

Abstract
The spatio-temporal properties of Ca2+ transients during excitation-contraction coupling and elementary Ca2+ release events (Ca2+ sparks) were studied in atrial and ventricular myocytes with ultra-fast confocal microscopy using a Zeiss LSM 5 LIVE system that allows sampling rates of up to 60 kHz. Ca2+ sparks which originated from subsarcolemmal junctional sarcoplasmic reticulum (j-SR) release sites in atrial myocytes were anisotropic and elongated in the longitudinal direction of the cell. Ca2+ sparks in atrial cells originating from non-junctional SR and in ventricular myocytes were symmetrical. Ca2+ spark recording in line scan mode at 40,000 lines/s uncovered step-like increases of [Ca2+]i. 2-D imaging of Ca2+ transients revealed an asynchronous activation of release sites and allowed the sequential recording of Ca2+ entry through surface membrane Ca2+ channels and subsequent activation of Ca2+-induced Ca2+ release. With a latency of 2.5 ms after application of an electrical stimulus, Ca2+ entry could be detected that was followed by SR Ca2+ release after an additional 3 ms delay. Maximum Ca2+ release was observed 4 ms after the beginning of release. The timing of Ca2+ entry and release was confirmed by simultaneous [Ca2+]i and membrane current measurements using the whole cell voltage-clamp technique. In atrial cells activation of discrete individual release sites of the j-SR led to spatially restricted Ca2+ release events that fused into a peripheral ring of elevated [Ca2+]i that subsequently propagated in a wave-like fashion towards the center of the cell. In ventricular myocytes asynchronous Ca2+ release signals from discrete sites with no preferential subcellular location preceded the whole-cell Ca2+ transient. In summary, ultra-fast confocal imaging allows investigation of Ca2+ signals with a time resolution similar to patch clamp technique, however in a less invasive fashion.
Introduction
Excitation-contraction coupling (ECC) refers to the process that links cell membrane depolarization to Ca2+ mobilization and development of contractile force in the heart [1]. Action potential (AP) depolarization activates voltage-gated L-type Ca2+ channels (LCC; also referred to as dihydropyridine receptors) in the surface membrane, resulting in localized (sub-membrane) increases in cytosolic Ca2+ ([Ca2+]i). Calcium that enters the cell activates sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptors, RyRs) through a process known as calcium-induced calcium release (CICR; [2]). RyRs and LCCs face each other in the diadic cleft formed by the close approximation of surface and SR membranes. RyRs assemble as clusters of channels in the SR membrane and form individual Ca2+ release units (CRU; [3]). Together with the diadic cleft and sarcolemmal Ca2+ channels CRUs are organized in discrete signaling domains or couplons [4]. In response to an AP hundreds of CRUs are activated essentially simultaneously. The spatial and temporal summation of Ca2+ release from individual CRUs gives rise to the whole-cell Ca2+ transient [5], [6], [7]. Because of the extensive transverse (t) tubular network - a 3-dimensional network of surface membrane invaginations that assure physical proximity of surface membrane Ca2+ channels and CRUs throughout the entire cell volume [8] - AP-induced Ca2+ release in ventricular myocytes is highly synchronized and spatially rather homogeneous.
In contrast, in atrial cells the t-tubular system is poorly developed or even entirely lacking [7], [9], [10], [11], [12]. In atrial cells two types of SR can be defined, based on their location relative to the surface membrane. Junctional SR (j-SR) is found in the cell periphery where it is organized in peripheral couplings, i.e. the SR membrane is found in close spatial association with the surface membrane, similar to the diadic cleft in ventricular myocytes [13], [14]. In contrast, non-junctional SR (nj-SR) is found in deeper regions of the cell and does not associate with the surface membrane. Both j-SR and nj-SR possess RyRs [10], [11], [12], [13], [15], [16], [17] which are organized in a 3-dimensional array of RyR clusters, and are capable of active SR Ca2+ release. As a consequence of these ultrastructural arrangements AP-induced Ca2+ release is spatially inhomogeneous (for review see e.g. [7], [18], [19]): AP-induced membrane depolarization activates Ca2+ entry through LCCs and CICR through RyRs of the j-SR. Elevation of peripheral [Ca2+]i propagates via CICR in a Ca2+ wave-like fashion in centripetal direction by a diffusion-reaction process or a ‘fire-diffuse-fire’ mechanism [20].
Elementary Ca2+ release events, arising from the activation of individual CRUs, have been termed Ca2+ sparks ([21]; for a recent comprehensive review see [22]), although Ca2+ release events of smaller magnitude, possibly arising from single RyRs, have been postulated [23], [24], [25]. To date Ca2+ sparks have been detected in a wide range of mammalian species (for review see [22]). In atrial cells Ca2+ sparks originate from j-SR and nj-SR, however with a high prevalence for the cell periphery [7], [16], [26], [27], [28].
While sparks have been demonstrated in cardiac myocytes already two decades ago [21], it has remained a matter of debate, how many RyRs within a CRU contribute to a spark. Available data on how many RyRs a CRU of a cardiac myocyte contains vary widely and range from less than ten to several hundred RyRs [29], [30], [31], [32], [33], [34], [35], [36]. However, it is still not established whether a fraction or all RyRs of a cluster are activated in a spark. Indeed, published reports on how many channels contribute to a cardiac spark vary widely and range from a single or only a few channels to the whole CRU, thus possibly involving several hundred channels [21], [32], [37], [38], [39], [40], [41], [42].
The fact that such critical questions have remained inconclusive is, at least in part, linked to methodological and technical limitations hampering the recording of locally restricted and rapidly changing subcellular Ca2+ signals. The last two decades have seen significant improvements in Ca2+ indicator dyes that have allowed the field to move forward at considerable pace [43]. Nonetheless, the vast majority of studies on local Ca2+ signaling have used line scan confocal microscopy in conjunction with fluorescent Ca2+ indicator dyes. While line scan imaging provides a reasonable temporal resolution (typical sampling rates used are in the 0.5–1 kHz range) it is severely limited by the fact that a cellular event such as a Ca2+ spark, a Ca2+ wave or the whole-cell Ca2+ transient occurs in a 3-dimensional space, however its optical recording by line scan imaging is reduced to one spatial dimension. Therefore, line scan data notoriously suffer from out-of-focus problems (the event of interest originates from a different optical plane than the recording plane) and how reliably complex 3-dimensional spatial properties can be captured with this approach. Recently, novel technical imaging tools, consisting of a fast 2-D (or x-y) confocal scanning mechanism (LSM 5 LIVE slit scanner) in conjunction with the ability to move the plane of focus vertically in rapid and reproducible manner, have enabled us to achieve x-y-z-t (or 4-D) imaging of sparks with a reasonable time resolution und to gain novel insight into the properties of cardiac Ca2+ sparks [41]. With this technique we were able to determine the true in-focus amplitude of Ca2+ sparks and make the observation that sparks had a modal amplitude distribution. Here, we extended fast confocal imaging to the study of AP-induced Ca2+ transients in atrial and ventricular myocytes with the goal to gain new insight into Ca2+ entry, local Ca2+ release and cellular spread of activation during ECC, and to explore Ca2+ sparks at high temporal resolution to reveal the characteristics of the underlying Ca2+ release and gating properties of the channels involved.
Materials and Methods
Myocyte Isolation
Single myocytes were isolated from cat [26], [44], [45], [46] and rabbit [47] hearts as described previously. The investigation conforms with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All procedures and protocols for animal handling and cell isolation were fully approved by the Institutional Animal Care and Use Committee of Loyola University Chicago (Permit number 05-016) and the Institutional Animal Care and Use Committee of Rush University Medical Center Chicago (Permit number 09-055). All efforts were made to minimize suffering. Briefly, animals were anesthetized with thiopental sodium (50 mg kg−1, I.P.). After thoracotomy, hearts were excised, mounted on a Langendorff apparatus, and retrogradely perfused via the aorta with oxygenated collagenase (cats) or Liberase Blendzyme TH (rabbits) containing solution (37°C). Myocytes were used for experimentation within 1–6 hours after isolation.
Chemicals, Solutions and Experimental Conditions
Chemicals were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. During experiments myocytes were superfused continuously with normal Tyrode solution (composition in mM: NaCl 135: KCl 4; CaCl2 2; MgCl2 1; D-glucose 10; HEPES 10; pH 7.4 adjusted with NaOH). All experiments were performed at room temperature (22–25°C). APs were elicited by electrical field stimulation by applying 1 ms voltage pulses of suprathreshold amplitude through a pair of platinum electrodes.
[Ca2+]i Measurements and Confocal Microscopy
Changes of [Ca2+]i were measured using fluorescence laser scanning confocal microscopy. Intact atrial and ventricular myocytes were loaded with the fluorescent Ca2+ indicator Fluo-4 by 20 min incubation in Tyrode solution containing 10 µM of the membrane permeant acetoxymethyl ester form of the indicator (Fluo-4/AM; Molecular Molecular Probes/Life Technologies, Grand Island, NY) at room temperature. 15–20 min were allowed for de-esterification of the dye. Fluo-4 was excited with the 488 nm line of an argon ion laser and emitted fluorescence was measured at wavelengths >515 nm. Confocal imaging at high temporal resolution was performed with a slit scanning confocal microscope (LSM 5 LIVE; Carl Zeiss, Oberkochen, Germany) equipped with a 63×, 1.20 n.a. water-immersion objective (C-Apochromat; Carl Zeiss). High-speed data acquisition was achieved by bi-directional scanning. 2-dimensional (2-D) imaging (also referred to as x-y imaging) yielded images of 512×53 or 512×31 pixels (pixel distance of 0.3 µm) recorded at 1038 and 1719 Hz, respectively. The x-dimension is defined as the longitudinal axis of the cell. The y-dimension refers to the transverse cell axis and the z-dimension defines the axial (or vertical) cell dimension. The point-spread function of the system had full-width at half maximum (FWHM) values of 0.52, 0.46, and 1.25 µm, respectively, in the x, y, and z directions [41]. Changes of [Ca2+]i are presented as background-subtracted normalized fluorescence (ΔF/F0) where F is the fluorescence intensity, F0 is resting fluorescence recorded under steady-state conditions at the beginning of an experiment, and ΔF = F–F0. Average data are presented as mean ± SEM.
Electrophysiology
Experiments for simultaneous [Ca2+]i and membrane Ca2+ current measurements were performed in rabbit ventricular myocytes using the voltage-clamp technique in the whole-cell ruptured patch configuration, using an Axopatch 200A amplifier, the Axon Digidata 1440A interface and pCLAMP 10.2 software (Molecular Devices, Sunnyvale, CA). The patch pipette solution contained (in mM): 120 L-aspartate; 120 CsOH; 20 TEA-Cl; 20 HEPES; 1 L-gluthathione (reduced); 1 DM-nitrophen (EMD Chemicals, Philadelphia, PA); 4 ATP-Na, 0.25 CaCl2 and 0.1 Fluo-4 pentapotassium salt (Molecular Probes); pH 7.2 adjusted with CsOH). Currents were recorded at 100 kHz and low-pass filtered at 5 kHz.
Results
Spatio-temporal Characteristics of Ca2+ Transients and Ca2+ Sparks in Atrial Myocytes
In a first set of experiments we investigated the spatio-temporal organization of Ca2+ transients during normal ECC in atrial myocytes. AP-dependent Ca2+ transients were elicited by electrical field stimulation (0.7 Hz). As shown previously cat atrial myocytes are devoid of a t-tubular network [7], however RyR Ca2+ release channels are present throughout the cell with the exception of the nuclear region [13]. Fig. 1A shows a sequence of 2-D [Ca2+]i images of the early phase of an electrically evoked Ca2+ transient in an atrial cell, recorded at 1719 Hz from a central axial plan, i.e. at equal distance from the top and bottom cell borders in the axial (z) dimension. Ca2+ release appeared spatially inhomogeneous and initiated at discrete sites in the periphery where the j-SR is located. Ca2+ release was also found to be asynchronous among peripheral j-SR release sites and required 3.0±0.1 ms (n = 7 atrial myocytes) for all detectable peripheral release sites to be activated (Fig. 1B). This is likely an underestimation of the time required to recruit all j-SR release sites because the appearance of new sites at later times becomes increasingly obscured by the overall increase of [Ca2+]i. Fig. 1C shows a single peripheral release site at higher magnification. The series of 2-D images reveals that from the point source of Ca2+ of the release site, Ca2+ diffuses laterally (x-dimension), and also centripetally (y-dimension). Fig. 1D shows the time course of the increase of [Ca2+]i at an individual j-SR release site and a peripheral subsarcolemmal (SS) non-release site at a lateral distance of ∼1 µm, revealing a time delay that was characteristic for subsarcolemmal Ca2+ diffusion. Traces representing the SS j-SR region were recorded within <1 µm from the edge of the cell. Ca2+ release from peripheral j-SR sites eventually gave rise to a rather homogeneous elevation of [Ca2+]i in the cell periphery, obscuring individual release sites. On average this homogeneous ‘ring’ of increased [Ca2+]i was complete 10.1±1.5 ms after application of the electrical stimulus. As shown in Fig. 1E [Ca2+]i started to rise with a delay at a central (CT) subcellular location distant from the cell membrane. At this location, which corresponds to the nj-SR, [Ca2+]i rose much more slowly and peaked at a level that was below peak [Ca2+]i in the cell periphery. As previously established [13], [26] activation of Ca2+ release from release units of the nj-SR in atrial cells occurs by propagating CICR or a ‘diffuse-fire-diffuse’ mechanism where Ca2+ released from a peripheral release site diffuses over a short distance (1–2 µm) to a neighboring more centrally located release site where it activates CICR. Through this sequence of events activation of Ca2+ release in atrial cells becomes spatially inhomogeneous and propagates from the periphery to the center of the myocyte in a Ca2+ wave-like fashion.
Figure 1. AP-induced Ca2+ transients in atrial myocytes.

A, series of confocal 2-D images of the early phase of a Ca2+ transient evoked by electrical field stimulation. White ovals mark the first appearance of individual Ca2+ release sites of the j-SR in the SS region. t = 0 indicates the image frame where the first activated CRU was detected. B, cumulative recruitment of individual j-SR CRUs. CRUs were detected in a SS region of 75 µm×2 µm and normalized to number of CRUs/100 µm2. C, Sequential 2-D images of an individual SS j-SR release site, marked by the white box in panel A, recorded at 0.58 ms time intervals. D, Evolution of [Ca2+]i at an individual SS CRU and an adjacent non-release site at a distance of ∼1 µm. The traces were recorded from a 1 pixel sized regions (0.09 µm2). E, comparison of SS (j-SR) and CT (nj-SR; recorded at a distance of ∼4 µm from the cell membrane) Ca2+ transients.
It is generally agreed that the AP-induced whole-cell Ca2+ transient is the result of spatial and temporal summation of elementary Ca2+ release events from individual CRU, termed Ca2+ sparks [21], [22]. To gain additional insight into the process that initiates the Ca2+ transient in atrial cells we investigated spontaneous Ca2+ sparks from peripheral j-SR CRUs. Fig. 2A shows a series of 2-D images of a spontaneous peripheral Ca2+ spark. Images were recorded at a sampling rate of 1038 Hz. Sparks in the cell periphery arising from atrial j-SR CRUs are highly anisotropic with a preferential spread in the longitudinal direction. FWHM in longitudinal (x-dimension) direction was 2.50±0.07 µm (n = 30 sparks analyzed), whereas the average FWHM in transverse direction was 1.51±0.05 µm. Below the panel of 2-D images, two line scan images (x-t- and y-t-images) are shown that were constructed from the stack of 2-D images. For the x-t image a single line of pixels in the x-dimension was extracted at a fixed value of y from each consecutive 2-D image and the lines were stacked from left to right along the time axis. Analogously, a single line of pixels in the y-dimension was extracted at a fixed value for x. While the x-t-image shows a symmetrical spread of [Ca2+]i from the site of initiation, the y-t-image clearly shows an anisotropic spread. The degree of anisotropy was quantified as eccentricity (e). Eccentricity was defined as the ratio of FWHM of Ca2+ sparks measured along two perpendicular axis within the focal plane and by convention the larger value was divided by the smaller measurement, or e = FWHMwide/FWHMnarrow. For peripheral j-SR Ca2+ sparks in atrial myocytes the average eccentricity was e = 1.71±0.05, i.e. peripheral sparks were approximately 70% wider along the longitudinal cell axis with all sparks analyzed extending more in the longitudinal dimension.
Figure 2. Ca2+ sparks in atrial myocytes.

A, Series of confocal 2-D images of a spontaneous Ca2+ spark originating from the SS j-SR, recorded at 0.96 ms intervals (top). Bottom: reconstructed line scan images (x-t and y-t) from the 2-D series. B, analogous to panel A: spontaneous Ca2+ spark recorded from a CT nj-SR Ca2+ release unit. C, movement of the x-y -coordinates of maximal fluorescence during a SS Ca2+ spark. Left: the spatial coordinates of peak fluorescence intensity were recorded along the dashed lines in x- and y-dimension. Right: x-t and y-t plots of the location of maximal fluorescence, indicating a preferential movement of the point source of Ca generating the spark in centripetal direction.
In contrast, sparks recorded from central (nj-SR) regions revealed a different geometry (Fig. 2B). Sparks were essentially symmetrical in x and y, with an average eccentricity of e = 1.12±0.03, i.e. on average FWHM measured along two perpendicular axes differed by 12% only with no preferential direction.
The structural arrangements of surface cell and SR membranes in the peripheral couplings of the j-SR can explain the preferential spread of Ca2+ when imaged in a single confocal plane cutting across such a coupling. As discussed above, during ECC Ca2+ release propagates by CICR from the cell periphery to the center of the cell. We therefore explored the spatio-temporal properties of peripheral Ca2+ sparks for patterns that indicate transverse or centripetal spread of Ca2+. For this purpose we determined frame-by-frame the x-y-coordinates of maximal fluorescence which identifies a point source of Ca2+ release. As shown in Fig. 2C the location of maximal fluorescence was stable in the longitudinal (x) direction, arguing for a symmetrical lateral dissipation of Ca2+ out of the diadic cleft of the peripheral couplings. In contrast, in transverse (y) direction the location of maximal fluorescence shifted by ∼0.5 µm from the cell membrane towards cell center over the time course of 20 ms, thus facilitating the spread of excitation in centripetal direction. This spatial pattern cannot be explained by simple Ca2+ diffusion away from a fixed point source of Ca2+ and may suggest a preferential direction of Ca2+ release propagation possibly within a peripheral CRU.
Spatio-temporal Characteristics of AP-induced Ca2+ Transients in Ventricular Myocytes
Ventricular myocytes are equipped with an extensive t-tubular membrane system that is organized in a sarcomeric pattern and functionally allows conduction of the AP into deeper regions of the myocyte. Transverse tubules provide the structural basis that CRUs are all in close contact with the cell membrane that hosts voltage-gated Ca2+ channels. Through this arrangement membrane depolarization by an AP allows the synchronous delivery of a triggering amount of Ca2+ into the diadic cleft (formed by t-tubular and SR membranes) to induce CICR. Because the t-tubular system guarantees synchronicity for CICR activation, the onset of Ca2+ release appears spatially and temporally rather homogeneous, although upon closer examination inhomogeneities have been observed [7], [26], [48], [49], [50].
Fig. 3A shows high-resolution 2-D images of the early phase of an AP-induced Ca2+ transient from a ventricular myocytes. The images were recorded from a central focal plane equidistant from the bottom and the top of the cell at a sampling rate of 1719 Hz. Several characteristic features of a ventricular Ca2+ transient become evident: (1) localized Ca2+ release signals from individual CRUs appeared randomly throughout the cell with no obvious preferential subcellular location; (2) recruitment of CRUs is not entirely synchronized. As shown in Fig. 3B the number of activated CRUs steadily increased over the course of ∼4 ms. On average, activation of all detectable release sites was reached after 3.9±0.1 ms (n = 9 ventricular myocytes). (3) Local increases of [Ca2+]i from peripheral and central CRUs that were recruited at the same time, were essentially indistinguishable (Fig. 3C).
Figure 3. Ca2+ release signals in ventricular myocytes.

A, series of confocal 2-D images of the early phase of a Ca2+ transient evoked by electrical field stimulation in a ventricular myocytes. White circles mark the first appearance of individual j-SR Ca2+ release sites. t = 0 indicates the image frame where the first activated CRU was detected. B, cumulative recruitment of individual j-SR CRUs. CRUs were detected in a region of interest encompassing the j-SR and measuring 60 µm×7.5 µm. Detected CRUs were normalized to number of CRUs/100 µm2. C, evolution of [Ca2+]i at individual SS and CT j-SR release sites. The traces were recorded from a 1 pixel sized regions (0.09 µm2). D, series of confocal 2-D images of the early phase of a Ca2+ transient evoked by electrical field stimulation in an atrial myocyte and recorded from a focal plane positioned in the SS j-SR space at the bottom of the cell. E, normalized Ca2+ transients recorded from ventricular j-SR (black), CT nj-SR atrial (blue) and SS j-SR atrial (red) release sites.
In atrial myocytes, the arrangement of surface membrane and j-SR membranes of the peripheral couplings have a certain resemblance with ventricular myocytes. The common features are that the peripheral couplings form diadic clefts were both membranes associate closely and voltage-gated Ca2+ channels face clusters of RyRs [13]. Peripheral couplings are found spaced approximately 2 µm apart. Therefore, we reasoned that the spatio-temporal pattern of CRU activation by an AP would be distinctly different from the one shown in Fig. 1A if we would be able to capture exclusively CRUs from the peripheral j-SR. For this purpose we positioned the focal plane at the bottom of the cell just above the bottom surface membrane. Fig. 3D shows a sequence of 2-D images of the recruitment of j-SR release sites induced by AP depolarization. Similar to ventricular myocytes, no preferential subcellular locations where activation started could be identified, recruitment of j-SR CRUs was temporally asynchronous and it required ∼10 ms for activation of all CRUs. Fig. 3E shows normalized local Ca2+ transients recorded from individual release sites of the atrial j-SR (SS), ventricular j-SR and atrial nj-SR (CT). The time course of the rapid rising phase of the ventricular and the atrial SS signals were virtually identical, whereas the rise of [Ca2+]i at the atrial CT site was significantly slower. Thus, in respect to the dynamics of local Ca2+ release ventricular and atrial j-SR sites behaved remarkably similar, whereas release from atrial nj-SR CRUs revealed a distinctly different spatio-temporal pattern.
Ultra-fast Line Scan Imaging of Ca2+ Sparks
It is generally agreed that Ca2+ sparks result from the concerted opening of a finite number of RyR Ca2+ release channels organized in clusters in the SR membrane. It is, however still a matter of debate how many individual release channels contribute to a spark signal, and the reported numbers vary widely, ranging from small numbers of less than a dozen to over a hundred such channels (see Introduction and Discussion).
Conventional confocal line scan imaging of Ca2+ sparks typically employs scanning speeds in the 0.5–1 kHz range. Such time resolution can impose severe limitations for the fidelity of recording of fast components of a Ca2+ spark such as its rising phase, and may hamper the interpretation of such signals in terms of the underlying channel gating and number of channels involved. Taking advantage of the high temporal resolution capabilities of the LSM 5 LIVE, we recorded Ca2+ sparks in line scan (x-t) mode with an acquisition rate of 40 kHz (25 µs/line) as shown in Fig. 4A. Fig. 4B (top) shows a single pixel-wide (0.3 µm) fluorescence profile (ΔF/F0) obtained from the center of the spark. The spark shown had a rise time (time-to-peak) of ∼6 ms. Detailed analysis of the rising phase of the Ca2+ spark revealed step-like increases of [Ca2+]i (marked by horizontal lines in Fig. 4B). To identify step-like changes of the rising phase of the spark the first derivative of the ΔF/F0 signal was calculated (d(ΔF/F0)/dt; Fig. 4B bottom). The first derivative of the ΔF/F0 signal represents an approximation of the underlying Ca2+ fluxes [26], [51]. During the rising phase of the spark the d(ΔF/F0)/dt revealed several discrete peaks identifying maxima of Ca2+ release flux. The timing of these peaks (vertical dashed lines) was used to identify discrete steps in the [Ca2+]i signal marked by solid black lines in Fig. 4B. In the example shown (similar observation were made in 7 myocytes) 9 distinct levels of the ΔF/F0 signal could be identified with this approach. The improved temporal resolution of Ca2+ spark recording may open new avenues to investigate RyR gating within a CRU in-situ by a non-invasive approach as will be discussed below.
Figure 4. Ultra-fast Ca2+ spark recordings.

A, x-t line scan image recorded at 40,000 lines/s. B, Top: one pixel-wide (0.3 µm) ΔF/F0 profile recorded from the center of the spark. For noise reduction data were averaged to 10,000 lines/s. The red trace represents a 5-point moving average. Bottom: d(ΔF/F0)/dt (s−1), first derivative of the ΔF/F0 signal of the rising phase of the spark. Vertical dashed lines mark maxima of the d(ΔF/F0)/dt signal identifying maxima of Ca2+ release flux. The discrete peaks of the d(ΔF/F0)/dt signal were used to identify step-like increases of the ΔF/F0 signal (marked by horizontal solid black lines). Grey bars indicate discrete d(ΔF/F0)/dt levels. A discrete d(ΔF/F0)/dt level was defined when at least two d(ΔF/F0)/dt peaks of the same amplitude were observed.
Ca2+ Fluxes During ECC Identified with High-speed Imaging
During ECC AP depolarization enables Ca2+ entry through voltage-gated Ca2+ channels that is followed by CICR from the SR. Thus, the global Ca2+ transient is the result of Ca2+ flux across the cell membrane and a much larger Ca2+ flux through RyR SR Ca2+ release channels. In the next set of experiments we set out to identify these two flux components with high speed Ca2+ imaging. Fig. 5Aa shows a whole cell global Ca2+ transient (ΔF/F0) induced by an AP in a ventricular cell and averaged over a region of interest of ∼550 µm2 (200×31 pixels) to obtain a low noise signal. Marker ‘1’ identifies the time when the electrical stimulus was applied to trigger an AP. Fig. 5Ab shows the first derivative of the [Ca2+]i signal (d(ΔF/F0)/dt) representing the underlying Ca2+ flux. The first derivative signal revealed three distinct phases. The first phase, defined by the time of application of the electrical stimulus (marker ‘1’) and the begin of the rise of the d(ΔF/F0)/dt signal (marker ‘2’) was defined as the latency period between electrical stimulus and activation of Ca2+ flux, presumably resulting from Ca2+ entry. The latency period was 2.5±0.3 ms (n = 9 ventricular myocytes). During the following phase (between markers ‘2’ and ‘3’) d(ΔF/F0)/dt increased steadily at a slow rate. At t = 5.5±0.2 ms (marker ‘3’) the flux rate rather abruptly increased and peaked (marker ‘4’) on average at d(ΔF/F0)/dt = 748±71 s−1, 9.9±0.7 ms after application of the electrical stimulus. Fig. 5Ac compares the Ca2+ fluxes in control ventricular cells and myocytes with a disabled SR. The SR was disabled by complete depletion with caffeine (10 mM) and prevention of refilling by treatment with the SERCA blocker thapsigargin (1 µM). Elimination of SR Ca2+ release abolished the large increase of Ca2+ flux normally observed after marker ‘3’, however the latency period (2.1±0.1 ms; n = 11 myocytes) and the subsequent slow rise of the d(ΔF/F0)/dt signal were preserved. The d(ΔF/F0)/dt signal peaked at 50±5 s−1, 7.7±0.7 ms after the electrical stimulus. In conclusion, these observations are consistent with the hypothesis that phase ‘1–2’ (latency) marks the time interval that is required for the spread of depolarization and activation of surface membrane ion channels. Phase ‘2–3’ represents Ca2+ entry flux, and was identical in control myocytes and cells with disabled SR function. Phase ‘3–4’ is identified as Ca2+ release flux from the SR, since it was completely abolished when the SR was eliminated.
Figure 5. Subcellular Ca2+ fluxes during ECC identified by fast Ca2+ imaging.

A, global Ca2+ transient (ΔF/F0) elicited by electrical field stimulation of a ventricular myocyte (panel a). Marker ‘1’ indicates the application of the electrical stimulus. Markers ‘2’ and ‘3’ were positioned according to the analysis in panel b. Panel b: first derivative of ΔF/F0 (s−1) from panel a, representing Ca2+ flux. Markers ‘2’ and ‘3’ indicate abrupt changes in Ca2+ flux rate, and marker ‘4’ indicates maximal Ca2+ flux. Panel c: spatially averaged Ca2+ flux under control conditions (Ctl) and during inhibition of SR function with thapsigargin (TG; 1 µM) and caffeine (Caff; 10 mM). B, subcellular Ca2+ transients (ΔF/F0) elicited by electrical field stimulation and recorded from subsarcolemmal (SS) j-SR and central (CT) nj-SR regions of an atrial myocyte (panel a). Panel b: first derivative of SS and CT Ca2+ signals representing subcellular Ca2+ flux rates. Markers ‘1’ to ‘4’ as in panel A. Panel c: subcellular (SS, CT) Ca2+ flux rates in control and in the presence of thapsigargin+caffeine. 2-D images were recorded 1719 Hz time resolution.
Fig. 5B shows an analogous analysis of data obtained in atrial myocytes. In atrial cells Ca2+ fluxes were further analyzed separately for SS (j-SR) and CT (nj-SR) regions. Fig. 5Ba shows the time course of the increase of [Ca2+]i (ΔF/F0) induced by electrical field stimulation applied at marker ‘1’, revealing the delayed and slower rise of [Ca2+]i in the CT region. Fig. 5Bb shows the first derivative of the ΔF/F0 representing the underlying Ca2+ fluxes. In the SS region, similar to ventricular cells, a latency period (3.6±0.7 ms; n = 6 atrial myocytes), was followed by a slow increase (starting at marker ‘2’) that changed to a fast increase of d(ΔF/F0)/dt at 6.4±0.3 ms after application of the electrical stimulus (marker ‘3’) and peaked at 1183±180 s−1 after 11.8±0.6 ms. In contrast the CT d(ΔF/F0)/dt signal did not reveal an obvious slow rising phase and did not start to rise until 2.2±0.3 ms after the first change of the SS signal (marker ‘2’) was observed. Maximal flux in the CT region was 3.5±0.3 times less than in the periphery and peaked 5.3±1.7 ms later. Fig. 5Bc shows regional (SS, CT) atrial Ca2+ fluxes in control and myocytes with disabled SR (TG+Caff). In the SS region d(ΔF/F0)/dt started to increase 2.1±0.1 ms (n = 6 atrial myocytes) after the electrical stimulus was applied and flux reached a maximum (164±30 s−1) after 6.5±0.5 ms, approximately at the time when in control cells SR Ca2+ release flux started. Ca2+ release flux in the CT region was 4.0±0.7 times smaller compared to the SS region.
In summary, fast 2-D Ca2+ imaging enabled us to identify Ca2+ fluxes associated with Ca2+ entry and Ca2+ release from the SR. Next, we attempted to validate the optically measured Ca2+ entry and release signals (Fig. 5) with simultaneous measurements of [Ca2+]i and membrane Ca2+ currents (ICa) recorded with the patch clamp technique. Spatially averaged [Ca2+]i and ICa measurements were obtained from single ventricular myocytes (Fig. 6). Cells where held at −40 mV and repetitively depolarized to +20 mV for 100 ms to elicit maximal LCCs. Fig. 6 shows from top to bottom, [Ca2+]i (ΔF/F0), Ca2+ flux (d(ΔF/F0)/dt), ICa and the command voltage. The Ca2+ flux signal showed a slow steady increase shortly after the begin of the depolarization pulse (marker ‘a’). The slow increase of Ca2+ flux abruptly increased at marker ‘b’, giving way to a rapid rise of the Ca2+ flux rate. The distinct change in flux rate (marker ‘b’) occurred 5.7 ms after the begin of the depolarization pulse and correlated well with the begin of SR Ca2+ release determined by optical methods (marker ‘3’ in Fig. 5A).
Figure 6. Simultaneous [Ca2+] and ICa measurements.
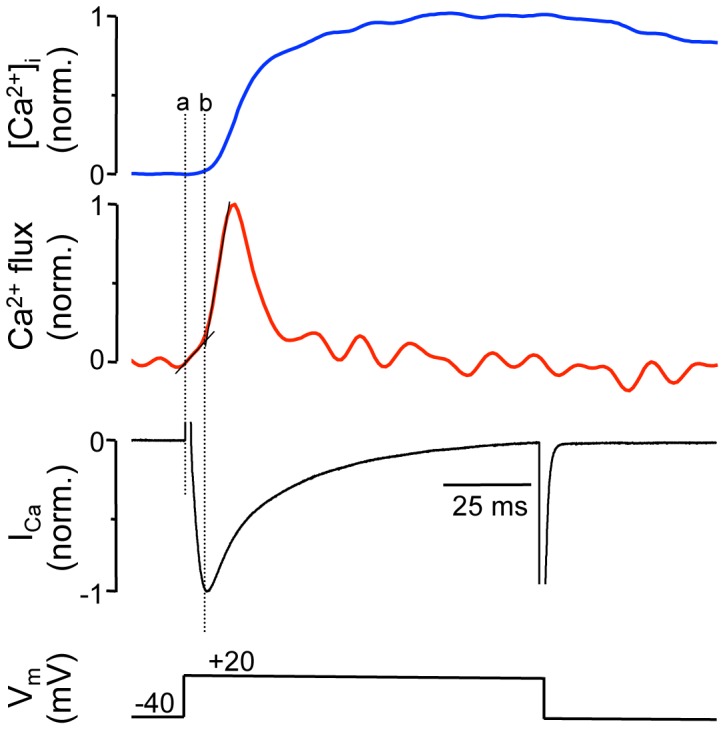
Simultaneous measurements of [Ca2+]i and ICa from a voltage-clamped (ruptured patch configuration) ventricular myocyte. Shown from top: [Ca2+]i (ΔF/F0), Ca2+ flux (d(ΔF/F0)/dt), ICa and the command voltage. Traces represent averages of 15 successive depolarization pulses applied to the same cell and were normalized to their respective maximal amplitude. In these experiments whole-cell Fluo-4 fluorescence was recorded using a photomultiplier tube. Marker ‘a’: begin of depolarization pulse; marker ‘b’: change in flux rate, presumably identifying activation of CICR from the SR.
In conclusion, the data presented in Figures 5 and 6 provide evidence that fast 2-D confocal Ca2+ imaging with sampling rates in the 1–2 kHz range can generate information on Ca2+ fluxes underlying ECC with a time resolution that is approaching the time resolution of the patch clamp technique, however has the advantage of lesser perturbation of the intracellular environment since the surface membrane of the cell remains intact.
Discussion
In this study, we investigated the behavior and interactions of CRUs during electrically evoked Ca2+ transients and individual elementary Ca2+ release events (Ca2+ sparks) in atrial and ventricular myocytes with 2-D and line scan confocal imaging at high recording speeds. The key findings of our investigation were as follows:
First, the recruitment of individual CRUs by AP depolarization was spatially inhomogeneous and temporally asynchronous. Spatial inhomogeneity was particularly pronounced in atrial myocytes that lack a t-tubular membrane system. Second, high-speed confocal Ca2+ imaging allowed to resolve a distinct sequency of events during a Ca2+ transient, consisting of a latency period, Ca2+ entry and SR Ca2+ release, by an optophysiological approach [22], that has a time resolution comparable to electrophysiological recording (patch clamp) methods, however, is considerably less invasive since the integrity of cell membrane and cytosolic environment remains intact. Third, ultra-fast line scan recordings revealed step-like changes of [Ca2+]i during the rising phase of Ca2+ sparks that may provide novel insights into RyR gating properties within individual CRUs.
Activation of SR Ca2+ release in atrial myocytes was asynchronous at two different levels. As has been described before [7], [10], [13], [26], [52], [53], due the lack (or poor development) of the t-tubular system, subsarcolemmal Ca2+ release originating at the peripheral couplings of the j-SR preceded Ca2+ release from the nj-SR in central regions of the cell, and peak CT [Ca2+]i lagged behind by tens of milliseconds during AP-triggered Ca2+ transients (Fig. 1E). However, the initial activation of j-SR Ca2+ release in the cell periphery was also asynchronous and spatially inhomogeneous (Fig. 1B). Recruitment of detectable peripheral CRUs extended over at least 3 ms. A similar asynchronous activation pattern was found in ventricular myocyes where recruitment of all discernable CRUs required ∼4 ms. These numbers are low-end estimates. At later times the progressing global increase of [Ca2+]i and Ca2+ diffusion away from activated release sites starts to obscure the recruitment of new sites and hampers their detectability at later times. Using line scan imaging numerous studies have found evidence for spatial inhomogeneities (e.g. [26], [48], [49]) that can be exacerbated in pathological conditions [50]. However, it is difficult to discern whether in line scan data apparent inhomogeneities arise from asynchronous activation of CRUs, silent or missing CRUs, or are the result of the inherent shortcoming of this recording mode that the scanned line potentially is missing CRUs (for example, when randomly placed between 2 neighboring Z-disks that are typically spaced ∼2 µm apart [13]). In 2-D imaging the problem is alleviated since the radial distance (i.e. the distance within the plane of a Z-disk) between CRUs is in the sub-micrometer range [29], [30], [32], [33], i.e. within the range of the spatial resolution of the microscope. In other words, upon AP stimulation all events recorded in 2-D mode originating from CRUs at the intersection of the focal plane and the plane of the Z-disk or from CRUs located on the Z-disk at distances that are within the point-spread function of the microscope (see Methods) will be detected. Consequently, every CRU that becomes activated during an AP within the resolved volume surrounding the focal plane will be captured. Thus, delays and asynchrony between activation of individual CRUs can more reliably be recorded.
Fast 2-D Ca2+ imaging allowed also to separate Ca2+ entry from SR Ca2+ release during an AP-induced Ca2+ transient by optical methods (Figures 5 and 6). By analyzing the first derivative of the Ca2+ transient three distinct phases could be discerned during the first ten milliseconds of the Ca2+ transient. First, a latency period of ∼2.5 ms between stimulus application and the first indication of an increase of [Ca2+]i was observed. The second phase, characterized by a slow increase of [Ca2+]i and lasting until 5.5 (ventricular) and 6.4 ms (atrial myocytes) after the begin of the pulse, represents Ca2+ entry across the surface membrane which was confirmed with simultaneous patch clamp and [Ca2+]i measurements (Fig. 6). This conclusion was supported by the fact, that the rise of [Ca2+]i was still observed when SR Ca2+ release was disabled, and the change in [Ca2+]i coincided well with the current recordings. The third phase, characterized by an abrupt increase of release flux by approximately an order of magnitude, peaked 10–12 ms after the pulse. Phase 3 was completely abolished when the SR was disabled. The data indicate that 2-D Ca2+ imaging with 1–2 kHz time resolution is capable of distinguishing between trigger and released Ca2+ in intact myocytes by an optophysiological approach, with the advantage that cell membrane and cytosolic environment are kept undisturbed.
Despite substantial progress made towards the understanding of Ca2+ release and ECC in atrial tissue, several key questions have remained unanswered. For example, with the lack of t-tubules the CRUs of the nj-SR (which account for a large majority of atrial CRUs) do not have the characteristics of a typical couplon. Specifically, the RyRs are not facing into a narrow diadic cleft where upon activation of LCCs [Ca2+]i rapidly increases to concentrations that are several orders magnitude higher than resting [Ca2+]i. With the inherent low Ca2+-sensitivity of the RyR [54], [55] it is difficult to reconcile how [Ca2+]i can rise sufficiently high in the vicinity of RyRs of the nj-SR to lead to channel opening and Ca2+ release. Furthermore, it has even been suggested that Ca2+ sequestering mitochondria in the subsarcolemmal space of atrial myocytes constitute a Ca2+ barrier [15], making the propagation of CICR from peripheral j-SR to nj-SR even more problematic. As shown previously [41], atrial sparks have a relatively large Ca2+ flux (∼11 pA) and large numbers of channels involved (20–30 RyRs) with an enhanced propensity to open and a diminished influence of negative feedback mechanisms as well as an extended spatial spread (see also [40]). All these features tend to facilitate propagation of release from nj-SR. Interestingly, in contrast to Ca2+ sparks recorded from nj-SR and sparks in ventricular cells, the location of maximal fluorescence identifying a point source of Ca2+ release of peripheral atrial sparks is not spatially fixed, but moves in centripetal direction during the evolution of the spark (Fig. 2C). Thus, peripheral atrial sparks reveal some inherent feature for directional propagation that facilitates the saltatory advancement of activation from j-SR to the nj-SR, although it remains to be determined what constitutes the structural or functional basis of this special feature.
The question how many RyRs are found in an individual CRUs has remained rather elusive. Earlier reports suggested the number to be in the range of 50–250 RyRs [23], [29], [30], [31], [32], [33], [35]. However, novel advanced technologies, including super-resolution imaging techniques [56] that go beyond diffraction-limited resolution, and 3-dimensional electron microscopy tomography [57], have revealed new aspects of the make up of the cardiac CRU [36], [58], [59]: CRUs are variable in size, of irregular geometry, lack the regular arrays of RyRs typical of skeletal muscle, and show a smaller density of RyRs than previously assumed. Furthermore, individual CRUs can assemble in groups to form ‘super-clusters’ resulting in variable degrees of coordination of Ca2+ release from sub-clusters. These recent studies that generated evidence of incomplete packaging of CRUs with RyRs and the existence of sub-domains of smaller sizes have corrected the number of RyRs per CRU towards lower values in the range of a few tens to less than ten RyRs [29], [34], [36]. While recent technological advances for studying CRUs have generated valuable novel insights, it is still far from being established how many RyRs actively contribute to Ca2+ release of a spark. The estimates, just for cardiac myocytes alone, range from possibly only one or only a very small fraction of the cluster (<10 RyRs), to whole-cluster activation with possibly contribution from several hundred channels [21], [32], [37], [38], [39], [40], [41], [42]. The ionic current or ionic flux underlying a spark is estimated to be in the range of 3–5 pA [38], [40], [60], [61], [62], [63], although it tends to be higher in atrial myocytes (11 pA; [41]). Ca2+ currents through single cardiac RyR channels reconstituted in lipid bilayer and recorded under physiological ionic conditions gave unitary current values of 0.3–0.7 pA [64]. Based on these estimates only ∼5–35 RyRs passing current simultaneously would participate in a single spark. This leads to the paradox that the estimated number of participating channels is nearly an order of magnitude smaller than the higher estimates for the number of RyRs per CRU. Recent results indicate that the current underlying a spark is quantal in nature. By using ‘sparklets’ - local Ca2+ signals of known magnitude arising from LCCs [39] - to calibrate Ca2+ sparks, it could be demonstrated that Ca2+ sparks have a quantal substructure and that Ca2+ spark release flux is made up of quanta of 1.2 pA. Furthermore, the number of quanta in a spark varies dynamically from one to ≤8 [62]. Whether single quanta reflect the opening of a single RyR or a group of tightly coupled channels is unclear. Cooperativity of activation and inactivation among release channels or ‘coupled gating’ [65], [66], [67] could enable entire groups of channel to generate a single quantum. In support of multichannel quanta is also the observation that the measured quantum size (1.2 pA) is larger than the estimates for RyR unitary currents (∼0.5 pA, lasting ∼10 ms) under physiological ionic conditions. Thus, quantal release with a low number of individual quanta does not necessarily rule out the involvement of a larger number of RyRs. In addition, the observation of RyR super-clusters raises the possibility that quantal release reflects the concerted activation of sub-clusters [56]. Since sub-clusters vary in size and number of RyRs this model could explain quantal release with quanta of variable sizes, however this would require a looser definition of the term ‘quantal’ to avoid violation of a stricter definition of ‘quanta’ which suggests entities of identical magnitude. Our data shown in Fig. 4 may point towards coordinated gating of an entire RyR cluster with a high degree of cooperativity. The d(ΔF/F0)/dt signal shown in Fig. 4B, at first glance, is reminiscent of single-channel gating with the peaks indicating channel openings. On the other hand, we have shown previously that in the same species atrial Ca2+ sparks involve release from 20–30 RyRs [41]. Coupled gating with a high degree of cooperativity within a CRU, where the entire RyR cluster behaves essentially as a single channel, could reconcile this apparent paradox. Upon closer examination, in the d(ΔF/F0)/dt signal in Fig. 4B at least three distinct amplitude levels can be identified (grey bars). Thus, the data could be consistent with coupled gating of the entire cluster or a small number of subclusters, but do not exclude a quantal nature of Ca2+ release since quantal release consisting of a small number of quanta (even 1 or 2 quanta only) has been demonstrated [62].
While entirely speculative at this point, the progressive activation of RyR sub-clusters within a CRU may also explain the centripetal propagation of Ca2+ release found in subsarcolemmal atrial CRUs (Fig. 2C). Furthermore, to reconcile the paradox of a presumably small fraction of active channels and a large number channels present in a CRU, several possibilities need to be considered. For one, the unitary RyR current could be much smaller (possibly in the femto-ampere range) during a spark than measured in the bilayer because the rapid rise of Ca2+ in the diadic cleft together with local SR depletion could significantly decrease the driving force for Ca2+ flux. Conceivably, powerful, however yet to be identified inhibitory mechanisms, negative cooperativity among RyRs (opening of a RyR reduces the open probability of its neighbors), strong Ca2+-dependent RyR inactivation or the recently proposed ‘pernicious attrition’ Ca2+ release termination mechanism [68] could prevent the activation of an entire CRU by intra-cluster CICR. As recently suggested, Mg2+ binding to the RyR could be responsible for such a strong inhibitory mechanism [69].
In summary, we demonstrate here that high-speed confocal imaging with sampling rates in the 1–40 kHz range in conjunction with high quantum yield fluorescent indicators allows to study Ca2+ dynamics and intricacies of single SR Ca2+ release units. With this technique we showed that dynamics of Ca2+ entry and CICR can reliably be distinguished at the level of individual CRUs in intact cells, that Ca2+ sparks of atrial myocytes have inherent properties that facilitate centripetal propagation of activation, and that the rising phase of Ca2+ sparks reveals step-like kinetics which bears potentially valuable information about intra-CRU channel gating. These findings have important ramifications for the understanding of the structural and functional organization of CRUs, CICR and ECC.
Funding Statement
This work was supported by the National Institutes of Health Grants HL62231, HL80101 and HL101235, and the Leducq Foundation (to LAB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415: 198–205. [DOI] [PubMed] [Google Scholar]
- 2. Fabiato A (1983) Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol 245: C1–14. [DOI] [PubMed] [Google Scholar]
- 3. Franzini-Armstrong C, Jorgensen AO (1994) Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol 56: 509–534. [DOI] [PubMed] [Google Scholar]
- 4. Stern MD, Song LS, Cheng H, Sham JS, Yang HT, et al. (1999) Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol 113: 469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng H, Cannell MB, Lederer WJ (1994) Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch 428: 415–417. [DOI] [PubMed] [Google Scholar]
- 6. Cheng H, Lederer MR, Xiao RP, Gomez AM, Zhou YY, et al. (1996) Excitation-contraction coupling in heart: new insights from Ca2+ sparks. Cell Calcium 20: 129–140. [DOI] [PubMed] [Google Scholar]
- 7. Huser J, Lipsius SL, Blatter LA (1996) Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol 494 (Pt 3): 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soeller C, Cannell MB (1999) Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res 84: 266–275. [DOI] [PubMed] [Google Scholar]
- 9. Cordeiro JM, Spitzer KW, Giles WR, Ershler PE, Cannell MB, et al. (2001) Location of the initiation site of calcium transients and sparks in rabbit heart Purkinje cells. J Physiol 531: 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mackenzie L, Bootman MD, Berridge MJ, Lipp P (2001) Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol 530: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, et al. (2010) Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell calcium 47: 210–223. [DOI] [PubMed] [Google Scholar]
- 12. Bootman MD, Higazi DR, Coombes S, Roderick HL (2006) Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. Journal of cell science 119: 3915–3925. [DOI] [PubMed] [Google Scholar]
- 13. Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, et al. (2001) Activation and propagation of Ca(2+) release during excitation-contraction coupling in atrial myocytes. Biophys J 81: 2590–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McNutt NS, Fawcett DW (1969) The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol 42: 46–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD (2004) The spatial pattern of atrial cardiomyocyte calcium signalling modulates contraction. J Cell Sci 117: 6327–6337. [DOI] [PubMed] [Google Scholar]
- 16. Woo SH, Cleemann L, Morad M (2003) Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res 92: e1–11. [DOI] [PubMed] [Google Scholar]
- 17. Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ Jr, et al. (1995) Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol 129: 672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL (2011) Atrial cardiomyocyte calcium signalling. Biochimica et biophysica acta 1813: 922–934. [DOI] [PubMed] [Google Scholar]
- 19. Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, et al. (2003) Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol 546: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keizer J, Smith GD, Ponce-Dawson S, Pearson JE (1998) Saltatory propagation of Ca2+ waves by Ca2+ sparks. Biophysical journal 75: 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng H, Lederer WJ, Cannell MB (1993) Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262: 740–744. [DOI] [PubMed] [Google Scholar]
- 22. Cheng H, Lederer WJ (2008) Calcium sparks. Physiological reviews 88: 1491–1545. [DOI] [PubMed] [Google Scholar]
- 23.Brochet DX, Xie W, Yang D, Cheng H, Lederer WJ (2010) Quarky Calcium Release in the Heart. Circ Res. [DOI] [PMC free article] [PubMed]
- 24. Lipp P, Niggli E (1996) Submicroscopic calcium signals as fundamental events of excitation–contraction coupling in guinea-pig cardiac myocytes. J Physiol 492 (Pt 1): 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lipp P, Niggli E (1998) Fundamental calcium release events revealed by two-photon excitation photolysis of caged calcium in Guinea-pig cardiac myocytes. J Physiol 508 (Pt 3): 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sheehan KA, Blatter LA (2003) Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J Physiol 546: 119–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Woo SH, Cleemann L, Morad M (2005) Diversity of atrial local Ca2+ signalling: evidence from 2-D confocal imaging in Ca2+-buffered rat atrial myocytes. J Physiol 567: 905–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sheehan KA, Zima AV, Blatter LA (2006) Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J Physiol 572: 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayashi T, Martone ME, Yu Z, Thor A, Doi M, et al. (2009) Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci 122: 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franzini-Armstrong C, Protasi F, Ramesh V (1999) Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophysical journal 77: 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Soeller C, Crossman D, Gilbert R, Cannell MB (2007) Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proceedings of the National Academy of Sciences of the United States of America 104: 14958–14963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scriven DR, Asghari P, Schulson MN, Moore ED (2010) Analysis of Cav1.2 and ryanodine receptor clusters in rat ventricular myocytes. Biophysical journal 99: 3923–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen-Izu Y, McCulle SL, Ward CW, Soeller C, Allen BM, et al. (2006) Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J 91: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jayasinghe ID, Baddeley D, Kong CH, Wehrens XH, Cannell MB, et al. (2012) Nanoscale organization of junctophilin-2 and ryanodine receptors within peripheral couplings of rat ventricular cardiomyocytes. Biophysical journal 102: L19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, et al. (2005) Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A 102: 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, et al. (2009) Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proceedings of the National Academy of Sciences of the United States of America 106: 22275–22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS (2002) Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J 83: 59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Izu LT, Mauban JR, Balke CW, Wier WG (2001) Large currents generate cardiac Ca2+ sparks. Biophys J 80: 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang SQ, Song LS, Lakatta EG, Cheng H (2001) Ca2+ signalling between single L-type Ca2+ channels and ryanodine receptors in heart cells. Nature 410: 592–596. [DOI] [PubMed] [Google Scholar]
- 40. Blatter LA, Huser J, Rios E (1997) Sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks in cardiac muscle. Proc Natl Acad Sci U S A 94: 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shkryl VM, Blatter LA, Rios E (2012) Properties of Ca2+ sparks revealed by four-dimensional confocal imaging of cardiac muscle. J Gen Physiol 139: 189–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bridge JH, Ershler PR, Cannell MB (1999) Properties of Ca2+ sparks evoked by action potentials in mouse ventricular myocytes. J Physiol 518 (Pt 2): 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bruton JD, Cheng AJ, Westerblad H (2012) Methods to detect Ca(2+) in living cells. Advances in experimental medicine and biology 740: 27–43. [DOI] [PubMed] [Google Scholar]
- 44. Wu JY, Vereecke J, Carmeliet E, Lipsius SL (1991) Ionic currents activated during hyperpolarization of single right atrial myocytes from cat heart. Circ Res 68: 1059–1069. [DOI] [PubMed] [Google Scholar]
- 45. Rubenstein DS, Lipsius SL (1995) Premature beats elicit a phase reversal of mechanoelectrical alternans in cat ventricular myocytes. A possible mechanism for reentrant arrhythmias. Circulation 91: 201–214. [DOI] [PubMed] [Google Scholar]
- 46. Kockskamper J, Blatter LA (2002) Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol 545: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Domeier TL, Blatter LA, Zima AV (2009) Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol 587: 5197–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng H, Cannell MB, Lederer WJ (1995) Partial inhibition of Ca2+ current by methoxyverapamil (D600) reveals spatial nonuniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Circ Res 76: 236–241. [DOI] [PubMed] [Google Scholar]
- 49. Heinzel FR, Bito V, Volders PG, Antoons G, Mubagwa K, et al. (2002) Spatial and temporal inhomogeneities during Ca2+ release from the sarcoplasmic reticulum in pig ventricular myocytes. Circulation research 91: 1023–1030. [DOI] [PubMed] [Google Scholar]
- 50. Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, et al. (2008) Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circulation research 102: 338–346. [DOI] [PubMed] [Google Scholar]
- 51. Sipido KR, Wier WG (1991) Flux of Ca2+ across the sarcoplasmic reticulum of guinea-pig cardiac cells during excitation-contraction coupling. J Physiol 435: 605–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Berlin JR (1995) Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells. Am J Physiol 269: H1165–1170. [DOI] [PubMed] [Google Scholar]
- 53. Woo SH, Cleemann L, Morad M (2002) Ca2+ current-gated focal and local Ca2+ release in rat atrial myocytes: evidence from rapid 2-D confocal imaging. J Physiol 543: 439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cannell MB, Soeller C (1997) Numerical analysis of ryanodine receptor activation by L-type channel activity in the cardiac muscle diad. Biophys J 73: 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meissner G, Henderson JS (1987) Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. The Journal of biological chemistry 262: 3065–3073. [PubMed] [Google Scholar]
- 56.Soeller C, Baddeley D (2012) Super-resolution imaging of EC coupling protein distribution in the heart. Journal of molecular and cellular cardiology. [DOI] [PubMed]
- 57.Das T, Hoshijima M (2012) Adding a new dimension to cardiac nano-architecture using electron microscopy: Coupling membrane excitation to calcium signaling. Journal of molecular and cellular cardiology. [DOI] [PubMed]
- 58. Hayashi H, Shiferaw Y, Sato D, Nihei M, Lin SF, et al. (2007) Dynamic origin of spatially discordant alternans in cardiac tissue. Biophys J 92: 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Asghari P, Scriven DR, Hoskins J, Fameli N, van Breemen C, et al. (2012) The structure and functioning of the couplon in the mammalian cardiomyocyte. Protoplasma 249 Suppl 1S31–38. [DOI] [PubMed] [Google Scholar]
- 60. Cheng H, Wang SQ (2002) Calcium signaling between sarcolemmal calcium channels and ryanodine receptors in heart cells. Front Biosci 7: d1867–1878. [DOI] [PubMed] [Google Scholar]
- 61. Soeller C, Cannell MB (2002) Estimation of the sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks. Biophys J 82: 2396–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang SQ, Stern MD, Rios E, Cheng H (2004) The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc Natl Acad Sci U S A 101: 3979–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Santiago DJ, Curran JW, Bers DM, Lederer WJ, Stern MD, et al. (2010) Ca Sparks Do Not Explain all Ryanodine Receptor-Mediated SR Ca Leak in Mouse Ventricular Myocytes. Biophys J 98: 2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kettlun C, Gonzalez A, Rios E, Fill M (2003) Unitary Ca2+ current through mammalian cardiac and amphibian skeletal muscle ryanodine receptor Channels under near-physiological ionic conditions. The Journal of general physiology 122: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, et al. (2001) Coupled gating between cardiac calcium release channels (ryanodine receptors). Circulation research 88: 1151–1158. [DOI] [PubMed] [Google Scholar]
- 66. Marx SO, Ondrias K, Marks AR (1998) Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281: 818–821. [DOI] [PubMed] [Google Scholar]
- 67. Porta M, Diaz-Sylvester PL, Neumann JT, Escobar AL, Fleischer S, et al. (2012) Coupled gating of skeletal muscle ryanodine receptors is modulated by Ca2+, Mg2+, and ATP. American journal of physiology Cell physiology 303: C682–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gillespie D, Fill M (2013) Pernicious attrition and inter-RyR2 CICR current control in cardiac muscle. Journal of molecular and cellular cardiology. [DOI] [PMC free article] [PubMed]
- 69. Zahradnikova A, Valent I, Zahradnik I (2010) Frequency and release flux of calcium sparks in rat cardiac myocytes: a relation to RYR gating. The Journal of general physiology 136: 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]