

Abstract
The excessive accumulation of adipocytes contributes to the development of obesity and obesity-related diseases. The interactions of several transcription factors, such as C/EBPβ, PPARγ, C/EBPα, Nrf2, and STAT3, are required for adipogenic differentiation. Dimethylfumarate (DMF), an immune modulator and antioxidant, may function as an inhibitor of STAT3 and an activator of Nrf2. This study examined whether DMF inhibits adipogenic differentiation of 3T3-L1 preadipocytes by inhibiting STAT3 or activating Nrf2. DMF suppressed 3T3-L1 preadipocyte differentiation to mature adipocytes in a dose-dependent manner as determined by Oil Red O staining. The mRNA and protein levels of adipogenic genes, including C/EBPβ, C/EBPα, PPARγ, SREBP-1c, FAS, and aP2, were significantly lower in DMF-treated 3T3-L1 preadipocytes. Suppression of adipogenic differentiation by DMF treatment resulted primarily from inhibition of the early stages of differentiation. DMF inhibits clonal expansion during adipogenic differentiation through induction of a G1 cell cycle arrest. Additionally, DMF regulates cell cycle-related proteins, such as p21, pRb, and cyclin D. DMF treatment markedly inhibited differentiation medium-induced STAT3 phosphorylation and inhibited STAT3 transcriptional activation of a reporter construct composed of four synthetic STAT3-response elements. Moreover, inhibition of endogenous Nrf2 activity using a dominant negative Nrf2 did not abolish the DMF-induced inhibition of adipogenic differentiation of 3T3-L1 preadipocytes. In summary, DMF is a negative regulator of adipogenic differentiation based on its regulation of adipogenic transcription factors and cell cycle proteins. This negative regulation by DMF is mediated by STAT3 inhibition, but is unlikely to involve Nrf2 activation.
Introduction
Adipose tissue contributes to the maintenance of energy homeostasis [1] and is considered to be an endocrine organ that contributes to the pathogenesis of obesity and obesity-related metabolic complications [1]. Excessive accumulation of adipose tissue in the body may cause the development of obesity and obesity-related diseases [2]. The accumulation of adipose tissue results from increases both in the size and number of adipocytes [3]. In addition, recent evidence has demonstrated that accelerated adipogenic differentiation is implicated in the excessive accumulation of body fat [4]. Adipogenic differentiation is a complex process accompanied by changes in cytoarchitecture, signaling pathways, and transcriptional regulation. The interactions of several transcription factors, such as peroxisome proliferator-activated receptor gamma (PPARγ), CCAATT enhancer binding proteins (C/EBP), and SREBP-1c, are required for adipogenic differentiation [4], [5].
In addition to these transcription factors, recent studies have shown that the signal transducer and activator of transcription 3 (STAT3) and NF-E2-related factor 2 (Nrf2) play important roles in adipogenic differentiation [6]–[9]. STAT3 is a transcription factor and is required for gp130-mediated cell survival and the G1/S transition in the cell cycle [10]. The transition from G1 to S phase in the cell cycle requires the activation of complexes of cyclin-dependent kinases (CDKs) [11]. In the HepG2 hepatoma cell line, STAT3 regulates the G1/S phase transition through interactions with p21, a potent CDK inhibitor [12]. In 3T3-L1 preadipocytes, STAT3 regulates adipogenesis via regulation of PPARγ and C/EBPβ [6], [7]. Adipogenic differentiation can be suppressed by STAT3 siRNA or a dominant negative STAT3 and the PPARγ agonist rescued adipogenesis in these treatments [6]. Recently, STAT3 was reported to regulate the transcription of C/EBPβ by binding the distal region of the C/EBPβ promoter [7]. By contrast, Nrf2, a basic leucine zipper (bZIP) transcription factor, induces the expression of genes including those related to antioxidant enzymes [13]. Several lines of evidence suggest that Nrf2 activation impairs lipid accumulation in adipose tissue and inhibits adipocyte differentiation [8], [9]. Nrf2 activation diminished during adipogenic differentiation of the bone marrow-derived ST2 cell line [14] and activation of Nrf2 was suggested to inhibit adipogenesis by modulating signaling by the aryl hydrocarbon receptor in experiments using a pharmacological activator of Nrf2 [8]. More recently, enhanced Nrf2 activity was shown to inhibit lipid accumulation in white adipose tissue in leptin-deficient mice [9].
DMF is the active ingredient of an oral formulation of fumaric acid esters with proven effectiveness in patients with chronic plaque psoriasis, a dermatological disorder associated with immune dysfunction [15], [16]. Since the 1950s, DMF has been proven effective in treatment of psoriasis, and several studies have revealed that DMF is also effective in treating multiple sclerosis, inflammatory lung disease, and other conditions [17], [18]. As an immune modulator, DMF decreased synthesis of proinflammatory mediators such as TNF-α, IL-1β, and IL-6 in microglial and astrocytic cells [19]. Because activation of STAT3 is induced by cytokines such as IL-6 and IL-10 [20], [21], DMF may have the potential to function as a STAT3 inhibitor. Moreover, recent reports have shown that DMF increases the expression of Nrf2, which is repressed by binding to the inhibitor Keap1 in the cytoplasm [22]–[24]. Collectively, these data suggest that DMF could modulate adipogenic differentiation.
Here, the potential role of DMF in adipogenic differentiation and the molecular mechanisms by which DMF inhibits adipogenic differentiation, either through inhibiting STAT3 or activating Nrf2, were investigated.
Results
DMF Inhibits Adipogenic Differentiation of 3T3-L1 Preadipocytes
To determine the effect of DMF on adipogenic differentiation, intracellular lipid accumulation was monitored with an Oil Red O staining assay. Post-confluent 3T3-L1 preadipocytes treated with differentiation medium (MDI), which contains a mixture of IBMX, dexamethasone, and insulin, initiated adipogenic differentiation and Oil Red O staining showed that intracellular lipid accumulation was marked by day 8, suggesting that 3T3-L1 preadipocytes differentiate into mature adipocytes. However, cells co-treated with 25 µM DMF for 8 days showed significant inhibition of MDI-induced lipid droplet accumulation, and 75 µM DMF almost completely blocked lipid droplet accumulation (p<0.001) (Fig. 1).
Figure 1. Effect of DMF on adipogenic differentiation.
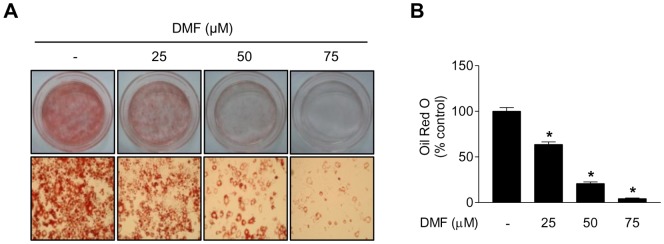
Two-day post-confluent 3T3-L1 preadipocytes were cultured with differentiation medium in the absence or presence of DMF (25–75 µM) for 8 days. (A) Representative micrographs showing cell culture dishes and the cell monolayers stained with Oil Red O (× 200). At day 8, intracellular lipid accumulation was determined by Oil Red O staining. (B) Quantitative analysis of Oil Red O staining. Oil Red O stained cells were extracted with isopropyl alcohol and the absorbance at 500 nm was measured. Data are presented as mean ± S.D. (n = 3); *p<0.001 vs. control.
DMF Inhibits Adipogenic Differentiation Associated Gene Expression
The effect of DMF on the expression levels of genes associated with adipogenic differentiation was investigated. Post-confluent 3T3-L1 preadipocytes were treated with various doses of DMF (0, 25, 50, and 75 µM) during MDI-induced adipogenic differentiation for 8 days. The effects of DMF on adipogenic gene expression were measured by real-time PCR. The mRNA levels of adipogenic genes including C/EBPβ, C/EBPα, PPARγ, SREBP-1c, FAS, and aP2 were significantly lower in DMF-treated 3T3-L1 preadipocytes. The reduction in mRNA levels of these genes was proportional to the DMF concentration (Fig. 2A). The protein levels of the gene products were examined by Western blot analysis. Consistent with the expression patterns, C/EBPβ protein levels were weakly detected and further decreased by DMF treatment. The protein levels of C/EBPα, PPARγ, SREBP-1c, and FAS in DMF-treated 3T3-L1 preadipocytes were decreased by DMF treatment in a dose-dependent manner (Fig. 2B).
Figure 2. Effect of DMF on adipogenic gene expression in 3T3-L1 cells.

Two-day post-confluent 3T3-L1 preadipocytes were subjected to adipogenic differentiation for 8 days in the absence or presence of various concentrations of DMF. (A) Real-time PCR was used to quantify the effect of DMF on adipogenic gene expression. On day 8, adipogenic markers were analyzed by real-time PCR using mRNA isolated from DMF-treated 3T3-L1 cells. (B) Representative Western blot analysis showing the effect of DMF on C/EBPβ, C/EBPα, PPARγ, and SREBP-1c levels. Data are presented as mean ± S.D. (n = 3); *p<0.05, **p<0.01, ***p<0.001 vs. control.
DMF Inhibits Differentiation of 3T3-L1 Preadipocytes during the Early Stages of Adipogenesis
To identify the critical stage of adipogenic differentiation affected by DMF treatment, differentiating 3T3-L1 preadipocytes were treated with 75 µM DMF at various time points during adipogenic differentiation, as illustrated in Figure 3A. After 8 days of differentiation, cells were subjected to Oil Red O staining and quantitative analysis of intracellular lipids. Treatment with DMF from day 0 to day 8 (treatment 4) showed the highest inhibition of intracellular lipid droplet accumulation. Treatment with DMF from day 0 to day 4 (treatment 2) or from day 0 to day 6 (treatment 3) showed comparatively low levels of intracellular lipid accumulation. Although the longer DMF treatment tended to decrease intracellular lipid accumulation more extensively, the amounts of inhibition were not statistically different between treatments 2, 3, and 4. Moreover, differentiating 3T3-L1 preadipocytes treated with DMF from days 4 to 8 (treatment 5) resulted in weaker inhibition of adipogenic differentiation compared with treatments 2 and 3 (Fig. 3B and C). These results suggested that DMF acts at an early stage of adipogenic differentiation.
Figure 3. Inhibitory effect of DMF during the early stages of adipogenic differentiation.

3T3-L1 cells were treated with 75 µM DMF for the indicated time periods during 3T3-L1 preadipocyte differentiation. (A) Schematic model depicting DMF treatment during 3T3-L1 preadipocyte differentiation. Thick lines indicate the treatment time at 75 µM DMF. 3T3-L1 cells were treated with 75 µM DMF for the indicated times during 3T3-L1 preadipocyte differentiation. (B) Representative micrographs showing cell culture dishes and cell monolayers stained with Oil Red O (× 200). (C) Quantitative analysis of Oil Red O staining. Oil Red O stained cells were extracted with isopropyl alcohol and the absorbance at 500 nm was measured. (D) Western blot analysis showing the protein levels of C/EBPβ, C/EBPα, PPARγ, SREBP-1c (precursor and mature), and FAS in 3T3-L1 cells after 0, 2, or 4 days of DMF treatment. The data are presented as mean ± S.D. (n = 3); *p<0.05, **p<0.001 vs. control.
To confirm the action of DMF on an early stage of adipogenic differentiation, the protein levels of adipogenic gene products were examined at day 2 and 4 after MDI treatment. As shown in Figure 3D, C/EBPβ expression on day 4 was still up-regulated, although the level was lower than on day 2 after MDI treatment. DMF decreased C/EBPβ expression on day 4 after MDI treatment but it did not decrease C/EBPβ expression on day 2 after MDI treatment. C/EBPα and PPARγ protein production was detected by day 2 and further increased by day 4 after MDI treatment, but these increases were inhibited by DMF treatment. The levels of mature SREBP-1c protein were minimal at day 2 but were clearly detected by day 4. FAS protein levels were markedly increased at day 4 after MDI treatment, but this increase was inhibited by DMF treatment (Fig. 3D).
DMF Inhibits 3T3-L1 Preadipocyte Clonal Expansion
Mitotic clonal expansion occurs during the early stages of preadipocyte differentiation. Therefore, whether the negative effect of DMF on adipogenic differentiation is the result of inhibition of mitotic clonal expansion was examined. Differentiating 3T3-L1 preadipocytes treated with the indicated concentrations of DMF (0, 50, and 75 µM) were subjected to flow cytometry. As shown in Figure 4A, mitotic clonal expansion in 3T3-L1 preadipocytes was induced by MDI, but DMF treatment effectively induced a G1 cell cycle arrest. The number of cells in the G2/M phase increased in 3T3-L1 preadipocytes in the MDI treatment. However, DMF-treated 3T3-L1 preadipocytes showed a dose-dependent increase in the number of cells in G1 phase and decrease in the number of cells in the G2/M phase (Fig. 4A).
Figure 4. Cell cycle analysis of DMF-treated 3T3-L1 cells during mitotic clonal expansion.

(A) Flow cytometric cell cycle analysis. Two-day post-confluent 3T3-L1 preadipocytes were incubated with MDI in the presence or absence of DMF. After 24 h, cells were stained with propidium iodide and subjected to flow cytometric cell cycle analysis (upper panel). The percentage of the cell population at different stages of the cell cycle were calculated from the data in (A) (lower panel). (B) Western blot analysis of cell cycle-related proteins. The time course of p21, p27, pRb, and cyclin D expression was analyzed after incubation in MDI with or without DMF treatment. (C) Relative cell numbers. Cell numbers were determined by hemocytometer counts after incubation with MDI in the presence or absence of DMF. The data are presented as mean ± S.D. (n = 3) *p<0.01.
Next, the effect of DMF on the levels of cell cycle-related proteins was examined. The level of p21, a key regulator of cell cycle progression during G1 phase [25], increased in 3T3-L1 preadipocytes treated with DMF for 48 h, compared with cells incubated in the absence of DMF (Fig. 4B). In addition, DMF reduced MDI-induced total cyclin D and phospho-Rb. The levels of p27 were unchanged (Fig. 4B). Consistent with these data, the relative cell numbers were increased by MDI treatment after 1 or 2 days in the medium, but DMF completely prevented the MDI-induced cell proliferation (Fig. 4C). Taken together, these data suggest that DMF inhibits clonal expansion during adipogenic differentiation through the induction of a G1 cell cycle arrest.
Nrf2 does not Mediate the Anti-adipogenic Differentiation Effect of DMF
DMF activates Nrf2, which is known to inhibit adipogenic differentiation [8], [9]. Therefore, whether the effect of DMF on adipogenic differentiation is Nrf2 dependent was examined using cells stably expressing dominant negative (DN) Nrf2. Oil Red O staining indicated that cells stably expressing Nrf2 suppressed adipogenic differentiation and cells stably expressing DN-Nrf2 showed augmented adipogenic differentiation (Fig. 5A). Interestingly, the DMF-induced inhibition of adipogenic differentiation was not abolished in DN-Nrf2 expressing cells, suggesting that DMF inhibition of preadipocyte differentiation is Nrf2 independent. The active function of DN-Nrf2 was confirmed by measuring the expression level of the Nrf2 target gene NQO-1. DMF-induced NQO-1 expression was not found in cells expressing DN-Nrf2 (Fig. 5A).
Figure 5. The effect of Nrf2 and STAT3 by DMF on adipogenic differentiation.

(A) RT-PCR analysis of 3T3-L1 preadipocytes stably expressing Vxy-puro, Vxy-Nrf2-puro, and Vxy-DN-Nrf2-puro shows the effects of DN-Nrf2 on expression of Nrf2 and its target gene, NQO-1 (upper panel). Oil Red O staining showed the effect of DMF (75 µM) on adipogenesis in Vxy-DN-Nrf2-puro overexpressing 3T3-L1 cells (lower panel). (B) Representative Western blot analysis showing the effect of DMF (75 µM) on STAT3 phosphorylation at Tyr705. Cells were incubated in MDI in the presence or absence of DMF. (C) Transient transfection study with the M67 luciferase reporter. AD-293 cells were transiently transfected with M67-luc and STAT3 expression plasmids. The cells were cultured for 24 h and treated with different concentrations DMF (25–75 µM) for 24 h. Luciferase activity was normalized by β-galactosidase for transfection efficiency. The data are presented as mean ± S.D. (n = 3) *p<0.05, **p<0.01, ***p<0.001 vs. STAT3 transfection.
STAT3 as a Target for the Anti-adipogenic Differentiation Effect of DMF
A regulatory role for STAT3 during the proliferative phases of adipogenesis has been reported previously [26]. Therefore, whether STAT3 is a target for the anti-adipogenic effect of DMF was examined. As shown in Figure 5B, MDI treatment induced STAT3 phosphorylation but DMF treatment markedly inhibited STAT3 phosphorylation (Fig. 5B). Using a reporter construct composed of four synthetic STAT-response elements (M67-luciferase), the effect of DMF on STAT3 transcriptional activity was examined. Consistent with the Western blot analysis of STAT3 phosphorylation, DMF markedly inhibited STAT3 transcriptional activity (Fig. 5C).
Discussion
In the present study, DMF was shown to inhibit 3T3-L1 preadipocyte differentiation to adipocytes through inhibition of clonal expansion by the induction of a G1 cell cycle arrest. DMF inhibits transcription factors involved in adipogenic differentiation, including C/EBPβ, C/EBPα, and PPARγ. It appears that DMF inhibits preadipocyte differentiation by down-regulation of STAT3 activity.
Studying adipogenic differentiation using 3T3-L1 preadipocytes is a well-established model system. When 3T3-L1 preadipocytes are incubated in the presence of hormonal inducers, adipogenesis is induced by adipogenesis-related transcription factors such as C/EBPβ, C/EBPα, and PPARγ [27]. C/EBPβ, C/EBPα, and PPARγ are induced by early, before the transcriptional activation of most adipogenic genes, and these factors enhance the expression of genes associated with adipogenic differentiation [28], [29]. This study showed that DMF inhibited 3T3-L1 preadipocyte differentiation and decreased adipogenic gene expression early in differentiation. After treatment of preadipocytes with MDI, a rapid and transient increase in the expression of C/EBPβ is observed. Within 2 days, C/EBPβ reaches its peak level and then begins to decrease, coincident with a rise in C/EBPα and PPARγ expression [30], [31]. C/EBPα is undetectable at the beginning of the differentiation process in preadipocytes, becomes detectable 2 days after MDI stimulation, and reaches its highest levels by 5 days after initiation of the adipogenic differentiation program [32], [33]. PPARγ is induced during the 2 days following induction of adipogenic differentiation and its expression peaks by day 3–4 [34]. Consistent with previous studies, our data showed the protein levels of C/EBPα and PPARγ were weakly detected at day 2, but became abundant by day 4 after MDI stimulation. The C/EBPβ protein level was strongly detected on day 2. The level on day 4 was still markedly up-regulated although it was slightly lower than on day 2. The increase in the C/EBPβ protein level was inhibited by DMF treatment. Collectively, these data suggest that the inhibition of adipogenic differentiation by DMF occurs primarily in the early stages of differentiation.
Preadipocytes differentiate into adipocytes through growth arrest, clonal expansion, early changes in gene expression, later events such as lipid accumulation, and are finally terminally differentiated [4]. In this chronology, mitotic clonal expansion occurs in the early stages of differentiation [4]. Mitotic clonal expansion consists of the two sequential rounds of mitosis which are completed 48–60 h after the induction of adipogenic differentiation [35]. Because the inhibitory effects of DMF occurred mainly within the first 4 days, we investigated the effect of DMF on mitotic clonal expansion of adipocytes. Flow cytometry showed that DMF induced a G1 cell cycle arrest. The present study showed that DMF-treated 3T3-L1 preadipocytes showed elevated levels of p21, which is a potent cyclin-dependent kinase inhibitor and functions as a regulator of the G1 phase of the cell cycle. DMF also inhibited pRb by down-regulation of the cyclin D-dependent kinase, which results in G1 arrest and inhibits clonal expansion.
STAT3 regulates cell growth and differentiation [10] through up-regulation of p21, which induces G1 arrest in the cell cycle [12]. Recent studies demonstrated that STAT3 is associated with adipogenic differentiation. The differentiation of preadipocytes into mature adipocytes was suppressed by inhibition of the STAT3 pathway through the regulation of adipogenic genes such as PPARγ and C/EBPß [6], [7], suggesting that STAT3 may play a role in mediating the effects of DMF on p21 induction and G1 cell cycle arrest. Indeed, DMF inhibited adipogenic MDI-stimulated STAT3 phosphorylation and STAT3 transcriptional activity, as shown using a reporter construct composed of four synthetic STAT-response elements. Because DMF is known to activate Nrf2 [22], the possibility that DMF inhibits adipogenic differentiation via Nrf2 activation was investigated. Inhibition of Nrf2 activity using cells stably expressing DN-Nrf2 increased 3T3-L1 preadipocyte differentiation. However, DN-Nrf2 expressing cells did not attenuate the effect of DMF on adipogenic differentiation, suggesting an Nrf2-independent mechanism.
Recently phase phase III clinical trials of DMF for multiple sclerosis, namely DEFINE and CONFIRM was completed [36], [37]. In these trials, DMF significantly reduced the relapse rates of multiple sclerosis, but changes in body weight were not investigated. In most animal studies of the beneficial effect of DMF, weight changes were not included as part of the data [22], [38]. Therefore, it is possible and necessary to evaluate the effects of DMF on body weight and adipogenesis in human with obesity, especially as DMF was generally well tolerated in previous clinical trials.
In summary, these data demonstrate that DMF is a negative regulator of preadipocyte differentiation through the regulation of adipogenic transcriptional factors and cell cycle proteins. This negative regulation by DMF is mediated by STAT3 inhibition but is unlikely to involve Nrf2 activation.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium was purchased from Hyclone (Logan, UT). Dimethylfumarate (DMF), insulin, 3-isobutyl-1-metylxanthine (IBMX), dexamethasone, and Oil Red O dye were purchased from Sigma-Aldrich (St Louis, MO, USA).
Cell Culture and Differentiation
Murine 3T3-L1 preadipocytes were cultured in high-glucose DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% bovine serum (Gibco, Auckland, NZ), penicillin (100 µg/ml), and streptomycin (100 µg/ml) in a humidified atmosphere of 5% CO2 at 37°C. Two days after reaching confluence, 3T3-L1 preadipocytes were induced to differentiate using medium supplemented with 1 µg/ml insulin, 1 µM dexamethasone, 0.5 mM IBMX, and 10% fetal bovine serum (FBS, Gibco, US). After 2 days, the medium was replaced with medium containing 10% FBS supplemented with 1 µg/ml insulin, and changed every 2 days thereafter with medium containing 10% FBS.
Oil Red O Staining
After day 8, differentiated 3T3-L1 preadipocytes were fixed with 10% formalin in PBS for 1 h and washed with 60% isopropyl alcohol. After drying at room temperature, cells were stained with filtered Oil Red O solution for 30 minutes. Stained cells were washed four times with distilled water. The phenotypic changes of adipogenic differentiation were observed using an inverted phase-contrast microscope (Olympus LX81 , Japan). To quantify the amount of Oil Red O stained lipids, stained cells were eluted with 100% isopropyl alcohol for 10min and the absorbance of the extracts was measured at 500 nm in a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Western Blot Analysis
At the indicated times, cells were washed with twice with PBS and lysed with lysis buffer [20 mM Tris (pH 7.4), 5 mM EDTA (pH 8.0), 10 mM Na4P2OH, 100 mM NaF, 2 mM Na3VO4, 1% NP-40 and 0.1 mM PMSF] containing proteinase and phosphatase inhibitors. Proteins were separated by SDS-PAGE, transferred to a PVDF membrane (Millipore, USA), and incubated with specific primary antibodies. The antibodies used in this study were directed against SREBP-1c, p21, p27 (BD Biosciences, San Jose, CA, USA), FAS, C/EBPα, PPARγ, Cyclin D, pRb, STAT3, phospho-STAT3 (Cell Signaling, Beverly, MA, USA), C/EBPβ (Santa Cruz Biotechnology, CA, USA). Detection of each protein was performed using ECL (Immobilon™ Western Chemiluminescent HRP Substrate, Millipore) according to the manufacturer’s instructions.
RT-PCR and Real-time PCR
Total RNA was extracted using the Trizol reagent (Invitrogen, Carlsbad, CA USA) and an aliquot (2 µg) of total RNA was reversed transcribed using the RevertAidTMFirst Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania) according to the manufacturer’s instructions. First strand cDNAs were amplified by PCR using gene-specific primers. Real-time PCR was carried out using SYBR Green (SYBR Green Master Mix, Applied Biosystems, Warrington, UK) with the StepOne™ Real-Time PCR system (Applied Biosystems). The sequences for the primers are shown in Table 1. The expression level of mouse 36B4 was used as the internal control.
Table 1. Primer sequences for RT-PCR and Real-time PCR.
| Gene | Sequences (5′→ 3′) of the RT-PCR primers |
| mouse Nrf2 | sense - CCATTTACGGAGACCCACCGC |
| antisense - GCCCAAGTCTTGCTCCAGCTC | |
| mouse NQO-1 | sense - TCGGAGAACTTTCAGTACCC |
| antisense - AGGCTAAGCTTGGGAAAAGAAA | |
| mouse 36B4 | sense –TGCCACACTCCATCATCAAT |
| antisense - CGAAGAGACCGAATCCCATA | |
| Gene | Sequences (5′→ 3′) of the Real-time PCR primers |
| mouse C/EBPβ | sense - AGCGGCTGCAGAAGAAGGT |
| antisense - GGCAGCTGCCTTGAACAAGTTC | |
| mouse C/EBPα | sense - GCGCAAGAGCCGAGATAAAG |
| antisense - CGGTCATTGTCACTGGTCAACT | |
| mouse PPARγ2 | sense - CACAAGAGCTGACCCAATGGT |
| antisense - GATCGCACTTTGGTATTCTTGGA | |
| mouse SREBP-1c | sense - CCCTACCGGTCTTCTATCAATGA |
| antisense - GCAGATTTATTCAGCTTTGCTTCA | |
| mouse FAS | sense - ACCTGGTAGACCACTGCATTGAC |
| antisense - CCTGATGAAACGACACATTCTCA | |
| mouse aP2 | sense - CCATCCGGTCAGAGAGTACTTTTAA |
| antisense - CGAATTCCACGCCCAGTT | |
| mouse 36B4 | sense - ACCTCCTTCTTCCAGGCTTT |
| antisense - CTCCAGTCTTTATCAGCTGC |
FACS Analysis
To determine the effect of DMF on cell cycle progression, two-day post-confluent preadipocytes were treated with an adipogenic cocktail in the presence or absence of 50 or 75 µM of DMF for 24 h. Cells were fixed with 70% cold ethanol overnight at –20°C and then washed twice with 1×PBS. Cells were analyzed using a FACS Calibur system (BD Bioscience, San Jose, CA, USA) after staining with 1 ml of cold propidiumiodide (PI) solution containing 40 µg/ml propidium iodide, 10 µg/ml of RNase A and 0.1% NP-40.
Retroviral Plasmids and Retroviral Infection
The pcDNA3-Nrf2 construct was the kind gift of Mi-Kyoung Kwak (Yeungnam University, Korea). The dominant negative mutant of Nrf2 (DN-Nrf2), lacking the N-terminal transcriptional activation domain, was generated by deleting amino acid residues 1–392 [39]. Full-length cDNAs of mouse Nrf2 and DN-Nrf2 were amplified by PCR and inserted into the XhoI and NotI site of the Vxy-puro retroviral vector. The constructs (Vxy-puro, Vxy-Nrf2-puro, and Vxy-DN-Nrf2-puro) were transfected into the Phoenix ecotropic packaging cells using Lipofectamine™ 2000 reagent (Invitrogen). Viral supernatants were collected after 48 h, clarified by filtration through 0.45 µm pore size syringe filters (Sartorius Stedim Biotech, Bohemia, NY, USA), and used for infection of 3T3-L1 preadipocytes in the presence of 5 µg/ml polybrene (Sigma) for 24 h. Infected 3T3-L1 cells were selected with 3 µg/ml puromycin (Sigma) for 7 days.
Luciferase Reporter Gene Assay
M67-luc and the constructs expressing wild type STAT3 (STAT3-FLAG) were the kind gifts of Dr. James E. Darnell (The Rockefeller University, New York, NY). For luciferase reporter gene assays, AD-293 cells plated in 24-well plates (70% confluence) were co-transfected with the STAT3 expression plasmid and 400 ng/well of M67-luc using the TransIT-LT1 transfection reagent (MirusBio Incorporation, Madison, WI). Cytomegalovirus (CMV)–β-galactosidase (50 ng/well) was co-transfected as an internal control for transfection efficiency. At 24 h post-transfection, the medium was replaced with 0.5% FBS medium with or without DMF and the cells were incubated for 24 h. Cell were lysed for luciferase and β-galactosidase assays and luciferase activity was normalized by the β-galactosidase activity.
Statistical Analysis
Data are expressed as mean ± S.D. Statistical analyses were performed using an unpaired Student’s t-test and a value of P<0.05 was considered to be significant.
Funding Statement
This work was supported by grants from the National Research Foundation (2012R1A2A1A03670452, 2012R1A2A2A01043867, WCU program R32-10064) and the Future-based Technology Development program (Bio Field 2010-0019514) funded by the Ministry of Education, Science, and Technology and a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A111345). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Wozniak SE, Gee LL, Wachtel MS, Frezza EE (2009) Adipose tissue: the new endocrine organ? A review article. Dig Dis Sci 54: 1847–1856. [DOI] [PubMed] [Google Scholar]
- 2. Pi-Sunyer FX (2002) The obesity epidemic: pathophysiology and consequences of obesity. Obesity research 10 Suppl 297S–104S. [DOI] [PubMed] [Google Scholar]
- 3. Jo J, Gavrilova O, Pack S, Jou W, Mullen S, et al. (2009) Hypertrophy and/or Hyperplasia: Dynamics of Adipose Tissue Growth. PLoS Computational Biology 5: e1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gregoire FM, Smas CM, Sul HS (1998) Understanding adipocyte differentiation. Physiological reviews 78: 783–809. [DOI] [PubMed] [Google Scholar]
- 5. Cristancho AG, Lazar MA (2011) Forming functional fat: a growing understanding of adipocyte differentiation. Nature reviews Molecular cell biology 12: 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang D, Zhou Y, Lei W, Zhang K, Shi J, et al. (2010) Signal transducer and activator of transcription 3 (STAT3) regulates adipocyte differentiation via peroxisome-proliferator-activated receptor gamma (PPARgamma). Biology of the cell/under the auspices of the European Cell Biology Organization 102: 1–12. [DOI] [PubMed] [Google Scholar]
- 7. Zhang K, Guo W, Yang Y, Wu J (2011) JAK2/STAT3 pathway is involved in the early stage of adipogenesis through regulating C/EBPbeta transcription. Journal of cellular biochemistry 112: 488–497. [DOI] [PubMed] [Google Scholar]
- 8. Shin S, Wakabayashi N, Misra V, Biswal S, Lee GH, et al. (2007) NRF2 modulates aryl hydrocarbon receptor signaling: influence on adipogenesis. Molecular and cellular biology 27: 7188–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL (2012) Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes 61: 3208–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirano T, Ishihara K, Hibi M (2000) Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 19: 2548–2556. [DOI] [PubMed] [Google Scholar]
- 11. Johnson DG, Walker CL (1999) Cyclins and cell cycle checkpoints. Annual review of pharmacology and toxicology 39: 295–312. [DOI] [PubMed] [Google Scholar]
- 12. Coqueret O, Gascan H (2000) Functional interaction of STAT3 transcription factor with the cell cycle inhibitor p21WAF1/CIP1/SDI1. J Biol Chem 275: 18794–18800. [DOI] [PubMed] [Google Scholar]
- 13. Nguyen T, Nioi P, Pickett CB (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284: 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chartoumpekis DV, Ziros PG, Sykiotis GP, Zaravinos A, Psyrogiannis AI, et al. (2011) Nrf2 activation diminishes during adipocyte differentiation of ST2 cells. International journal of molecular medicine 28: 823–828. [DOI] [PubMed] [Google Scholar]
- 15. Altmeyer PJ, Matthes U, Pawlak F, Hoffmann K, Frosch PJ, et al. (1994) Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. Journal of the American Academy of Dermatology 30: 977–981. [DOI] [PubMed] [Google Scholar]
- 16. Mrowietz U, Christophers E, Altmeyer P (1999) Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. The British journal of dermatology 141: 424–429. [DOI] [PubMed] [Google Scholar]
- 17. Seidel P, Merfort I, Tamm M, Roth M (2010) Inhibition of NF-kappaB and AP-1 by dimethylfumarate correlates with down-regulated IL-6 secretion and proliferation in human lung fibroblasts. Swiss medical weekly 140: w13132. [DOI] [PubMed] [Google Scholar]
- 18. Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, et al. (2008) Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 372: 1463–1472. [DOI] [PubMed] [Google Scholar]
- 19. Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, et al. (2010) Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. Journal of neuroinflammation 7: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niemand C, Nimmesgern A, Haan S, Fischer P, Schaper F, et al. (2003) Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. Journal of immunology (Baltimore, Md : 1950) 170: 3263–3272. [DOI] [PubMed] [Google Scholar]
- 21. Wehinger J, Gouilleux F, Groner B, Finke J, Mertelsmann R, et al. (1996) IL-10 induces DNA binding activity of three STAT proteins (Stat1, Stat3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS letters 394: 365–370. [DOI] [PubMed] [Google Scholar]
- 22. Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, et al. (2011) Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain : a journal of neurology 134: 678–692. [DOI] [PubMed] [Google Scholar]
- 23. Oh CJ, Kim JY, Choi YK, Kim HJ, Jeong JY, et al. (2012) Dimethylfumarate attenuates renal fibrosis via NF-E2-related factor 2-mediated inhibition of transforming growth factor-beta/Smad signaling. PLoS ONE 7: e45870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellrichmann G, Petrasch-Parwez E, Lee DH, Reick C, Arning L, et al. (2011) Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 6: e16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes & development 13: 1501–1512. [DOI] [PubMed] [Google Scholar]
- 26. Deng J, Hua K, Lesser SS, Harp JB (2000) Activation of signal transducer and activator of transcription-3 during proliferative phases of 3T3-L1 adipogenesis. Endocrinology 141: 2370–2376. [DOI] [PubMed] [Google Scholar]
- 27. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes & development 14: 1293–1307. [PubMed] [Google Scholar]
- 28. Mandrup S, Lane MD (1997) Regulating adipogenesis. J Biol Chem 272: 5367–5370. [DOI] [PubMed] [Google Scholar]
- 29. Rosen ED (2005) The transcriptional basis of adipocyte development. Prostaglandins, leukotrienes, and essential fatty acids 73: 31–34. [DOI] [PubMed] [Google Scholar]
- 30. Brun RP, Kim JB, Hu E, Altiok S, Spiegelman BM (1996) Adipocyte differentiation: a transcriptional regulatory cascade. Current opinion in cell biology 8: 826–832. [DOI] [PubMed] [Google Scholar]
- 31. Darlington GJ, Ross SE, MacDougald OA (1998) The role of C/EBP genes in adipocyte differentiation. J Biol Chem 273: 30057–30060. [DOI] [PubMed] [Google Scholar]
- 32. Christy RJ, Kaestner KH, Geiman DE, Lane MD (1991) CCAAT/enhancer binding protein gene promoter: binding of nuclear factors during differentiation of 3T3-L1 preadipocytes. Proceedings of the National Academy of Sciences of the United States of America 88: 2593–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin FT, Lane MD (1994) CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3-L1 adipocyte differentiation program. Proceedings of the National Academy of Sciences of the United States of America 91: 8757–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clarke SL, Robinson CE, Gimble JM (1997) CAAT/enhancer binding proteins directly modulate transcription from the peroxisome proliferator-activated receptor gamma 2 promoter. Biochemical and biophysical research communications 240: 99–103. [DOI] [PubMed] [Google Scholar]
- 35. Tang QQ, Otto TC, Lane MD (2003) Mitotic clonal expansion: a synchronous process required for adipogenesis. Proceedings of the National Academy of Sciences of the United States of America 100: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, et al. (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. The New England journal of medicine 367: 1098–1107. [DOI] [PubMed] [Google Scholar]
- 37. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, et al. (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. The New England journal of medicine 367: 1087–1097. [DOI] [PubMed] [Google Scholar]
- 38. Ghoreschi K, Bruck J, Kellerer C, Deng C, Peng H, et al. (2011) Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. The Journal of experimental medicine 208: 2291–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chui DH, Tang W, Orkin SH (1995) cDNA cloning of murine Nrf 2 gene, coding for a p45 NF-E2 related transcription factor. Biochemical and biophysical research communications 209: 40–46. [DOI] [PubMed] [Google Scholar]