6 mM sucrose, 0.5 mM EDTA, 0.5 mM dithiothreitol and protease inhibitors), sonicated 1 min and centrifuged at 500 xg for five min to remove nuclei. The supernatant (cytosolic fraction) was separated from the membrane fraction by centrifugation at 100,000 xg for one h. The membrane fraction was solubilized with cold buffer A containing 0.5% Triton X-100 and non-solubilized material was removed by centrifugation at 100,000 xg. Each fraction was applied to a DEAE-cellulose column and PKC activity was eluted with 20 mM Tris, 2 mM EDTA, 5 mM EGTA, 2 mM DTT and 100 mM NaCl. PKC activity was measured by transfer of 32P from [γ-32P]-ATP to 300 μM α-peptide (Amersham, IL). Peptide was collected onto phosphocellulose squares and associated radioactivity quantified by liquid scintillation counting. PKC activity was defined as the difference between the kinase activity in incubations containing Ca2+/phosphatidylserine vs. incubations containing EDTA and lacking Ca2+/phosphatidylserine.
Quantification of apoptosis
Morphologic changes in nuclear chromatin of cells undergoing apoptosis were detected by staining with the DNA-binding fluorochrome bis-benzimide trihydrochloride, as described.15 Cells were visually inspected using an Olympus BH2 fluorescence microscope equipped with a Dich mirror cube filter (BH2-DMU2UV). Cells displaying at least three apoptotic bodies were scored as apoptotic.
Analysis of ATM protein expression
Cells were lysed in RIPA buffer (50 mM TRIS-HCl (pH 6.8), 1% Triton X100, 0.5% Sodium deoxycholate, 150 mM NaCl and 5 mM EDTA), containing one complete mini EDTA-free protease inhibitor tablet (Roche, IN) per 10 ml and 1 mM PMSF. Cell lysates were centrifuged at 18,000 g for 10 min to remove membranes and nuclei. Mouse prostates were dissected and homogenized using a Dounce homogenizer and RIPA buffer containing protease inhibitors, followed by centrifugation at 18,000 g for 10 min. Protein concentration was determined using the BCA reagent (Pierce Co., Rockford, IL). Samples were boiled in 4x denaturing Laemmli sample buffer and stored at -80°C. ATM was resolved using a 5% SDS-polyacrylamide gel. Monoclonal anti-ATM antibody MAT3 (kindly provided by Prof. Y Shiloh, Tel-Aviv University, Tel-Aviv, Israel) was used at 1:1000 dilution while anti-mouse HRP secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used at 1:1000 dilution. Detection of bands was performed with the Western Lightning chemiluminescence reagent (Perkin Elmer, MA). For loading controls, membranes were first stripped using 100 mM TRIS-HCl, pH 6.8, 100 mM β-mercaptoethanol and 2% SDS for 1 h at 50°C, followed by washing 3x in TBS containing 0.1% Tween-20. Membrane was re-probed using an anti-DNA-PKcs mAb (Neomarkers, CA) at 1:200 dilution and an anti-mouse HRP-conjugated secondary antibody. Bands corresponding to ATM were scanned and analyzed using ImageJ v1.29 image analysis software.
Transfection experiments
A plasmid encoding either constitutively active (CA) PKCα (a gift of Peter J. Parker, Cancer Research UK) or dominant negative kinase dead (KD) mutant of PKCα25 was transfected (4 μg) into LNCaP cells using Lipofectamine PLUS (Gibco BRL). For apoptosis experiments, each plasmid was -co-transfected with 0.4 μg of GFP-containing plasmid (pcDNA3.1/CT-GFP, Invitrogen) and only cells displaying green fluorescence were scored. Cells were used for experiments 24 h following transfection. The stably ATM-transfected and empty-vector transfected LNCaP cell lines were produced as described previously.26
Ceramide measurement
Ceramide levels were quantified using the diacylglycerol kinase assay as described previously.16
Electrophoretic mobility shift assay
Binding to the Sp-1 promoter was determined by a modification of a method described previously.27 Briefly, 6 x 106 cells were washed in ice-cold PBS before resuspension in 10 mM HEPES pH 7.9 buffer containing 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.2 mM PMSF. After 15 min on ice, 25 μl of 10% NP-40 was added, the sample was vortexed for 10 sec and centrifuged at 18,000 xg for 1 min. To elute the nuclear proteins, the pellet was resuspended in 40 μl of 20 mM HEPES pH 7.9 containing 400 mM KCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1 mM DTT and 0.2 mM PMSF and rotated for 30 min at 4°C. After centrifugation at 18,000 xg for 5 min, the supernatant was flash-frozen in liquid nitrogen in 15 μl aliquots and stored at -80°C. Approximately 35 fmol of 32P-labeled (using T4 polynucleotide kinase) Sp-1 consensus sequence (5′-ATT CGA TCG GGG CGG GGC GAGC-3′) was added to 10 μg of nuclear extract in the presence of 1 μg of herring sperm DNA, 10 μg of BSA and binding buffer consisting of 10 mM HEPES pH 7.9, 50 mM KCl, 0.5 mM EDTA, 10% glycerol, 0.1% NP-40, 5 mM DTT and 0.2 mM PMSF. The binding reaction proceeded at room temperature for 25 min and was followed by electrophoresis on a native 5% polyacrylamide gel to resolve the bands.
Chromatin immunoprecipitation
Sp-1 bound chromatin was immunoprecipitated and prepared for PCR using the ChIP-IT kit (Active Motif, CA) according to the manufacturer’s instructions. Briefly, chromatin was enzymatically sheared followed by Sp-1 immunoprecipitation, as well as use of an irrelevant Ab of the same isotype (rabbit IgG). For a positive control some of the sheared DNA (input DNA) was stored before any Ab was added. After protein digestion and DNA purification, PCR was performed to amplify a 200 bp region surrounding the Sp-1 consensus sequence in the ATM promoter. As a final control, the reaction was performed using water instead of DNA. 36 cycles of 20 sec 95°C, 30 sec 58°C and 30 sec 72°C, with a 6 min extension step at the end using probes with the sequence 5′-CGT CCG CGC TTA CCC AAT ACAA-3′ and 5′-ATC CCC GCC CCT CCA AGT CTG A-3′ were performed and run on a 2.5% agarose gels before visualization.
Colony forming assay
LNCaP cells were transfected with either a control plasmid or with CA-PKCα and 48 h later irradiated with 0, 2, 3 and 4 Gy. Cells were trypsinized and replated in triplicate at various cell concentrations for colony formation. After two weeks in culture, colonies were stained with 1% crystal violet in 0.2 M citric acid. Colonies with at least 50 cells, representing at least six cell divisions, were scored and then analyzed using the CellFit v2.5 program.28 The single hit multiple target model was used to determine survival values.
Orthotopic prostate cancer model
Eight to ten week-old male Swiss nude (nu/nu) mice were implanted orthotopically with 3.0 x 106 LNCaP cells. Immediately before tumor implantation, cultured LNCaP cells were trypsinized and resuspended in RPMI 1640 with 10% FBS and viability determined by trypan blue exclusion. Only single cell suspensions with > 90% viability were used for in vivo injection. Mice were anesthetized with 12.5 mg Ketamine + 1.25 mg Xylazine (i.p., intraperitoneal) and orthotopic tumor implantation was performed as described.29 Briefly, a low midline abdominal incision was made with a #15 blade (Bard Parker). The peritoneal cavity was entered by sharply incising the linea alba. The bladder and seminal vesicles were identified and gently raised, thus exposing the dorsal lobes of the mouse prostate. Approximately 3.0 x 106 LNCaP cells were injected in 0.1 ml of medium using a 26 gauge needle. Proper implantation of cell suspension was indicated by blebbing under the prostatic capsule. Visceral contents were then replaced into the abdominal cavity and the wound closed with surgical autoclips (Becton Dickinson). Mice were monitored during the postoperative period according to animal care facility guidelines. Injected mice were housed (five mice/cage) in a pathogen-free environment, using filtered, laminar airflow hoods in standard vinyl cages with air filter tops. Cages, bedding and water were autoclaved before use.
Orthotopically-transplanted LNCaP tumors in nude mice secrete PSA that can be detected histochemically in tumor cells and by radioimmunassay in mouse serum.30 To assess LNCaP tumor take and tumor volume after intra-prostatic transplantation, mice were anesthetized with 12.5 mg Ketamine + 1.25 mg Xylazine (i.p.). Phlebotomy was performed by accessing the retro-orbital venous plexus with a microcapillary pipette (Fisher). Serum PSA determinations were performed by radioimmunoassay (Hybritech) according to the recommendations of the manufacturer. Using serum PSA determinations as an indicator of tumor size,16 mice were only utilized in experiments when PSA levels ranged from 5.0–15.0 ng/ml representing tumors with a mass in the range of 70–150 mg. Mice were divided into four treatment groups: (a) Control, DMSO i.p.; (b) TPA at 40 μg/kg in DMSO (MTD dose) i.p.; (c) DMSO 16 h before each fraction of 5 x 4 Gy; (d) TPA 16 h before each fraction of 5 x 4 Gy. In the experiments using the DAG-Lactone (JH-131E-153), 1 Gy radiation was delivered daily 5 times per week for two weeks to a total dose of 10 Gy. Mice received daily i.p. injections of JH-131E-153 (12 mg/kg) 16 h prior to each radiation treatment during first week, whereas during the second week only two additional injections were delivered at a 48 h interval with the first injection given on Monday. Mice were irradiated by exposure to a small pelvic RT field using a specially-designed Lucite jig. All procedures and post-operative care with animals were in accordance with NIH guidelines and received prior approval by the Institutional Animal Care and Use Committee at Memorial Sloan-Kettering Cancer Center.
Statistics
Student’s t-test were performed on pairs of data points for apoptotic values while the Wilcoxon rank sum test was performed on the animal data. Both tests were calculated using Prism v5 software.
Acknowledgments
We thank John Fuller for assistance with the ChIP assay. This work was supported by NIH RO1 #CA105125 to A.H-F., NIH ROI #CA85704 to R.K and a gift from the The Virginia and D.K. Ludwig Fund for Cancer Research to Z.F. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research to V.E.M.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/7119
References
- 1.Truman JP, Gueven N, Lavin M, Leibel S, Kolesnick R, Fuks Z, et al. Down-regulation of ATM protein sensitizes human prostate cancer cells to radiation-induced apoptosis. J Biol Chem. 2005;280:23262–72. doi: 10.1074/jbc.M503701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garcia-Bermejo ML, Leskow FC, Fujii T, Wang Q, Blumberg PM, Ohba M, et al. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCalpha. J Biol Chem. 2002;277:645–55. doi: 10.1074/jbc.M107639200. [DOI] [PubMed] [Google Scholar]
- 3.Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem. 1995;270:28495–8. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 4.Dekker LV, Parker PJ. Protein kinase C--a question of specificity. Trends Biochem Sci. 1994;19:73–7. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 5.Blobe GC, Obeid LM, Hannun YA. Regulation of protein kinase C and role in cancer biology. Cancer Metastasis Rev. 1994;13:411–31. doi: 10.1007/BF00666107. [DOI] [PubMed] [Google Scholar]
- 6.Jarvis WD, Turner AJ, Povirk LF, Traylor RS, Grant S. Induction of apoptotic DNA fragmentation and cell death in HL-60 human promyelocytic leukemia cells by pharmacological inhibitors of protein kinase C. Cancer Res. 1994;54:1707–14. [PubMed] [Google Scholar]
- 7.Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–71. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 8.Grant S, Jarvis WD, Swerdlow PS, Turner AJ, Traylor RS, Wallace HJ, et al. Potentiation of the activity of 1-β-D-arabinofuranosylcytosine by the protein kinase C activator bryostatin 1 in HL-60 cells: association with enhanced fragmentation of mature DNA. Cancer Res. 1992;52:6270–8. [PubMed] [Google Scholar]
- 9.Lotem J, Cragoe EJ, Jr., Sachs L. Rescue from programmed cell death in leukemic and normal myeloid cells. Blood. 1991;78:953–60. [PubMed] [Google Scholar]
- 10.Haimovitz-Friedman A, Balaban NA, McLoughlin M, Ehleiter D, Michaeli J, Vlodavsky I, et al. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 1994;54:2591–7. [PubMed] [Google Scholar]
- 11.Ishii HH, Gobé GC. Epstein-Barr virus infection is associated with increased apoptosis in untreated and phorbol ester-treated human Burkitt’s lymphoma (AW-Ramos) cells. Biochem Biophys Res Commun. 1993;192:1415–23. doi: 10.1006/bbrc.1993.1574. [DOI] [PubMed] [Google Scholar]
- 12.Kizaki H, Tadakuma T, Odaka C, Muramatsu J, Ishimura Y. Activation of a suicide process of thymocytes through DNA fragmentation by calcium ionophores and phorbol esters. J Immunol. 1989;143:1790–4. [PubMed] [Google Scholar]
- 13.Reyland ME, Barzen KA, Anderson SM, Quissell DO, Matassa AA. Activation of PKC is sufficient to induce an apoptotic program in salivary gland acinar cells. Cell Death Differ. 2000;7:1200–9. doi: 10.1038/sj.cdd.4400744. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Bhuiyan M, Mohammad RM, Sarkar FH. Induction of apoptosis in breast cancer cells by TPA. Oncogene. 1998;17:2915–20. doi: 10.1038/sj.onc.1202218. [DOI] [PubMed] [Google Scholar]
- 15.Garzotto M, White-Jones M, Jiang Y, Ehleiter D, Liao WC, Haimovitz-Friedman A, et al. 12-O-tetradecanoylphorbol-13-acetate-induced apoptosis in LNCaP cells is mediated through ceramide synthase. Cancer Res. 1998;58:2260–4. [PubMed] [Google Scholar]
- 16.Garzotto M, Haimovitz-Friedman A, Liao WC, White-Jones M, Huryk R, Heston WD, et al. Reversal of radiation resistance in LNCaP cells by targeting apoptosis through ceramide synthase. Cancer Res. 1999;59:5194–201. [PubMed] [Google Scholar]
- 17.Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569:123–32. doi: 10.1016/j.mrfmmm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Liao W-C, Haimovitz-Friedman A, Persaud RS, McLoughlin M, Ehleiter D, Zhang N, et al. Ataxia telangiectasia-mutated gene product inhibits DNA damage-induced apoptosis via ceramide synthase. J Biol Chem. 1999;274:17908–17. doi: 10.1074/jbc.274.25.17908. [DOI] [PubMed] [Google Scholar]
- 19.Kang JH, Siddiqui MA, Sigano DM, Krajewski K, Lewin NE, Pu Y, et al. Conformationally constrained analogues of diacylglycerol. 24. Asymmetric synthesis of a chiral (R)-DAG-lactone template as a versatile precursor for highly functionalized DAG-lactones. Org Lett. 2004;6:2413–6. doi: 10.1021/ol0492041. [DOI] [PubMed] [Google Scholar]
- 20.Choi Y, Kang JH, Lewin NE, Blumberg PM, Lee J, Marquez VE. Conformationally constrained analogues of diacylglycerol. 19. Synthesis and protein kinase C binding affinity of diacylglycerol lactones bearing an N-hydroxylamide side chain. J Med Chem. 2003;46:2790–3. doi: 10.1021/jm030082c. [DOI] [PubMed] [Google Scholar]
- 21.Sramkoski RM, Pretlow TG, 2nd, Giaconia JM, Pretlow TP, Schwartz S, Sy MS, et al. A new human prostate carcinoma cell line, 22Rv1. In Vitro Cell Dev Biol Anim. 1999;35:403–9. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 22.Haimovitz-Friedman A, Balaban N, McLoughlin M, Ehleiter D, Michaeli J, Vlodavsky I, et al. Protein kinase C mediates basic fibroblast growth factor protection of endothelial cells against radiation-induced apoptosis. Cancer Res. 1994;54:2591–7. [PubMed] [Google Scholar]
- 23.Makowske M, Rosen OM. Complete activation of protein kinase C by an antipeptide antibody directed against the pseudosubstrate prototope. J Biol Chem. 1989;264:16155–9. [PubMed] [Google Scholar]
- 24.Niles RM, Loewy BP. Induction of protein kinase C in mouse melanoma cells by retinoic acid. Cancer Res. 1989;49:4483–7. [PubMed] [Google Scholar]
- 25.Sullivan RM, Stone M, Marshall JF, Uberall F, Rotenberg SA. Photo-induced inactivation of protein kinase calpha by dequalinium inhibits motility of murine melanoma cells. Mol Pharmacol. 2000;58:729–37. doi: 10.1124/mol.58.4.729. [DOI] [PubMed] [Google Scholar]
- 26.Truman J, Liao W-C, Ehleiter D, Kolesnick R, Lavin M, Leibel SA, et al. Transcriptional downregulation of Ataxia-Telangiectasia Mutated protein (ATM) radiosensitizes human prostate cancer cells. Proceedings of the AACR 2004; 45. [Google Scholar]
- 27.Keating KE, Gueven N, Watters D, Rodemann HP, Lavin MF. Transcriptional downregulation of ATM by EGF is defective in ataxia-telangiectasia cells expressing mutant protein. Oncogene. 2001;20:4281–90. doi: 10.1038/sj.onc.1204527. [DOI] [PubMed] [Google Scholar]
- 28.Albright N. Computer programs for the analysis of cellular survival data. Radiat Res. 1987;112:331–40. doi: 10.2307/3577260. [DOI] [PubMed] [Google Scholar]
- 29.Stephenson RA, Dinney CP, Gohji K, Ordóñez NG, Killion JJ, Fidler IJ. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst. 1992;84:951–7. doi: 10.1093/jnci/84.12.951. [DOI] [PubMed] [Google Scholar]
- 30.Gleave ME, Hsieh JT, Wu HC, von Eschenbach AC, Chung LW. Serum prostate specific antigen levels in mice bearing human prostate LNCaP tumors are determined by tumor volume and endocrine and growth factors. Cancer Res. 1992;52:1598–605. [PubMed] [Google Scholar]
- 31.Powell CT, Brittis NJ, Stec D, Hug H, Heston WD, Fair WR. Persistent membrane translocation of protein kinase C alpha during 12-0-tetradecanoylphorbol-13-acetate-induced apoptosis of LNCaP human prostate cancer cells. Cell Growth Differ. 1996;7:419–28. [PubMed] [Google Scholar]
- 32.Berenblum I. A re-evaluation of the concept of cocarciongenesis. Prog Exp Tumor Res. 1969;11:21–30. doi: 10.1159/000391387. [DOI] [PubMed] [Google Scholar]
- 33.Slaga TJ, Fischer SM, Nelson K, Gleason GL. Studies on the mechanism of skin tumor promotion: evidence for several stages in promotion. Proc Natl Acad Sci U S A. 1980;77:3659–63. doi: 10.1073/pnas.77.6.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fürstenberger G, Berry DL, Sorg B, Marks F. Skin tumor promotion by phorbol esters is a two-stage process. Proc Natl Acad Sci U S A. 1981;78:7722–6. doi: 10.1073/pnas.78.12.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han ZT, Zhu XX, Yang RY, Sun JZ, Tian GF, Liu XJ, et al. Effect of intravenous infusions of 12-O-tetradecanoylphorbol-13-acetate (TPA) in patients with myelocytic leukemia: preliminary studies on therapeutic efficacy and toxicity. Proc Natl Acad Sci U S A. 1998;95:5357–61. doi: 10.1073/pnas.95.9.5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han ZT, Tong YK, He LM, Zhang Y, Sun JZ, Wang TY, et al. 12-O-Tetradecanoylphorbol-13-acetate (TPA)-induced increase in depressed white blood cell counts in patients treated with cytotoxic cancer chemotherapeutic drugs. Proc Natl Acad Sci U S A. 1998;95:5362–5. doi: 10.1073/pnas.95.9.5362. [DOI] [PMC free article] [PubMed] [Google Scholar]