

Summary
Protein folding and aggregation inevitably compete with one another. This competition is even keener for proteins with frustrated landscapes, such as those rich in β-structure. Interestingly, despite their rugged energy landscapes and high β-sheet content, intracellular lipid-binding proteins (iLBPs) appear to successfully avoid aggregation, as they are not implicated in aggregation diseases. In this study, we used a canonical iLBP, cellular retinoic acid-binding protein 1 (CRABP1), to better understand how folding is favored over aggregation. Analysis of folding kinetics of point mutants reveals that the folding pathway of CRABP1 involves early barrel closure. This folding mechanism protects sequences in CRABP1 that comprise cores of aggregates as identified by NMR. The amino acid conservation pattern in other iLBPs suggests that early barrel closure may be a general strategy for successful folding and minimization of aggregation. We suggest that folding mechanisms more broadly may incorporate steps that disfavor aggregation.
INTRODUCTION
The unavoidable competition between protein folding and aggregation links the energy landscapes of these two processes (Clark, 2004; Hartl et al., 2011; Jahn and Radford, 2008; Vendruscolo, 2012). Yet in large measure, we lack adequately detailed descriptions of these landscapes to gain insights into vulnerabilities of proteins to aggregation and how to reduce the inherent risks. Predominantly β-sheet proteins are a particularly apt structural class for examining molecular details of the folding/aggregation balance as they have rugged folding energy landscapes with a high degree of frustration and consequent population of intermediate states and increased susceptibility to aggregation (Chavez et al., 2004; Plaxco et al., 1998). Nonetheless, there are several large families of β-rich proteins with no association with amyloid diseases, arguing that mechanisms exist to enable them to favor folding over aggregation. Some protection from aggregation may be attributable to chaperone action, but we hypothesize that intrinsic folding mechanisms must also incorporate strategies to minimize aggregation.
Intracellular lipid-binding proteins (iLBPs), a large family of successful folders (Banaszak et al., 1994), offer an excellent model system to explore this hypothesis. Despite their complex folding landscapes and extremely diverse sequence space, these ten-stranded β-barrel-containing proteins (Fig. 1A) have not been implicated in any misfolding diseases. This observation suggests that iLBPs have evolved a robust folding mechanism with built-in aggregation protection. Here we searched for topology-determining structural motifs as signatures of this mechanism. We used a model iLBP, cellular retinoic acid-binding protein 1 (CRABP1). Its rugged folding landscape is characterized by two on-pathway intermediates – an early collapsed state (I1) and a later β-molten globule-like state with native topology (I2) – and thus exemplifies β-barrel frustration (Clark et al., 1997; Clark et al., 1998).
Figure 1. Structural and topological features of CRABP1.
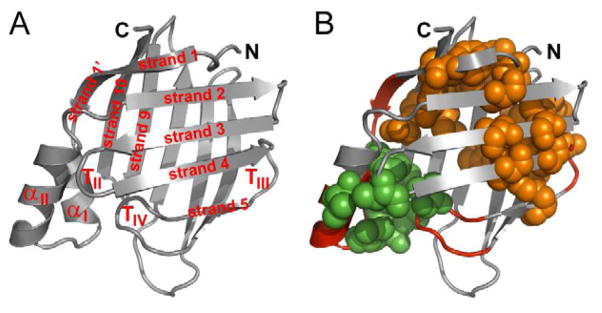
(A) Secondary structure elements of CRABP1 (shown on the structure of holo-CRABP1, PDB: 1CBR, with ligand omitted; W.L. DeLano, http://www.pymol.org (2002)). Helices are designated αI and αII, and turns II, III and IV are designated as TII, TIII and TIV, respectively. Strands are designated as ‘strand i’ where i represents strand number; note that strands 9 and 10 belong to the back sheet.
(B) Proposed topological determinants in CRABP1. Shown in red backbone are the helix-turn-helix motif and turns III and IV – local sequence elements in CRABP1 found to adopt native-like structures in peptide models (Rotondi and Gierasch, 2003b; Sukumar and Gierasch, 1997). The major hydrophobic core is shown in orange spheres, and the minor core in green spheres.
The β-barrels of iLBPs are formed via several medium-range contacts between strands, and long-range contacts that link N- and C-terminal sequences to close the barrel (Fig. 1A). The major conserved hydrophobic core of iLBPs comprises several highly networked conserved long-range interactions between the front and back sheet (Banaszak et al., 1994; Gunasekaran et al., 2004; Kleywegt et al., 1994; Marcelino et al., 2006) (orange spacefill in Fig. 1B). In addition, there is a smaller cluster of primarily hydrophobic interactions that was identified in a study of conserved pairwise interactions (green spacefill in Fig. 1B) (Gunasekaran et al., 2004). This minor core comprises portions of the helix-turn-helix motif that caps the β-barrel, β-strands 1′ and 10, and turns II and IV. Intriguingly, the minor core involves interactions between two local structural motifs, helix-turn-helix and turn IV, that have been shown (along with turn III) to populate native-like conformations in peptide studies (backbone in red, Fig. 1B) (Rotondi and Gierasch, 2003b; Sukumar and Gierasch, 1997). This propensity to form native structure as isolated fragments led to the proposal that these local sequences adopted structure early in folding, perhaps even in the unfolded ensemble. These features in fact have been predicted to act as folding-initiating nuclei in iLBPs (Nikiforovich and Frieden, 2002).
Two alternative mechanisms for the formation of native topology during iLBP folding can thus be envisioned: Either, as previously reported for the immunoglobulin fold (Cota et al., 2001; Fowler and Clarke, 2001; Hamill et al., 2000), residues participating in the conserved and highly networked major hydrophobic core act as a folding nucleus and specify the sheet topology. Or alternatively, local structural features that interact across the β-barrel closure region form early and restrict conformational space in a manner that is uniquely advantageous to the β-clam fold. Either way, the absence of iLBP-related misfolding diseases implies that their folding, whether driven by consolidation of the hydrophobic core or interaction of locally encoded structural features, is robust enough to bypass aggregation.
To distinguish between these two possibilities and uncover features that provide aggregation protection, we introduced single residue substitutions at 33 sites in the CRABP1 sequence with extensive coverage of structural elements including the minor hydrophobic core near the barrel closure region. Observing the impact of these mutations on CRABP1 stability and unfolding kinetics revealed that its rate-determining transition state (TS) is highly polarized, with barrel closure interactions forming before the TS, and interactions in the major hydrophobic core developing only after the TS. Strikingly, barrel closure provides partner interactions for edge strand 10, and in so doing protects regions of high predicted aggregation propensity, including strands 3, 4, 9 and 10, which we have found to form the core of isolated CRABP1 aggregates. We speculate that this folding mechanism may be common among other iLBP family members and that it exemplifies an intrinsic aggregation-protection strategy.
RESULTS AND DISCUSSION
β-Barrel Closure Occurs Before the Rate-Determining Transition State of CRABP1, But Packing of the Major Hydrophobic Core Occurs Post-Transition State
To characterize the rate-determining TS of CRABP1 we used protein engineering methods (Fersht et al., 1992). Because CRABP1 folds via multiple states, we used the established strategy of comparing the effect of mutations on the unfolding barrier, ΔΔG‡-N, to their effect on native state stability, ΔΔGU-N (assuming the unfolded state energy is negligibly perturbed), thus generating unfolding φ-values (φU), to report on whether an interaction is present in the TS or only forms post-TS (Bueno et al., 2006; Connell et al., 2009; Huysmans et al., 2010). Since it is conventional to report φ-values in the folding direction, we converted the φU’s to φ’s (φ = 1 − φU). ΔΔGU-N values for mutant forms of CRABP1 were extracted from two-state fits of equilibrium urea denaturation curves, and ΔΔG‡-N values were obtained by linear extrapolation of plots of ln kU versus [urea] to 0 M urea. Representative examples of equilibrium and kinetic data are shown in Fig. 2A and B. The stabilities of the CRABP1 variants varied from 5.2 ± 0.2 kcal/mol for the most destabilized variant, Y133S, to 9.8 ± 0.1 kcal/mol for the minimally perturbed V58A variant (compare to 10.0 ± 0.2 kcal/mol for WT CRABP1) (see Table 1). All substitutions were destabilizing except for G70A in turn III (ΔΔGU-N = 0.7 ± 0.2 kcal/mol); this variant was included in the analysis due to consistent φ-values of the neighboring residues. The increased stability of G70A may arise because the dihedral angles of the backbone at this position are compatible with Ala, and the entropy cost is lower for folding to native with an Ala residue than Gly.
Figure 2. Equilibrium stability, unfolding kinetics and the properties of the rate-determining TS of CRABP1.

(A, C) Representative curves for urea-induced equilibrium denaturation for CRABP1 WT and its variants at 25 °C. The solid lines are fits of the data to a two-state model using a fixed equilibrium m-value of −2.0 kcal/mol·M.
(B, D) Representative curves for the dependence of the logarithm of the unfolding rates on [urea] for CRABP1 WT and its variants at 25 °C; the solid lines on the ln kU plots represent linear fits.
Panels (A) and (B) show the data for the residues that yield φ-values < 0.5, panels (C) and (D) – for the residues with φ-values > 0.5.
(E) The interactions present in the rate-determining TS mapped onto the native structure of CRABP1. Residues subjected to perturbation analysis are shown in spacefill and colored according to their φ-values from blue (0) to red (1); those caused no or minor perturbation are colored in gray.
(F) Sequence histogram with φ-values for the CRABP1 variants under study. The secondary structural features of CRABP1 are shown below.
See also Figures S1 and S2.
Table 1.
Thermodynamic and kinetic parameters of CRABP1 variantsa
| Variant | Locationb | ΔΔGU-N (kcal/mol)c | ln kUd | mU (kcal/mol·M)d | ΔΔG‡-N (kcal/mol)e | φ-valuef |
|---|---|---|---|---|---|---|
| wild typeg | n/a | −11.0 ± 0.1 | 0.64 ± 0.02 | n/a | n/a | |
| F3I+ h | Strand 1 | −3.6 ± 0.2 | −6.5 ± 0.1 | 0.80 ± 0.03 | −2.6 ± 0.1 | 0.26 ± 0.05 |
| W7Y | Strand 1 | −2.2 ± 0.2 | −9.7 ± 0.2 | 0.68 ± 0.02 | −0.8 ± 0.1 | 0.65 ± 0.07 |
| M9A+ | Strand 1′ | −1.8 ± 0.2 | −11.5 ± 0.2 | 0.87 ± 0.02 | 0.3 ± 0.2 | 1.17 ± 0.09 |
| S12G+ | Strand 1′ | −1.8 ± 0.2 | −10.38 ± 0.08 | 0.706 ± 0.008 | −0.34 ± 0.08 | 0.82 ± 0.05 |
| F15A | Helix I | −2.9 ± 0.2 | −8.5 ± 0.2 | 0.65 ± 0.01 | −1.25 ± 0.08 | 0.57 ± 0.04 |
| L18A+ | Helix I | −3.5 ± 0.2 | −9.5 ± 0.4 | 0.88 ± 0.05 | −0.9 ± 0.2 | 0.75 ± 0.07 |
| L19A− i | Helix I | −3.3 ± 0.2 | −7.58 ± 0.07 | 0.547 ± 0.005 | −2.00 ± 0.09 | 0.39 ± 0.05 |
| L22A+ | Helix I | −2.7 ± 0.2 | −10.43 ± 0.02 | 0.859 ± 0.002 | −0.31 ± 0.08 | 0.88 ± 0.03 |
| R29A− | Helix II | −0.8 ± 0.3 | −9.17 ± 0.04 | 0.458 ± 0.003 | −1.06 ± 0.08 | n/a |
| V31A | Helix II | −0.6 ± 0.3 | −10.6 ± 0.2 | 0.587 ± 0.008 | −0.2 ± 0.1 | n/a |
| V33A− | Helix II | −0.4 ± 0.3 | −9.9 ± 0.5 | 0.54 ± 0.05 | −0.6 ± 0.3 | n/a |
| I43V | Strand 2 | −1.6 ± 0.2 | −8.9 ± 0.1 | 0.61 ± 0.05 | −1.2 ± 0.1 | 0.3 ± 0.1 |
| F50I | Strand 3 | −1.7 ± 0.2 | −8.51 ± 0.04 | 0.66 ± 0.01 | −1.45 ± 0.08 | 0.1 ± 0.1 |
| V58A | Turn II | −0.2 ± 0.2 | −10.9 ± 0.2 | 0.642 ± 0.002 | 0.0 ± 0.1 | n/a |
| K66A− | Turn III | −1.8 ± 0.3 | −7.85 ± 0.07 | 0.48 ± 0.01 | −1.84 ± 0.09 | 0.0 ± 0.2 |
| V67A | Turn III | −3.3 ± 0.2 | −6.5 ± 0.2 | 0.70 ± 0.03 | −2.7 ± 0.1 | 0.18 ± 0.06 |
| G68A− | Turn III | −2.6 ± 0.2 | −6.9 ± 0.2 | 0.50 ± 0.03 | −2.4 ± 0.2 | 0.06 ± 0.09 |
| E69A− | Turn III | −1.3 ± 0.2 | −9.02 ± 0.07 | 0.51 ± 0.01 | −1.15 ± 0.09 | 0.1 ± 0.1 |
| G70A | Turn III | 0.7 ± 0.2 | −11.9 ± 0.1 | 0.69 ± 0.01 | 0.6 ± 0.1 | 0.2 ± 0.2 |
| T75A− | Turn IV | −2.4 ± 0.3 | −9.0 ± 0.1 | 0.54 ± 0.05 | −1.2 ± 0.1 | 0.51 ± 0.08 |
| V76A | Turn IV | −1.5 ± 0.3 | −10.3 ± 0.1 | 0.675 ± 0.007 | −0.4 ± 0.1 | 0.74 ± 0.08 |
| D77A− | Turn IV | −1.0 ± 0.3 | −9.56 ± 0.07 | 0.529 ± 0.001 | −0.83 ± 0.09 | 0.2 ± 0.2 |
| G78A− | Turn IV | −2.5 ± 0.4 | −9.53 ± 0.04 | 0.547 ± 0.009 | −0.85 ± 0.08 | 0.66 ± 0.06 |
| R79A− | Turn IV | −3.1 ± 0.3 | −8.6 ± 0.2 | 0.46 ± 0.02 | −1.4 ± 0.1 | 0.55 ± 0.06 |
| W87F | Strand 6 | −2.6 ± 0.4 | −7.7 ± 0.1 | 0.59 ± 0.02 | −1.9 ± 0.1 | 0.2 ± 0.1 |
| I93V− | Strand 7 | −1.8 ± 0.2 | −8.49 ± 0.05 | 0.567 ± 0.004 | −1.46 ± 0.08 | 0.2 ± 0.1 |
| W109I− | Strand 8 | −2.9 ± 0.2 | −7.6 ± 0.1 | 0.37 ± 0.02 | −2.0 ± 0.1 | 0.31 ± 0.07 |
| L113V | Strand 8 | −0.8 ± 0.2 | −10.48 ± 0.02 | 0.66 ± 0.01 | −0.28 ± 0.08 | n/a |
| L118V+ | Strand 9 | −2.3 ± 0.4 | −9.9 ± 0.2 | 0.79 ± 0.03 | −0.7 ± 0.2 | 0.72 ± 0.08 |
| C129A− | Strand 10 | −0.6 ± 0.2 | −9.8 ± 0.1 | 0.55 ± 0.01 | −0.68 ± 0.09 | n/a |
| Q131A− | Strand 10 | −2.0 ± 0.2 | −8.15 ± 0.02 | 0.516 ± 0.006 | −1.66 ± 0.08 | 0.17 ± 0.09 |
| Y133S+ | Strand 10 | −4.8 ± 0.2 | −9.32 ± 0.02 | 0.950 ± 0.001 | −0.97 ± 0.08 | 0.80 ± 0.02 |
| R135G+ | Strand 10 | −3.9 ± 0.3 | −8.7 ± 0.1 | 0.71 ± 0.01 | −1.34 ± 0.09 | 0.66 ± 0.04 |
The average values from at least two independent measurements and the standard deviation are reported; all values reported are extrapolated to 0 M denaturant
Secondary structural location of the selected variant residue
The energetic effect of mutations on the free energy of the unfolded state (U) with respect to the native state (N) calculated as ΔΔGU-N = ΔGU-NWT−ΔGU-Nmut where ΔGU-NWT was determined to be 10.0 ± 0.2 kcal/mol, and ΔGU-N values were obtained by fitting equilibrium urea unfolding curves using meq = −2.0 kcal/mol·M (Fig. 2A,C)
Obtained from linear extrapolation of ln kU versus [urea] (Fig. 2B,D)
The energetic effect of mutation on the free energy of the transition state (‡) with respect to the native state (N), calculated as ΔΔG‡-N = −RT ln (kUWT/kUmut)
Calculated as φF = 1 − φU
His-tagged CRABP1 with a stabilizing R131Q mutation was used as WT
Designates variants with mU > 0.7 kcal/mol·M (mU+)
Designates variants with mU < 0.57 kcal/mol·M (mU−)
We did not anticipate significant perturbations in the denatured state of CRABP1 as a result of the substitutions except when the original residue was a Gly. In these cases restricted conformational flexibility due to the introduction of a bulkier side chain may affect protein’s denatured state ensemble. In order to assess the effect of substitutions, the 1H-15N heteronuclear single quantum coherence spectra (HSQC) for the Gly to Ala variants (G68A, G70A and G78A) were compared to that of the WT protein under denaturing conditions. Only a few peaks were affected by the substitutions, as shown for the G78A variant in Fig. S1, arguing that the ensemble of structures sampled by the chain is very similar.
φ-values for most of the residues belonging to the major, intersheet hydrophobic core and adjacent turn III were between 0 and 0.3, consistent with specific packing of this region only after the TS (Fig. 2A–B and Table 1). In a previous amide hydrogen/deuterium (H/D) exchange study, we showed that interstrand H-bonds formed only during the rate-determining folding step of CRABP1 (Clark et al., 1997). The current data make it clear that specific side chain packing in the major hydrophobic core is also a late event in CRABP1 folding.
In contrast, residues in the minor hydrophobic core and along the strand 1′ to strand 10 connection displayed high φ-values (Fig. 2A–B and Table 1), suggesting that side-chain specific interactions that are implicated in β-barrel closure form before the rate-determining TS. Residues showing high φ-values include F15, L18 and L22 from helix I and V76, G78 and R79 from turn IV, the two local structural features previously demonstrated to form in peptide fragments (Rotondi and Gierasch, 2003b; Sukumar and Gierasch, 1997), as well as W7 from strand 1, M9 and S12 from strand 1′, L118 from strand 9, Y133 and R135 from the C-terminus of strand 10. The early involvement of W7 was suggested by our previous observation that its fluorescence changed in the dead time in stopped-flow mix refolding experiments (ca. 3 ms, (Clark et al., 1998)). Altogether, residues with high φ-values, indicating formation of their interactions before the rate-determining TS, comprise a structurally contiguous group in the native protein and participate in some of the longest-range interactions in the native structure of CRABP1 (Fig. 2A). Finally, mutations in helix II (R29A, V31A, V33A) and turn II (V58A) minimally perturbed the native state stability and did not change CRABP1 unfolding kinetics (Table 1), despite formally contributing to the smaller hydrophobic core. This is consistent with the dynamic nature of helix II (Krishnan et al., 2000) and lack of native-like structural propensity in isolated turn II (Rotondi and Gierasch, 2003b) in CRABP1.
Overall, the rate-determining TS of CRABP1 is highly polarized with a folding nucleus encompassing helix I, turn IV, strands 1, 1′ and 10 (Fig. 2E–F) that is formed in the TS. This nucleus resembles the ‘two-strand-helix’ nucleation motif shared by many α/β proteins (Lindberg and Oliveberg, 2007). Importantly, for CRABP1 β-barrel this nucleation mechanism implies early barrel closure.
How Early in Folding Does Barrel Closure Occur?
To probe the timing of barrel closure we examined the refolding kinetics of selected variants with substitutions in the barrel-closing region. Refolding of WT CRABP1 is triphasic, with the fastest phase (< 3 ms) reporting on the formation of the hydrophobically collapsed I1, followed by a fast phase (ca. 200 ms) corresponding to formation of the topologically native but highly solvated intermediate I2, and a 1 s phase corresponding to the formation of N (Clark et al., 1997; Clark et al., 1998). [Note: Folding of a small population of CRABP1 molecules (about 20%) is limited cis-trans isomerization of the L84-P85 bond, leading to a low amplitude slow (15 – 20 s) phase (Eyles and Gierasch, 2000).] All CRABP1 variants with substitutions in the folding nucleus as determined by perturbation analysis (W7Y, M9A, S12G, F15A, L18A, L22A and L118V, red spheres in Fig. 3A) showed significantly retarded formation of both I2 intermediate and N (Fig. 3B and Table S1). The fact that they retained three refolding kinetic phases, like WT CRABP1, argues that they fold by the same mechanism, albeit with modest perturbation due to the mutations. The Y133S and R135G variants exhibited the slowest refolding kinetics (data not shown). Slower formation of I2 implies structural and energetic perturbation of the TS barrier between I1 and I2. In an earlier study from our group the fast phase in CRABP1 folding was attributed to docking of the N- and C-termini based on the fluorescence signature of W7 (Clark et al., 1998). The present data extends this model to implicate barrel closure, including docking of N- and C-termini, in the early stage of CRABP1 folding. It is noteworthy that the C129A substitution, which caused only minor destabilization of the native state (ΔΔGU-N = −0.6 ± 0.2 kcal/mol) and did not formally ‘qualify’ for φ-value analysis, showed substantially retarded formation of I2. Thus, C129 is most likely a peripheral part of the barrel-closing patch via its contacts with S12, F15 and L18. We speculate that early docking and formation of specific interactions between the terminal β-strands may also help in simultaneous assembly of the front and the back sheets and thereby aid in rapid folding of the barrel.
Figure 3. Refolding kinetics for selected CRABP1 variants.
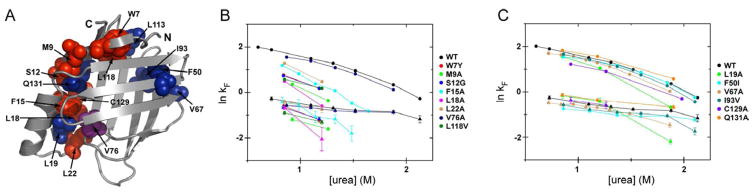
(A) CRABP1 variants used in refolding kinetics experiments are shown on the CRABP1 structure, with red spheres representing variants with φ-values > 0.5 and significant retardation of refolding kinetics, and blue spheres representing variants with φ-values < 0.5 and WT-like refolding behavior. V76A, which has a φ value of 0.74, but WT-like stopped-flow refolding, is shown in violet, and C129A, which perturbs the fast phase, is shown in red.
(B) Dependence of the logarithm of the rates of I2 and N formation (fast and medium kinetic phases) on [urea] at 25 °C for CRABP1 WT and selected variants with φ-values > 0.5. Shown are plots of ln kF vs. [urea] for the fast (in circles) and medium phases (in triangles).
(C) Same as panel (B) for the CRABP1 variants with φ-values < 0.5.
Colors correspond to those in Fig. 1A–D except for the C129A variant added in this figure.
See also Table S1.
Overall, analysis of the folding kinetics for residues with high φ-values paints a consistent picture of barrel closure occurring between the I1 and I2 intermediates, well before the rate-determining TS. Intriguingly, one class of residue that presents a high φ-value deviates from the pattern described above: The folding phases that are detectable by stopped-flow mixing kinetics experiments were essentially unperturbed from WT for turn IV variants such as V76A (Fig. 3B and Table S1, also T75A, G78A and R79A at 0.73 M [urea]). We conclude that turn IV formation occurs very early. In fact, the corresponding backbone region may sample turn conformations in the unfolded state as suggested by previous peptide studies (Rotondi and Gierasch, 2003b) while specific interactions between side chains may occur in the I1. Indeed, another study on the turn IV peptide model has shown that V76A, D77A and R79A substitutions did not fully reduce turn propensity (Rotondi and Gierasch, 2003a). Experiments are underway to test this possibility in the context of the full-length CRABP1. Note that turn IV has two key roles in folding: It joins residues from helix 2 and strands 1 and 1′ to form the β-barrel closure cluster, and it is in a topologically strategic position splitting front and back sheets via a β-bulge.
The primary perturbation associated with residues that display low φ-values is expected to be destabilization of the native state, with little change in folding kinetics. Consistent with their φ-values, CRABP1 variants F50I, V67A, I93V, L113V and Q131A (blue spheres in Fig. 3A) showed essentially unperturbed WT-like refolding behavior (Fig. 3C and Table S1). Thus, our data strongly argue for the formation of specific side chain interactions in the barrel-closing region as early as in I1 intermediate; while the rest of the protein including helix II and most of the major conserved hydrophobic core remains plastic until after the rate-determining TS.
The Rate-Determining Transition State of CRABP1 Is Malleable
Several CRABP1 variants exhibited unfolding m-values (mU-values) more than 10% different from that of the WT protein (0.64 kcal/mol·M). These variants were classified as mU+ (mU > 0.7 kcal/mol·M) or mU− (mU < 0.57 kcal/mol·M). The dependence of the difference between free energies of the native and the denatured states on [denaturant] is proportional to the accessible surface area (ASA) change between these states (Myers et al., 1995); the dependence of the logarithm of the unfolding rate constant (ln kU) on [denaturant] is proportional to the ASA change between the native state and the rate-determining TS. Generally, the change in mU may be explained in terms of movement of the TS, perturbation of the native state, or change of the folding pathway (Fowler and Clarke, 2001; Matouschek and Fersht, 1993; Otzen and Oliveberg, 2002). Since refolding behavior of all CRABP1 variants tested was triphasic similar to the WT, we favor the interpretation that it is the movement of the rate-determining TS that accounts for the observed mU changes. The observation of unperturbed equilibrium m-values strongly supports this conclusion suggesting that for all variants used in this study both the denatured and native states resemble WT CRABP1 in terms of ASA, as simultaneous and equal perturbation of ASA of both states is unlikely. Therefore, the altered mU-values likely report on the changes in the ASA of the corresponding TS, rather than on the native state. Indeed, for both mU+ and mU− variants we observed curvature in plots of the log of the unfolding rate constant as a function of [urea] at high denaturant (Fig. S2), thus indicating that the rate-determining TS of CRABP1 is also malleable with respect to the [denaturant]. We were unable to discern any curvature in the log unfolding rate plot of WT protein over the experimentally accessible [urea] range. The stronger denaturant dependence of the unfolding rate for the mU+ variants argues that the structure of their TS is expanded relative to that of the WT CRABP1. Conversely, the mU− variants have relatively more compact TS. We cannot, however, rule out selective destabilization of the native state by some mutations, which would lead to the accumulation of an expanded native-like intermediate in the burst phase of the unfolding reaction (Otzen and Oliveberg, 2002; Seeliger et al., 2003).
Interestingly, the distribution of φ-values qualitatively correlates with that of mU-values. Seven out of eight mU+ variants (M9A, S12G, L18A, L22A, L118V, Y133S and R135G) are in the early folding core. The expanded TS of these variants can be explained by looser packing due to substitution of a residue with a bulky side chain by one with a small side chain at critical positions where specific side chain packing in the TS occurs. Most of the mU− variants are polar to non-polar substitutions (R29A, K66A, E69A, T75A, D77A, R79A, W109I, C129A and Q131A). Here removal of electrostatic interactions and introduction of a hydrophobic side chain may lead to tighter, but non-specific packing, which results in compaction of the TS. The same argument holds for G68A and G78A substitutions, which increased side chain hydrophobicity. The L19A, V33A and I93V variants are in regions that lack specific side chain packing; these mU− substitutions mirror the effects of similar mU+ ones in the early folding core.
This deeper analysis of the effects of substitutions on denaturant-dependence of unfolding kinetics reveals intriguing aspects of the TS ensemble and its packing properties. The rate-determining TS is at once malleable, as substitutions alter its compaction/expansion, and robust, in that all of our observations can be interpreted in terms of a single self-consistent TS ensemble.
β-Barrel Closure Protects Aggregation-Prone Regions of CRABP1
While no iLBP has been implicated in a misfolding disease, CRABP1 and variants aggregate to varying extents in vitro or in E. coli expression systems, indicating that aggregation of this protein indeed competes with its folding. Multiple sequence-based aggregation prediction algorithms including Zyggregator (Tartaglia and Vendruscolo, 2008) and PASTA (Trovato et al., 2007), consistently identify several sequence stretches in the β-strands of CRABP1 as aggregation-prone (Fig. 4A). To experimentally test these predictions, we identified sequences that form the core of CRABP1 aggregates using NMR spectroscopy (Hoshino et al., 2002). Specifically, we determined the amide H/D exchange profile of in vitro-prepared aggregates of a highly aggregation-prone CRABP1 variant (F71A). The overlay of 1H-15N HSQC spectra of CRABP1 F71A aggregates dissolved in DMSO before and after four weeks of incubation in D2O is shown in Fig. 4B. Consecutive stretches including residues 51–65, which corresponds to strand 3-turn II-strand 4 with the exception of S55, residues 119–123 (strand 9), and 127–134 (strand 10) are highly protected even after four weeks of exchange (magenta bars in Fig. 4A and regions on structure in Fig. 4C). We conclude that these 27 residues constitute the core of CRABP1 aggregates. These experimentally determined core sequences agree well with the Zyggregator (Tartaglia and Vendruscolo, 2008) and PASTA (Trovato et al., 2007) predictions. It is noteworthy that strand 10 belongs to the aggregation core. This strand can be categorized as an ‘edge strand’ in terms of the architecture of CRABP1. Early barrel closure in the folding mechanism of CRABP1 would protect strand 10 by providing it a set of partner interactions, thus mitigating its vulnerability as an unpartnered ‘edge strand’ (Richardson and Richardson, 2002). Additionally, inspection of the location of the high φ residues in the CRABP1 folding mechanism and the aggregation-prone regions suggests a more general protection of these regions by the early closure of the barrel. For example, intrinsically labile strand 9 (Krishnan et al., 2000; Xiao and Kaltashov, 2005) also benefits from protection, which in this case may be provided by strand 10 once barrel closure occurs. Thus, we hypothesize that protection of aggregation-prone regions in CRABP1 by structural features and barrel closure occurs early in folding and significantly reduces its risk of aggregation. In other words, the folding pathway is driven by topology so as to avoid misfolding, rather than by a specific hydrophobic collapse, which was the previously accepted folding mechanism in this structural family. Furthermore, to the best of our knowledge, this is the first direct evidence connecting the folding and aggregation landscapes of a protein.
Figure 4. Predicted and experimentally identified aggregation-prone regions in CRABP1.

(A) Regions of CRABP1 predicted to be aggregation-prone by the Zyggregator (Tartaglia and Vendruscolo, 2008) and PASTA (Trovato et al., 2007) algorithms shown in red and cyan bars, respectively. Magenta bars indicate the actual aggregation cores determined by NMR.
(B) 1H-15N HSQC spectra of CRABP1 F71A aggregates dissolved in 95% d6-DMSO/5% D2O. Resonances in black correspond to unexchanged aggregates, while resonances in magenta correspond to aggregates that have been H/D-exchanged for four weeks. Peaks that retained significant intensity in the exchanged sample are identified.
(C) Regions of CRABP1 F71A found to be protected in the H/D exchange experiment (less than 50% drop in concentration-normalized peak intensity) are illustrated on the structure of CRABP1.
See also Figure S3.
Is Early Barrel Closure a General Feature of iLBP Folding?
Can our findings on CRABP1 be extrapolated to other iLBP family members or other protein structural classes? While data for other iLBPs are not as extensive as those available for CRABP1, there is some evidence supporting the generality of early barrel closure as a feature on iLBP folding landscapes. First, most of the residues identified in this study as parts of the folding nucleus of CRABP1 are highly conserved among iLBPs (Fig. 5A). Moreover, the majority of the highly conserved residues in iLBPs are localized to two spatially contiguous structural regions that define a β-barrel topology: the turn III-strand 5-turn IV element that connects the two sheets, and barrel-closing strands 1 and 10. In addition, the very C-terminus has been shown to be a critical topological determinant. Deletion of three C-terminal residues in rat intestinal fatty acid-binding protein (IFABP) produced a swollen molten globule with no specific aromatic tertiary interactions and little residual secondary structure (Clerico et al., 2000). Analogous deletion in CRABP1 also yields a molten globule-like non-native state (unpublished data). Substitution of the helix-turn-helix motif with a short linker turns causes complete loss of native structure in the apo-form of rabbit ileal lipid binding protein (ILBP) (Kouvatsos et al., 2007). Somewhat surprisingly, removal of the helical domain from rat IFABP is tolerated, but significantly retards refolding (Kim et al., 1996). More recent folding studies on rat IFABP suggest early formation of native-like structure around F2 and F17 (corresponding to F3 and L18 in CRABP1) (Li and Frieden, 2007), consistent with our findings on CRABP1. The folding pathway of rat IFABP deduced from equilibrium denaturation NMR experiments (Hodsdon and Frieden, 2001; Ropson and Frieden, 1992) provides additional parallels. Proposed initial folding steps for IFABP involve structure consolidation around the turn between two helices, turn III and the turn between strands 9 and 10. Similarly, a number of the main hydrophobic core residues in the back sheet were found to participate in the IFABP equilibrium intermediate observed between 5.0 and 6.0 M urea (Hodsdon and Frieden, 2001). This intermediate is likely to represent a sub-ms burst phase kinetic intermediate observed for CRABP1 and involves non-specific collapse of the main hydrophobic core. It is important to note here that perturbation analysis data report on specific side chain interactions while equilibrium denaturation NMR experiments report on the environment of amide backbones; the comparison between them may not always be straightforward (Zarrine-Afsar et al., 2012). This may explain why the authors have not observed early formation of turn IV, but did observe that of turn III in IFABP. Taken together, our results and evidence from previous studies on iLBPs strongly argue that early barrel closure is a mechanistic step likely to be shared by several iLBP family members. Prediction of aggregation-prone regions of other members of iLBP family (Fig. 5B) identifies similar regions to those predicted for CRABP1 and suggests that the early closure of the barrel could indeed be a general mechanism to increase the probability of productive folding.
Figure 5. Residues that form contacts early in the folding (pre-TS) and constitute aggregation-prone regions in CRABP1 are evolutionarily conserved in the iLBP family.
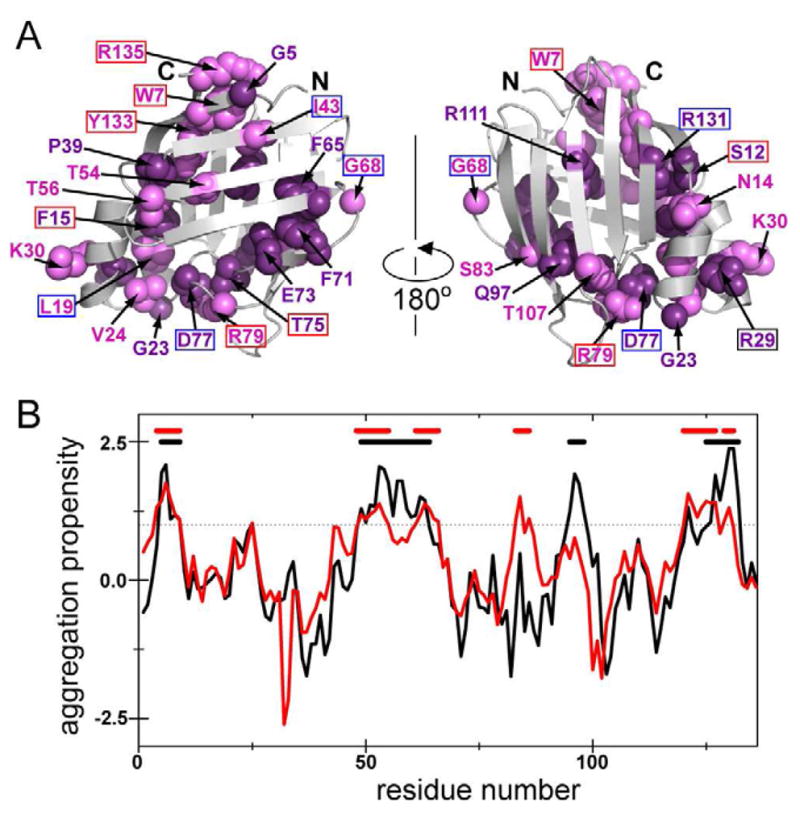
(A) Sequence conservation residues in the iLBP family. Multiple sequence alignment of iLBPs to CRABP1 was carried out using the ConSurf server (Ashkenazy et al., 2010). Residues shown in dark purple spheres on the CRABP1 structure have the highest ConSurf score (9) and those shown in light purple have the next highest (8). Residue numbering corresponds to that in the CRABP1 sequence. Boxed residues were subjected to perturbation analysis in this study, and the box color corresponds to φ-values (red for φ > 0.5 and blue for φ < 0.5).
(B) Aggregation propensity profiles for CRABP1 and iLBP family average predicted by Zyggregator (Tartaglia and Vendruscolo, 2008). The aggregation propensity profile for CRABP1 WT is shown in black, and the average iLBP aggregation profile in red. The horizontal dotted line represents the 1.0 threshold for significant aggregation propensity; continuous stretches of residues with aggregation score above the threshold are considered aggregation-prone and marked as black and red horizontal bars, respectively.
Folding and aggregation landscapes are inevitably overlapping (Clark, 2004; Hartl et al., 2011; Jahn and Radford, 2008; Vendruscolo, 2012). Proteins have evolved to avoid aggregation through the stabilization of the native state and/or destabilization of risky intermediates (Chiti and Dobson, 2009), presence of residual structure in the aggregation-prone segments in the unfolded state ensemble (Hamada et al., 2009; Routledge et al., 2009), or even functional protein-protein interactions (Masino et al., 2011). Another strategy is implied by our studies of CRABP1 where it appears that folding steps may protect aggregation-prone regions. This mechanism is likely to be general, even beyond iLBPs and other β-barrel proteins. We note a couple of examples other than the iLBPs. Cold shock proteins (Csp’s) adopt the five-stranded β-barrel OB-fold. While strands 1 and 3 of CspA are implicated in its aggregation (Alexandrescu and Rathgeb-Szabo, 1999), the TS of the related CspB has been mapped and reveals that strands 1 and 4 are involved in pre-TS non-local specific interactions (Garcia-Mira et al., 2004). Linking these two studies using the assumption that the folding mechanism of CspA resembles that of CspB suggests early folding events may act to diminish risk of aggregation in this family as well. Additionally, a surface loop in interleukin 1β has been proposed to form a hydrophobically structured microdomain (Chrunyk and Wetzel, 1993) and speculated to pack against strands 6–10 to prevent self-association (Finke et al., 2000), while terminal strands remain exposed until barrel closure late in folding (Chavez et al., 2006). There are counter examples: For example, a recent study of the FynSH3 domain elegantly described an aggregation precursor in which the carboxyl terminal β-strand 5 is unstructured (Neudecker et al., 2012). This disrupts its interactions with β-strand 1, which is an aggregation-prone region. However, based on earlier studies (Northey et al., 2002), neither of these terminal strands exhibits specific interactions in the rate-determining TS. Thus, the folding mechanism in this case does not appear to protect against aggregation via strand 1. In addition, the aggregation-prone intermediate is only sparsely populated, suggesting that there are other strategies in play to minimize aggregation. Thus, we suspect that in-depth studies of additional proteins will reveal that there are indeed multiple strategies that arise from the evolutionary pressures on folding mechanisms to suppress the likelihood of aggregation.
EXPERIMENTAL PROCEDURES
Protein design, expression and purification
A variant of murine CRABP1 with an N-terminal (His)10-tag and a stabilizing R131Q mutation (Clark et al., 1998), referred to as wild type (CRABP1 WT) here, was used as a template for mutagenesis. The single site variants were generated by site-directed mutagenesis using a QuikChange protocol (Stratagene) and confirmed by DNA sequencing. The WT and mutant proteins were expressed in E. coli strain BL21(DE3) (Novagen). In the case of variants, 30 min prior to induction, L-Pro and NaCl were added to the growing culture to a final concentration of 20 mM and 0.3 M, respectively, to improve protein solubility (Ignatova and Gierasch, 2006). Protein expression was induced with 0.4 mM IPTG, and the cells were allowed to grow for 4 h at 30 °C. All proteins were purified upon cell lysis from the soluble fraction of the cell extract by Ni-NTA (Qiagen) affinity chromatography. Protein concentration was determined using a molar extinction coefficient of ε280 = 20,970 M−1cm−1. The mutations were also validated by ESI mass-spectrometry of pure proteins.
Equilibrium denaturation
Protein samples (about 5 μM protein concentration) in 10 mM Tris-HCl pH 8.0 containing 1 mM dithiothreitol (DTT) and varying urea concentrations were equilibrated overnight (16–18 h) at 25 °C. Unfolding transitions were monitored by Trp fluorescence (excitation at 280 nm, emission at 350 nm). The data were analyzed by a two-state model using the linear extrapolation method (Pace, 1986). The equilibrium m-values (meq) for all variants were within ±10% of that of WT CRABP1. Therefore, the meq was fixed to −2.0 kcal/mol·M to reduce errors in the determination of ΔGU-N.
Unfolding kinetics
Protein samples in 10 mM Tris-HCl pH 8.0, 1 mM DTT were manually added to urea solutions in the same buffer to a final concentration of about 5 μM. Unfolding kinetics was monitored by following Trp fluorescence (excitation at 280 nm, emission at 350 nm) at 25 °C. The rate constant of unfolding (kU) was determined from data fitted to a single-exponential equation with a linear component introduced to account for photobleaching.
Refolding kinetics
Refolding kinetics was followed by Trp fluorescence (excitation at 280 nm, emission at 350 nm) using an SFM-400 stopped-flow device (BioLogic) at 25 °C. Urea-denatured protein (about 100 μM) in 20 mM Tris-HCl pH 8.0, 1 mM DTT was refolded by dilution into refolding buffer (20 mM Tris-HCl pH 8.0, 1 mM DTT containing varying urea concentrations). A cuvette with a pathlength of 0.8 mm was used. The dead time of the instrument was 2.4 ms. Kinetics traces were averaged and fit to a multi-exponential equation using Origin (OriginLab) or SigmaPlot (Systat Software).
Perturbation analysis
Perturbation analysis, also known as φ-value analysis (Fersht et al., 1992), was performed on 33 variants of CRABP1. Typically, φ-values report on the onset of specific interactions with respect to the rate-determining TS. Unfolding φ-values (φU-values) were calculated as a ratio between the energetic perturbation of the TS (ΔΔG‡-N) and the native state (ΔΔGU-N) upon mutation. Only those mutations that caused significant perturbation (ΔΔGU-N > 0.8 kcal/mol) were considered for analyses. φU-values were converted to the folding φF-values (φ) as φ = 1 − φU. Accordingly, φ = 0 was interpreted as an indication of formation of residue-specific native-like interactions after the rate-determining TS and φ = 1 as an indication of formation of those before the rate-determining TS. Note that all φ-values reported in the text are folding φF-values and referred to as φ-values for the sake of simplicity.
Preparation of CRABP1 aggregates
The15N/13C-labeled F71A variant of CRABP1 WT was overexpressed in BL21(DE3) E. coli cells, and the protein was purified from the inclusion bodies as reported previously (Clark et al., 1998) with minor modification. Cells were lysed, and the pellet fraction was dissolved in 300 mM NaCl, 50 mM NaPi pH 8.0 containing 8 M urea. The protein was applied to a Ni-NTA agarose column in 8 M urea in 300 mM NaCl, 20 mM Tris-HCl pH 8.0 and eluted using an imidazole gradient (from 50 mM to 300 mM). Aggregates were prepared by overnight dialysis of the urea-denatured protein against 150 mM NaCl, 10 mM NaPi pH 7.0, 5 mM DTT at 37 °C. Aggregates formed upon dialysis were collected by centrifugation and resuspended in D2O containing 0.025% (w/v) NaN3.
H/D exchange measurements
The regions of CRABP1 aggregates that constitute its aggregation core were identified by hydrogen-deuterium (H/D) exchange monitored by solution NMR spectroscopy as described previously (Hoshino et al., 2002). Briefly, in vitro aggregates of 15N/13C-labeled CRABP1 F71A were resuspended in D2O. After four weeks of exchange at 4 °C, aggregates were collected, lyophilized and then resuspended in d6-DMSO containing 0.1% (v/v) TFA, 50 mM DTT and 5% (v/v) D2O (pD 3.0) to a final protein concentration of 200 μM. The solution was immediately transferred to an NMR tube, and the heteronuclear single quantum coherence (HSQC) spectrum was recorded at 26 °C on a 600-MHz Bruker Avance spectrometer using a TXI cryoprobe. For unexchanged samples, aggregates were resuspended in water containing 0.025% (w/v) NaN3 and incubated at 4 °C for the same amount of time. Data were processed using NMRPipe (Delaglio et al., 1995) and CARA (Keller, 2004). Backbone assignments of CRABP1 aggregates dissolved in DMSO were obtained using a standard set of triple resonance experiments, including HNCACB, CBCA(CO)NH, HBHANH, HNCO, and HNCACO.
Sequence analysis
Conservation analysis on the iLBP family was performed using the ConSurf server (Ashkenazy et al., 2010) on 263 unique sequences out of 469 PSI-BLAST hits with murine CRABP1 as a query. For aggregation propensity analysis in the iLBP family the sequences were retrieved by ExPasy BLAST (Artimo et al., 2012) again using murine CRABP1 as a query. Data set redundancy was decreased by filtering for sequences with 30–90% identity and with no significant gaps and/or insertions. The sequences were submitted to the Zyggregator server to predict aggregation propensities for each iLBP. The aggregation propensity score was calculated for each position corresponding to the CRABP1 WT sequence and normalized by the total number of residues occurring at corresponding position according to a MAFFT alignment (Katoh and Toh, 2008).
Supplementary Material
Highlights.
The folding pathway of a β-barrel protein protects it from aggregation
The rate-determining transition state of CRABP1 is polarized and malleable
Regions constituting the aggregate core of CRABP1 are protected early in folding
Early barrel closure in iLBPs may offer a general strategy for productive folding
Acknowledgments
This work was supported by a grant from the National Institutes of Health (OD000945).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrescu AT, Rathgeb-Szabo K. An NMR investigation of solution aggregation reactions preceding the misassembly of acid-denatured cold shock protein A into fibrils. J Mol Biol. 1999;291:1191–1206. doi: 10.1006/jmbi.1999.3039. [DOI] [PubMed] [Google Scholar]
- Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, Duvaud S, Flegel V, Fortier A, Gasteiger E, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszak L, Winter N, Xu Z, Bernlohr DA, Cowan S, Jones TA. Lipid-binding proteins: a family of fatty acid and retinoid transport proteins. Adv Protein Chem. 1994;45:89–151. doi: 10.1016/s0065-3233(08)60639-7. [DOI] [PubMed] [Google Scholar]
- Bueno M, Ayuso-Tejedor S, Sancho J. Do proteins with similar folds have similar transition state structures? A diffuse transition state of the 169 residue apoflavodoxin. J Mol Biol. 2006;359:813–824. doi: 10.1016/j.jmb.2006.03.067. [DOI] [PubMed] [Google Scholar]
- Chavez LL, Gosavi S, Jennings PA, Onuchic JN. Multiple routes lead to the native state in the energy landscape of the beta-trefoil family. Proc Natl Acad Sci U S A. 2006;103:10254–10258. doi: 10.1073/pnas.0510110103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez LL, Onuchic JN, Clementi C. Quantifying the roughness on the free energy landscape: entropic bottlenecks and protein folding rates. J Am Chem Soc. 2004;126:8426–8432. doi: 10.1021/ja049510+. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Chrunyk BA, Wetzel R. Breakdown in the relationship between thermal and thermodynamic stability in an interleukin-1 beta point mutant modified in a surface loop. Protein Eng. 1993;6:733–738. doi: 10.1093/protein/6.7.733. [DOI] [PubMed] [Google Scholar]
- Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Clark PL, Liu ZP, Rizo J, Gierasch LM. Cavity formation before stable hydrogen bonding in the folding of a beta-clam protein. Nat Struct Biol. 1997;4:883–886. doi: 10.1038/nsb1197-883. [DOI] [PubMed] [Google Scholar]
- Clark PL, Weston BF, Gierasch LM. Probing the folding pathway of a beta-clam protein with single-tryptophan constructs. Fold Des. 1998;3:401–412. doi: 10.1016/s1359-0278(98)00053-4. [DOI] [PubMed] [Google Scholar]
- Clerico EM, Peisajovich SG, Ceolin M, Ghiringhelli PD, Ermacora MR. Engineering a compact non-native state of intestinal fatty acid-binding protein. Biochim Biophys Acta. 2000;1476:203–218. doi: 10.1016/s0167-4838(99)00247-2. [DOI] [PubMed] [Google Scholar]
- Connell KB, Miller EJ, Marqusee S. The folding trajectory of RNase H is dominated by its topology and not local stability: a protein engineering study of variants that fold via two-state and three-state mechanisms. J Mol Biol. 2009;391:450–460. doi: 10.1016/j.jmb.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota E, Steward A, Fowler SB, Clarke J. The folding nucleus of a fibronectin type III domain is composed of core residues of the immunoglobulin-like fold. J Mol Biol. 2001;305:1185–1194. doi: 10.1006/jmbi.2000.4378. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Eyles SJ, Gierasch LM. Multiple roles of prolyl residues in structure and folding. J Mol Biol. 2000;301:737–747. doi: 10.1006/jmbi.2000.4002. [DOI] [PubMed] [Google Scholar]
- Fersht AR, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J Mol Biol. 1992;224:771–782. doi: 10.1016/0022-2836(92)90561-w. [DOI] [PubMed] [Google Scholar]
- Finke JM, Roy M, Zimm BH, Jennings PA. Aggregation events occur prior to stable intermediate formation during refolding of interleukin 1beta. Biochemistry. 2000;39:575–583. doi: 10.1021/bi991518m. [DOI] [PubMed] [Google Scholar]
- Fowler SB, Clarke J. Mapping the folding pathway of an immunoglobulin domain: structural detail from Phi value analysis and movement of the transition state. Structure. 2001;9:355–366. doi: 10.1016/s0969-2126(01)00596-2. [DOI] [PubMed] [Google Scholar]
- Garcia-Mira MM, Boehringer D, Schmid FX. The folding transition state of the cold shock protein is strongly polarized. J Mol Biol. 2004;339:555–569. doi: 10.1016/j.jmb.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Gunasekaran K, Hagler AT, Gierasch LM. Sequence and structural analysis of cellular retinoic acid-binding proteins reveals a network of conserved hydrophobic interactions. Proteins. 2004;54:179–194. doi: 10.1002/prot.10520. [DOI] [PubMed] [Google Scholar]
- Hamada D, Tanaka T, Tartaglia GG, Pawar A, Vendruscolo M, Kawamura M, Tamura A, Tanaka N, Dobson CM. Competition between folding, native-state dimerisation and amyloid aggregation in beta-lactoglobulin. J Mol Biol. 2009;386:878–890. doi: 10.1016/j.jmb.2008.12.038. [DOI] [PubMed] [Google Scholar]
- Hamill SJ, Steward A, Clarke J. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J Mol Biol. 2000;297:165–178. doi: 10.1006/jmbi.2000.3517. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hodsdon ME, Frieden C. Intestinal fatty acid binding protein: the folding mechanism as determined by NMR studies. Biochemistry. 2001;40:732–742. doi: 10.1021/bi001518i. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Katou H, Hagihara Y, Hasegawa K, Naiki H, Goto Y. Mapping the core of the beta(2)-microglobulin amyloid fibril by H/D exchange. Nat Struct Biol. 2002;9:332–336. doi: 10.1038/nsb792. [DOI] [PubMed] [Google Scholar]
- Huysmans GH, Baldwin SA, Brockwell DJ, Radford SE. The transition state for folding of an outer membrane protein. Proc Natl Acad Sci U S A. 2010;107:4099–4104. doi: 10.1073/pnas.0911904107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova Z, Gierasch LM. Inhibition of protein aggregation in vitro and in vivo by a natural osmoprotectant. Proc Natl Acad Sci U S A. 2006;103:13357–13361. doi: 10.1073/pnas.0603772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Keller R. Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. Zurich: Swiss Federal Institute of Technology; 2004. [Google Scholar]
- Kim K, Cistola DP, Frieden C. Intestinal fatty acid-binding protein: the structure and stability of a helix-less variant. Biochemistry. 1996;35:7553–7558. doi: 10.1021/bi9529115. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Bergfors T, Senn H, Le Motte P, Gsell B, Shudo K, Jones TA. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans-retinoic acid and a synthetic retinoid. Structure. 1994;2:1241–1258. doi: 10.1016/s0969-2126(94)00125-1. [DOI] [PubMed] [Google Scholar]
- Kouvatsos N, Thurston V, Ball K, Oldham NJ, Thomas NR, Searle MS. Bile acid interactions with rabbit ileal lipid binding protein and an engineered helixless variant reveal novel ligand binding properties of a versatile beta-clam shell protein scaffold. J Mol Biol. 2007;371:1365–1377. doi: 10.1016/j.jmb.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Krishnan VV, Sukumar M, Gierasch LM, Cosman M. Dynamics of cellular retinoic acid binding protein I on multiple time scales with implications for ligand binding. Biochemistry. 2000;39:9119–9129. doi: 10.1021/bi000296l. [DOI] [PubMed] [Google Scholar]
- Li H, Frieden C. Observation of sequential steps in the folding of intestinal fatty acid binding protein using a slow folding mutant and 19F NMR. Proc Natl Acad Sci U S A. 2007;104:11993–11998. doi: 10.1073/pnas.0705253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MO, Oliveberg M. Malleability of protein folding pathways: a simple reason for complex behaviour. Curr Opin Struct Biol. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Marcelino AM, Smock RG, Gierasch LM. Evolutionary coupling of structural and functional sequence information in the intracellular lipid-binding protein family. Proteins. 2006;63:373–384. doi: 10.1002/prot.20860. [DOI] [PubMed] [Google Scholar]
- Masino L, Nicastro G, Calder L, Vendruscolo M, Pastore A. Functional interactions as a survival strategy against abnormal aggregation. FASEB J. 2011;25:45–54. doi: 10.1096/fj.10-161208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouschek A, Fersht AR. Application of physical organic chemistry to engineered mutants of proteins: Hammond postulate behavior in the transition state of protein folding. Proc Natl Acad Sci U S A. 1993;90:7814–7818. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundstrom P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, Kay LE. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336:362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- Nikiforovich GV, Frieden C. The search for local native-like nucleation centers in the unfolded state of beta -sheet proteins. Proc Natl Acad Sci U S A. 2002;99:10388–10393. doi: 10.1073/pnas.162362199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northey JG, Di Nardo AA, Davidson AR. Hydrophobic core packing in the SH3 domain folding transition state. Nat Struct Biol. 2002;9:126–130. doi: 10.1038/nsb748. [DOI] [PubMed] [Google Scholar]
- Otzen DE, Oliveberg M. Conformational plasticity in folding of the split beta-alpha-beta protein S6: evidence for burst-phase disruption of the native state. J Mol Biol. 2002;317:613–627. doi: 10.1006/jmbi.2002.5423. [DOI] [PubMed] [Google Scholar]
- Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropson IJ, Frieden C. Dynamic NMR spectral analysis and protein folding: identification of a highly populated folding intermediate of rat intestinal fatty acid-binding protein by 19F NMR. Proc Natl Acad Sci U S A. 1992;89:7222–7226. doi: 10.1073/pnas.89.15.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotondi KS, Gierasch LM. Local sequence information in cellular retinoic acid-binding protein I: specific residue roles in beta-turns. Biopolymers. 2003a;71:638–651. doi: 10.1002/bip.10592. [DOI] [PubMed] [Google Scholar]
- Rotondi KS, Gierasch LM. Role of local sequence in the folding of cellular retinoic abinding protein I: structural propensities of reverse turns. Biochemistry. 2003b;42:7976–7985. doi: 10.1021/bi034304k. [DOI] [PubMed] [Google Scholar]
- Routledge KE, Tartaglia GG, Platt GW, Vendruscolo M, Radford SE. Competition between intramolecular and intermolecular interactions in an amyloid-forming protein. J Mol Biol. 2009;389:776–786. doi: 10.1016/j.jmb.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeliger MA, Breward SE, Itzhaki LS. Weak cooperativity in the core causes a switch in folding mechanism between two proteins of the cks family. J Mol Biol. 2003;325:189–199. doi: 10.1016/s0022-2836(02)01202-0. [DOI] [PubMed] [Google Scholar]
- Sukumar M, Gierasch LM. Local interactions in a Schellman motif dictate interhelical arrangement in a protein fragment. Fold Des. 1997;2:211–222. doi: 10.1016/S1359-0278(97)00030-8. [DOI] [PubMed] [Google Scholar]
- Tartaglia GG, Vendruscolo M. The Zyggregator method for predicting protein aggregation propensities. Chem Soc Rev. 2008;37:1395–1401. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- Trovato A, Seno F, Tosatto SC. The PASTA server for protein aggregation prediction. Protein Eng Des Sel. 2007;20:521–523. doi: 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- Vendruscolo M. Proteome folding and aggregation. Curr Opin Struct Biol. 2012;22:138–143. doi: 10.1016/j.sbi.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Xiao H, Kaltashov IA. Transient structural disorder as a facilitator of protein-ligand binding: native H/D exchange-mass spectrometry study of cellular retinoic acid binding protein I. J Am Soc Mass Spectrom. 2005;16:869–879. doi: 10.1016/j.jasms.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Zarrine-Afsar A, Dahesh S, Davidson AR. A residue in helical conformation in the native state adopts a beta-strand conformation in the folding transition state despite its high and canonical Phi-value. Proteins. 2012;80:1343–1349. doi: 10.1002/prot.24030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.