

Abstract
Adenomatous Polyposis Coli (APC) is best known for its crucial role in colorectal cancer suppression. Rodent models with various Apc mutations have enabled experimental validation of different Apc functions in tumors and normal tissues. Since the development of the first mouse model with a germline Apc mutation in the early 1990s, twenty other Apc mouse and rat models have been generated. This article compares and contrasts currently available Apc rodent models with particular emphasis on providing potential explanations for their reported variation in three areas: 1) intestinal polyp multiplicity, 2) intestinal polyp distribution, and 3) extra intestinal phenotypes.
Introduction
Tumor suppressor Adenomatous polyposis coli (APC) is critical for maintaining cellular homeostasis in the intestine (1, 2). APC is a large (2843 amino acids), multi-domain protein that has been implicated in many cellular functions including cellular proliferation, differentiation, cytoskeleton regulation, migration and apoptosis (3). Mechanistically, APC is best known for its ability to antagonize Wnt signaling by targeting the oncoprotein β-catenin for proteasomal degradation (4).
Acquiring a somatic APC mutation is an early, if not initiating event in the great majority of colorectal tumors (5). Inheriting a germline APC mutation results in the development of hundreds to thousands of colonic polyps, a condition termed familial adenomatous polyposis (FAP). These precancerous polyps are thought to initiate following a somatic mutation in the wild-type APC allele (6, 7). To avoid the progression of these polyps into invasive carcinoma, prophylactic colon removal is recommended for FAP (8). There are no reports of humans with germline mutation of both APC alleles, consistent with early developmental lethality associated with complete loss of APC function (9–11). Germline and somatic APC mutations typically result in premature APC protein truncation and group between codons 1250 and 1464, a region termed the “mutation cluster region”, MCR (12).
A meta-analysis of genotype-phenotype correlation in FAP patients showed that germline mutations in the MCR result in the most severe intestinal polyposis phenotype, with up to 5000 polyps (13). Mutations on either side of the MCR are associated with an intermediate intestinal polyposis phenotype, while mutations that result in a truncation in APC after amino acid (a.a.) 1595 or before a.a. 157 are associated with an attenuated phenotype (AFAP), characterized by development of only a few polyps (13). Complete deletion of APC has been reported only rarely and results in an intermediate phenotype (14, 15).
Over two-thirds of FAP patients also have extra-colonic manifestations (13). Chronic hypertrophy of retinal pigment epithelium (CHRPE) is the most frequent phenotype, associated with APC truncation between a.a. 311–1446. Desmoid tumors, on the other hand, are associated with APC truncations 3′ to the MCR, after a.a. 1400. Duodenal and gastric tumors have been associated with APC mutations in two different regions, downstream of codon 1395 and between codons 564–1465 (13). It is important to note that these genotype-phenotype correlations are not rigid or complete, suggesting roles for other genetic and environmental factors in tumor development (13, 16).
For the past two decades, rodent models have been valuable for analysis of APC functions in intestinal homeostasis and tumor suppression (17, 18). APC is well-conserved between human and rodent, with 92% similarity at the amino acid level (9, 19). Furthermore, some rodent models with germline Apc mutations that result in Apc protein truncation develop intestinal polyposis similar to that seen in FAP patients (18). A brief summary of all published rodent models with germline Apc mutations appears in Tables 1–3, with a schematic provided in figure 1.
Table 1.
Summary of rodent models with germline Apc mutations before MCR *#
| Model (Ref) | Apc mutation | Intestinal phenotype | Polyp distribution | Extra-intestinal phenotype |
|---|---|---|---|---|
| ApcΔe1-15/+ (34) |
|
|
|
|
| ApcΔ242/+ (55) |
|
|
|
|
| ApcΔ474/+ (86) |
|
|
|
|
| ApcΔ580/+ (75) |
|
|
|
|
| ApcΔ14/+ (86) |
|
|
|
|
| Apc580D/+ (93) |
|
|
|
|
| ApcΔ15/+ (76) |
|
|
|
|
| ApcΔ716/+ (10, 40) |
|
|
|
|
| ApcMin/+ (19, 67, 79) |
|
|
|
|
| PIRC rat (9, 94) |
|
|
|
|
Table 3.
Summary of mouse models with other germline Apc mutations *
| Model (Ref) | Apc mutation | Intestinal phenotype | Polyp distribution | Extra-intestinal phenotype |
|---|---|---|---|---|
| ApcmNLS/mNLS (39) |
|
|
N/A | NR |
| ApcΔSAMP (65) |
|
|
|
|
| ApcNeoR and ApcNeoF (36, 37) |
|
|
|
|
| ApcΔ716/+/+ (98) |
|
|
NR |
|
| ApcΔ716/Δ716/+ (98) |
|
|
|
|
Apc mouse models reported in this table are on C57Bl/6 background however, with different backcross isogenicity from N2 to > N20
All models are mouse models
NR: not reported
NeoR: Neomycin resistance gene
Figure 1. Sites of Apc mutations in different Apc mouse models relative to Apc domains.
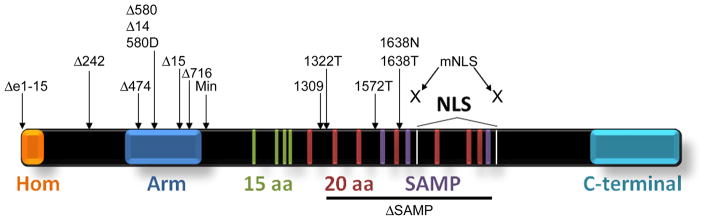
Domains of Apc are indicated as follows: Hom = homodimerization, Arm = Armadillo repeats, 15 aa = 15 amino acid repeats, 20 aa = 20 amino acid repeats, Serine-Alanine-Methionine-Proline (SAMP)= axin binding, NLS=nuclear localization signals, and C-terminal includes microtubule, EB1 and PDZ binding domains. The Mutation Cluster Region (MCR) is between codons 1250 and 1464.
Characterization of the many available Apc mouse and rat models has aided in discovery of various pathways important in colon carcinogenesis. Apc rodent models were also useful for elucidating the effect of various environmental and genetic factors on intestinal tumorigenesis, and for testing potential chemoprevention and therapeutic agents. The many positive contributions of Apc mouse models have been reviewed previously (20, 21). As with most experimental systems, studies of the Apc models have also led to unanswered questions, particularly regarding phenotypic variation among the different models. Here we review some of these variations, provide potential explanations, and pose challenges for future investigation.
I- Variation in intestinal polyp multiplicity
As shown in table 1, the average number of polyps varies greatly between different mouse models with germline Apc mutations. In addition, the number of polyps also varies in the same Apc mouse model maintained in different laboratories (17). These variations in intestinal polyp number in different models likely stem from the nature of the Apc mutations as well as environmental and genetic factors (17, 18). We propose that the number of intestinal tumors that develop in different Apc models and in the same model analyzed by different laboratories is influenced by one or more of the following factors:
1- Different rates and mechanisms of wildtype Apc allele loss (e.g. LOH, mutation of wildtype Apc, gene silencing)
In both FAP patients and rodent models with germline Apc mutations, loss or inactivation of the wildtype APC/Apc allele is required for polyp formation (22, 23). The mechanism by which the second wildtype Apc allele is lost appears to depend on the Apc mouse model (24). Because this second Apc “hit” is essential for polyp initiation (10, 22, 25), the rate at which second “hit” occurs will directly affect the number of intestinal polyps. Increasing the expected rate of these second “hits” through introduction of genomic instability, X-ray exposure, or injection with a mutagen significantly increases the number of polyps in ApcMin/+ and Apc1638N mice (26–30). It has been suggested that certain Apc mutations might lead to chromosomal instability, which could affect the rate of wildtype Apc loss (31).
Apc1638N/+ mice develop relatively few intestinal polyps and the second Apc “hit” is usually inactivation of the wildtype Apc allele, predicted to be a rare event (24). On the other hand, ApcMin/+ mice, where the wildtype Apc allele is lost by means of a more frequent LOH event, develop considerably more polyps (24). Loss of the wildtype Apc allele in both ApcMin/+ and Apc1322T/+ mice, however, is reported to occur via LOH, yet these two mouse models have widely different polyp numbers (32). Although the rate and underlying mechanism of wildtype Apc allele loss might contribute to intestinal polyp numbers in Apc mouse models, it is unlikely that these are sole defining parameters.
2- Different rates of polyp growth due to differences in Wnt signaling
Polyps must reach a certain size to be detectable. If two polyps are initiated at the same time, a more rapidly growing polyp should be detectable earlier than a slower growing polyp. The most recognized function of Apc is to antagonize the Wnt signaling pathway through inhibition of β-catenin’s activity as a transcription co-factor (4). As Wnt signaling can drive cellular proliferation, we might expect that different Apc mutations would lead to different levels of Wnt signal activation and different corresponding changes in cellular proliferation. In FAP patients, mutations in the MCR are associated with the most severe intestinal phenotypes while mutations outside the MCR lead to reduced polyp multiplicity (13). Notably, APC mutations 5′ and 3′ to the MCR result in higher and lower activation of Wnt signaling, respectively (33). This observation has led to the proposal that submaximal upregulation of Wnt signaling promotes more polyp growth than higher or lower elevation of Wnt signaling; the “just right” hypothesis (34, 35).
Wnt signaling has been assessed in many Apc mouse models. Some models have high polyp multiplicity and show elevated Wnt signaling in these polyps (ApcMin/+, Apc Δ716/+, Apc1322T/+ and ApcΔe1-15/+) (10, 34, 35). Wnt signaling is also elevated in the few polyps that develop in ApcNeoR/+ and ApcNeoF/+ mice (36, 37). ApcmNLS/mNLS mice have elevated Wnt signaling in intestinal epithelial cells (38, 39). Apc1572T/1572T embryonic stem cells also have elevated Wnt signaling (38, 39). Neither ApcmNLS/mNLS nor Apc1572T/+ mice develop intestinal polyps (38, 39).
The “just right” hypothesis is supported by reports of increased polyp multiplicity in Apc1322T/+ and ApcΔe1-15/+ mice relative to ApcMin/+ mice (34, 35). Compared to ApcMin, Apc1322T protein retains one 20 a.a. repeat which can bind to β-catenin and decrease Wnt signaling (34, 35). The ApcΔe1-15 allele results in complete deletion of Apc and polyps in ApcΔe1-15/+ mice also display less Wnt signaling than polyps in ApcMin/+ mice (34). However, the “just right” hypothesis does not readily explain why Apc Δ716/+ mice show higher activation of Wnt signaling and more polyps than ApcMin/+ mice (40). In addition, several groups have reported that although loss of both Apc alleles is required to activate Wnt signaling (as assessed by nuclear translocation of β-catenin), this Apc loss is not sufficient for full Wnt signal activation (11, 41, 42). To establish the extent to which Wnt signaling and polyp growth contribute to phenotypic variation, Wnt signaling activities and proliferation rates must be directly compared in different Apc mouse models.
3- Different abilities to evade growth inhibitory effects
Another explanation of variation in polyp number among different Apc mouse models is negative selection of particular Apc genotypes. This negative selection could contribute to the “Just right” hypothesis. Support for negative selection contributing to polyp phenotypes is provided by the observation that addition of Cdx2 or BubR1 mutations to ApcΔ716/+ or ApcMin/+ mice, respectively, results in reduced polyp multiplicity and increased apoptotic indices in the small intestines, despite the increased proliferation index in these cells (43, 44). Similarly, induction of a conditional Apc mutation in hematopoietic stem cells results in upregulation of Wnt signaling and increased stem cell proliferation with increased apoptosis and eventual exhaustion of the stem cell population (45). If this phenotype holds true for intestinal tissues, the “just right” hypothesis might explain the increased stem cell number in polyps from Apc1322T/+ mice relative to those from ApcMin/+, despite lower Wnt signaling in polyps from the former model relative to those from ApcMin/+ mice.
4- Distinctive effects on differentiation
It is possible that the effect of Apc genotypes on enterocyte differentiation contributes to differences in intestinal polyp number. For instance, compared to ApcMin/+ mice, Apc1322T/+ mice have a higher proportion of Paneth cells and cells that express stem cell markers (Lgr5, Bmi1, Msi1 and CD44), not only in adenomas but also in apparently normal intestinal epithelial cells (35). Cell fates that result from different Apc genotypes might alter tumor initiation or growth. Again, Wnt signaling is one of several factors proposed to affect differentiation.
5- Contributions of genetic modifiers or environmental factors
It is well established that genetic and environmental factors affect intestinal polyp multiplicity in Apc mouse models. Polyp multiplicity in ApcMin/+ mice varies greatly between laboratories (20–100/mouse) (17, 18). This inconsistency might result from variations in diet, emergence of genetic modifiers, and even from different methods of polyp detection. A genetic modifier is a genetic locus that modifies the effect produced by a non-allelic locus. Modifier genes are present in different mouse strains and can even emerge in what is considered a congenic strain (46). Several modifier loci have been found to affect intestinal polyposis in ApcMin/+ mice and are named modifier of min (Mom) (reviewed in (18)). Some modifiers are single genes, others are thought to represent contiguous genes, and some remain less well-defined (47). The modifiers appear to function as recessive, dominant or semi-dominant loci (17). The first identified modifier gene, Mom-1 (Pla2g2a), works in a cell-non-autonomous manner, possibly by reducing inflammatory response in the gut (48–50). The Mom-2 (Atp5a1) allele is on the same chromosome as Apc (chromosome 18) and appears to inhibit loss of the wild-type Apc allele (48, 51). The mechanisms of action of other modifiers such as Mom-3, Mom-7, Mom-12 and Mom-13 are not understood (52–54).
Though identified in ApcMin/+ mice, Mom genes likely also affect phenotypes of other Apc mouse models. For instance, the C3H/HeJ mouse strain carries at least one Mom allele that is absent from the C57BL/6 strain, Mom-1 (48). Both ApcMin/+ and ApcΔ242/+ mice show reduced polyp multiplicity in the first generation mixed C57BL/6: C3H/HeJ mice compared to in C57BL/6 mice (55). At present, there appears to be no direct examination of the effect of specific modifiers of Min on different Apc mouse models.
Environmental factors, such as intestinal flora, might also contribute to phenotypic variation (56). While intestinal flora appear to increase the number of polyps in ApcMin/+ mice (57), ApcΔ14/+ mice raised in pathogen-free conditions showed significant increases in intestinal polyp number (58).
Diet is another major environmental factor that clearly impacts the mouse phenotype (59–61). Although typically defined, the concentration of various vitamins, fiber, and total fat varies greatly between laboratory mouse diets. In our own experience, switching the mouse diet had a dramatic effect on polyp multiplicity in our ApcMin/+ mouse colony. We found that the polyp burden per mouse significantly increased from 45.9±4.5 in 10 ApcMin/+ mice on Lab diet 5001 (Purina) to 81±9.3 in 25 age-matched ApcMin/+ mice on Harlan 2018 diet (p= 0.0006). Notably, the new diet (Harlan 2018) has a 24% increase in fat and decreased fiber, vitamin D, and folic acid by 42%, 67%, and 44%, respectively. Unfortunately, these inter-laboratory variables such as diet confound direct comparison of the phenotypes of Apc mouse models studied in different laboratories.
6- Differences in cellular migration and adhesion
APC interaction with cytoskeletal components, including actin filaments and microtubules, is thought to affect cell adhesion and migration (62, 63). Decreased cellular adhesion and migration in cells with APC mutations is expected to contribute to tumor formation (64). APC interacts with cytoskeletal proteins through its C-terminal region, which is absent in Apc from most mouse models (figure 1). Adding the C-terminal Apc region to Apc1322T (as in ApcΔSAMP mice) did not change the phenotype (65). However, it is possible that cytoskeletal alterations affect later stages of tumor progression such as invasion and metastasis, which do not occur in most Apc mouse models (66). Currently, evidence supporting a direct role of the Apc C-terminus in intestinal phenotype variation among different Apc mouse models is lacking.
7-Differences in technologies used to generate the mouse model
Apc rodent models have been generated using 3 different technologies; chemical mutagenesis screen, insertion of an antibiotic-resistance gene and Cre-lox induced DNA excision. The ApcMin/+ mouse, PIRC rat and KAD rat were generated by chemical mutagenesis which resulted in a single base-pair change in the Apc gene (9, 67, 68). Many other models, such as Apc1309, Apc1638N and Apc1638T, were generated through insertion of an antibiotic-resistance gene into the Apc gene, thus introducing a nonsense mutation (69–71). In ApcneoF and ApcneoR alleles, the antibiotic-resistance gene disrupts an enhancer sequence in intron 13 (36, 37). Other mouse models with Apc truncation including Apc1322T/+ and ApcΔe1-15 were generated using Cre-lox mediated-deletion of specific Apc regions. The later technology allowed removal of most exogenous DNA sequences originating from the targeting vector including the antibiotic-resistance gene. The ApcmNLS model contains mutations “knocked into” the Apc gene, with the antibiotic-resistance gene subsequently removed by Cre-Lox-mediated deletion (39).
The Apc1638N/+ and Apc1638T/+ models, which differ only by orientation of the inserted neomycin-resistance gene, provide clear evidence for the contribution of extraneous DNA to phenotypic variation (69). Apc1638N/+ mice express so little truncated Apc protein that they might be considered virtually null (69, 72); yet the described phenotype of Apc1638N/+ mice is not similar to that of the ApcΔe1-15 model, which has a complete deletion of the Apc gene (34, 72). The neomycin-resistance gene clearly affects the phenotypes of these mice and if inserted in reverse orientation, might affect not only Apc expression, but also expression of genes upstream of Apc. It is possible that the 6-fold difference in intestinal polyp number between Apc1322T/+ and Apc1309/+ mice, which differ by only 13 amino acids, stems from the different technology used in their generation; Cre-lox-mediated deletion in Apc1322T/+ versus insertion of an antibiotic resistance cassette in Apc1309/+. However, other genetic and environmental factors may contribute to the variation between these two mouse models as well (32, 70). A final illustration of the challenges in generation of Apc mouse models is the ApcΔ474/+ mice, which have a duplication of Apc exons 7–10. This feature complicates dissection of the contribution of exon duplication to the phenotype (73).
8-Differences in expression of the mutant allele
When analyzing the phenotypes of different Apc mouse models, another consideration is the level of expression of the mutant allele. Apc is a large multi-domain protein. Truncations of Apc in most FAP patients and rodent models leave N-terminal domains intact, figure 1. Although normal expression levels of truncated Apc protein have been verified in ApcΔ716, ApcMin/+, Apc1322T, and Apc1638T mice, this is not universally the case (32, 69, 74). In Apc580D, ApcΔ14, ApcΔ474, and ApcΔ242 models, the truncating mutation occurs before the final exon (15), and thus there is the possibility of a nonsense-mediated RNA decay. Truncated Apc was not detected in intestinal polyps from ApcΔ580/+ mice or ES cells from ApcΔ15/+ mice (75, 76), which suggests that these alleles might also be virtually null. A related consideration is the effect of the introduced mutation (and possibly the antibiotic selection cassette) on Apc folding. Although most of Apc is thought to be natively unfolded (77), the effects of mutations on inherently folded domains of Apc, and the consequences of potential folding defects in relation to phenotype, are not understood.
II- Variation in polyp distribution
Tumors in most Apc mouse models occur mainly in the small intestine, while germline mutations of APC in humans result in tumors predominantly in the large intestine (21, 78). The PIRC Apc rat model has tumors in both small and large intestines (9, 13, 79). A pig model with germline Apc mutations was recently reported to develop polyps in the colon (80). In addition to this inter-species variation, mouse models with different germline Apc mutations show different distributions of intestinal polyps. Analysis of ApcMin/+ mice with different genetic backgrounds has led to the hypothesis that polyp distribution is somehow linked to the mechanism by which the wildtype Apc allele is lost (24). Haigis et al. showed that in a B6 background, ApcMin/+ mice develop polyps mainly in the distal half of the small intestine, and loss of the wildtype Apc allele occurs by means of LOH. In an AKR background, ApcMin/+ mice develop polyps predominantly at the ileo-cecal junction, and inactivation of the wildtype Apc allele is achieved through allelic silencing. In the B6 background, ApcMin/+ mice with additional mutations that inactivate the mismatch repair gene Mlh develop polyps all over the small intestine, and loss of the wildtype Apc allele is achieved through a point mutation. Apc1638N/+ mice develop polyps in a similar distribution, and appear to retain the wildtype Apc allele (24).
Mechanistically, two models have been proposed to explain the connection between polyp distribution and loss of the wildtype Apc allele. In the first model, the molecular machinery in different intestinal regions determines the mechanism of the second Apc “hit” and hence the distribution of polyps. This model is supported by the finding that mice in which the wildtype Apc allele is inactivated by the same mechanism (eg. ApcMin/+/Mlh−/−, and Apc1638N/+) have similar polyp distributions (24). However, the finding that both Apc1322T/+ and ApcMin/+ mice lose the wildtype Apc allele through LOH, yet have different polyp distributions, does not support this model. A second model proposes that polyp growth is dictated by the Apc status but also by the particular environment of the different intestinal regions, independent of the mechanism of the second Apc mutation. Supporting this hypothesis, ApcΔ716/+ mice with an additional mutation of Cdx2 exhibit more colonic and fewer small intestinal polyps. Yet, loss of the wildtype Apc allele occurs via LOH regardless of Cdx2 status (44). Similarly, a colonic shift of polyps has been described in ApcMin/+ mice with an additional BubR1 mutation, although the mechanism of loss of the wildtype Apc allele in these mice was not reported (43). Mutation of both Cdx2 and BubR1 increases chromosomal instability and changes the proliferation and apoptotic indices in intestines of ApcΔ714/+ and ApcMin/+ mice, respectively (43, 44). Further support for the second model comes from ApcMin/+ mice in a 129/Sv background, where additional mutations that inactivate Smad3 result in increased colonic tumors; yet in both cases, loss of the wildtype Apc allele is achieved through LOH (81). Finally, PPARγ agonists increase colonic but not small intestinal tumors in ApcMin/+ mice (82, 83). PPARγ is expressed in higher quantities in the colon and cecum relative to the small intestine, that might account for this differential effect (83).
An expansion of the “just right” hypothesis has been proposed to explain the variation in polyp distribution among FAP patients, ApcMin/+ and Apc1322T/+ mice. The basal level of Wnt signaling is not the same in different intestinal regions. It was proposed that changes in Wnt signaling that result from specific Apc mutations cause optimal Wnt signaling for polyp growth only in certain intestinal regions. On the other hand, in other intestinal regions, these same Apc mutations will result in a higher or lower Wnt signaling level than what is optimal for tumor growth (84).
Perhaps some of these mechanisms can be clarified by studying ApcMin-FCCC mice which were generated by mating C57Bl/6J ApcMin/+ males with Apc+/+ females from an independent colony of C57Bl/6 mice maintained at Fox Chase Cancer Center. ApcMin-FCCC/+ mice develop more colon polyps than do ApcMin/+ mice, but the molecular basis behind this polyp shift has not been determined (85). Further clarification of the underlying mechanisms that control polyp distribution might also be achieved through careful analysis of ApcΔ14/+ and Apc580D/+ mice, which carry similar mutations (truncating the Apc protein at amino acid 580) but appear to have different polyp distributions. ApcΔ14/+ mice develop more colonic polyps than do ApcMin/+ mice. Apc580D/+ mice develop a similar number of colonic polyps as ApcMin/+ mice, although direct comparison of Apc580D/+ and either ApcΔ14/+ or ApcMin/+ mice has not been reported (75, 86).
III- Variation in extra-intestinal phenotypes
Although best known for its role to suppress colorectal tumorigenesis, APC mutations have been seen in other tumors including breast and liver carcinomas (4). In addition, both FAP patients and rodent models with germline Apc mutations develop extra-intestinal phenotypes (see table 1). As with the intestinal phenotype, the underlying mechanism for variation in extra-intestinal phenotypes between FAP patients and Apc rodent models as well as among different Apc rodent models is not completely understood. FAP patients have increased susceptibility to hepatic, pancreatic, thyroid and brain tumors. They also develop desmoid tumors, dental anomalies, and congenital hypertrophy of retinal pigment epithelium. It is important to note that the penetrance of these extra-intestinal phenotypes is variable in FAP patients (16, 87). The basis behind this variation is not completely understood, although it seems to correlate with the APC germline as well as the acquired somatic mutations. (16, 33).
Apc rodent models also develop some of these extra-intestinal manifestations, for example, Apc1638N/+ mice develop desmoid tumors (72) and PIRC rats show mandibular osteoma (9). Other phenotypes described in FAP patients have not been reported for Apc rodent models. The short life span of most Apc rodent models could prevent the full expression of some of these phenotypes. On the other hand, Apc rodent models manifest some other extra-intestinal phenotypes that have not been described in FAP patients (table 1). For example, many mouse models with germline Apc mutations develop mammary tumors. Although APC mutations and promoter methylation have been found in up to 70% of sporadic human breast cancers, FAP patients do not appear at an increased risk for breast tumors (88–90). In addition, adenoacanthoma is a common type of mammary tumor that develops in Apc mouse models but it has not been reported in humans (91). Other extra-intestinal phenotypes described in Apc rodent models include; splenomegaly, abnormal hematopoiesis, changes in the serum lipid profile, gonadal changes, cutaneous cysts, and thyroid abnormalities. Differences in physiology, life span and genetic content between human, mouse and rat could be underlying causes.
Among different Apc mouse models, some extra-intestinal phenotypes, such as anemia and splenomegaly, seem to correlate with the severity of intestinal polyposis. In contrast, mammary gland tumors in Apc mouse models appear to correlate with the severity of polyposis in only a few cases, such as in the ApcMin/+ and ApcΔ474/+ models. Very few ApcMin/+ mice develop mammary tumors, whereas ApcΔ474/+ mice develop mammary tumors at a rate that is almost double that seen in ApcMin/+ mice (73, 91).. In contrast, there are no reports of mammary tumor development in Apc mouse models with the most severe intestinal polyposis (ApcΔ714, Apc1322T, and ApcΔSAMP) (32, 40, 65). Perhaps mice with severe polyposis die too early, before mammary tumors have a chance to develop. Apc1572T/+ mice, which develop no intestinal polyps, have a fully-penetrant mammary tumor phenotype in females. K14-cre-ApcCKO/+ mice are a conditional model in which the ApcΔ580 allele is expressed only in ectoderm-derived tissues including the mammary gland (75, 92). Mammary tumors from these mice have mutations in the wildtype Apc allele that cluster around codon 1530 consistent with the requirement of an optimal level of Wnt signaling for mammary tumorigenesis (38). It is likely that some of the genetic and environmental factors previously described also account for the variability in extra-intestinal phenotypes among different Apc rodent models.
Conclusions and future directions
APC research has benefitted greatly from different rodent models with germline Apc mutations. However genotype/phenotype correlation of these different models is confounded by many genetic and environmental factors. Use of standardized genetic backgrounds and environmental conditions in different laboratories should enable reliable genotype/phenotype analysis of these animals. This standardization will also shed light on the role of different Apc mutations in tumorigenesis. When possible, a direct comparative analysis of different models in the same laboratory will illuminate the contribution of many factors described in this review to phenotypic variation in rodent models with germline Apc mutations.
Table 2.
| Model (Ref) | Apc mutation | Intestinal phenotype | Polyp distribution | Extra-intestinal phenotype |
|---|---|---|---|---|
| Apc1309/+ (70, 95, 96) |
|
|
|
|
| Apc1322T/+ (32, 35) |
|
|
|
|
| Apc1572T/+ (38) |
|
None |
|
|
| Apc1638T/1638T (69, 97) |
|
None | N/A |
|
| Apc1638N/+ (71) |
|
|
|
|
| KAD rat (68) |
|
|
|
|
Apc mouse models reported in this table are on C57Bl/6 background however, with different backcross isogenicity from N2 to > N20
Apc rat models reported in the table are on F344 background
Apc models are mouse models unless otherwise noted
NR: not reported
NeoR: Neomycin resistance gene
Acknowledgments
This work was supported by grants from the National Cancer Institute (RO1 CA109220), the National Center for Research Resources (P20 RR016475), and the National Institute of General Medical Sciences (P20 GM103418) from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
References
- 1.Neufeld KL. Nuclear APC. Adv Exp Med Biol. 2009;656:13–29. doi: 10.1007/978-1-4419-1145-2_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Sayans M, Suarez-Penaranda JM, Herranz-Carnero M, Gayoso-Diz P, Barros-Angueira F, Gandara-Rey JM, et al. The role of the adenomatous polyposis coli (APC) in oral squamous cell carcinoma. Oral Oncol. 2011 [Google Scholar]
- 3.Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: How do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101. doi: 10.1186/1476-4598-10-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–51. [PubMed] [Google Scholar]
- 5.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–33. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 6.Jorde LB, Carey HC, Bamshad MJ. Medical genetics. 4. Philadelphia PA: Mosby ELSEVIER; 2010. [Google Scholar]
- 7.Weinberg RA. The biology of cancer. New York: Garland Science, Taylor & Francis Group LLC; 2007. [Google Scholar]
- 8.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 9.Amos-Landgraf JM, Kwong LN, Kendziorski CM, Reichelderfer M, Torrealba J, Weichert J, et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc Natl Acad Sci U S A. 2007;104:4036–41. doi: 10.1073/pnas.0611690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, Taketo M. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci U S A. 1995;92:4482–6. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–34. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohler EM, Derungs A, Daum G, Behrens J, Schneikert J. Functional definition of the mutation cluster region of adenomatous polyposis coli in colorectal tumours. Hum Mol Genet. 2008;17:1978–87. doi: 10.1093/hmg/ddn095. [DOI] [PubMed] [Google Scholar]
- 13.Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Critical reviews in oncology/hematology. 2007;61:153–61. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Herrera L, Kakati S, Gibas L, Pietrzak E, Sandberg AA. Gardner syndrome in a man with an interstitial deletion of 5q. Am J Med Genet. 1986;25:473–6. doi: 10.1002/ajmg.1320250309. [DOI] [PubMed] [Google Scholar]
- 15.Sieber OM, Lamlum H, Crabtree MD, Rowan AJ, Barclay E, Lipton L, et al. Whole-gene APC deletions cause classical familial adenomatous polyposis, but not attenuated polyposis or “multiple” colorectal adenomas. Proc Natl Acad Sci U S A. 2002;99:2954–8. doi: 10.1073/pnas.042699199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisgaard ML, Bulow S. Familial adenomatous polyposis (FAP): genotype correlation to FAP phenotype with osteomas and sebaceous cysts. American journal of medical genetics Part A. 2006;140:200–4. doi: 10.1002/ajmg.a.31010. [DOI] [PubMed] [Google Scholar]
- 17.Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009;656:85–106. doi: 10.1007/978-1-4419-1145-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCart AE, Vickaryous NK, Silver A. Apc mice: models, modifiers and mutants. Pathol Res Pract. 2008;204:479–90. doi: 10.1016/j.prp.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–70. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 20.Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136:780–98. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- 21.Corpet DE, Pierre F. How good are rodent models of carcinogenesis in predicting efficacy in humans? A systematic review and meta-analysis of colon chemoprevention in rats, mice and men. Eur J Cancer. 2005;41:1911–22. doi: 10.1016/j.ejca.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 22.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–52. [PubMed] [Google Scholar]
- 23.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet journal of rare diseases. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haigis KM, Hoff PD, White A, Shoemaker AR, Halberg RB, Dove WF. Tumor regionality in the mouse intestine reflects the mechanism of loss of Apc function. Proc Natl Acad Sci U S A. 2004;101:9769–73. doi: 10.1073/pnas.0403338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smits R, Kartheuser A, Jagmohan-Changur S, Leblanc V, Breukel C, de Vries A, et al. Loss of Apc and the entire chromosome 18 but absence of mutations at the Ras and Tp53 genes in intestinal tumors from Apc1638N, a mouse model for Apc-driven carcinogenesis. Carcinogenesis. 1997;18:321–7. doi: 10.1093/carcin/18.2.321. [DOI] [PubMed] [Google Scholar]
- 26.Sieber OM, Howarth KM, Thirlwell C, Rowan A, Mandir N, Goodlad RA, et al. Myh deficiency enhances intestinal tumorigenesis in multiple intestinal neoplasia (ApcMin/+) mice. Cancer Res. 2004;64:8876–81. doi: 10.1158/0008-5472.CAN-04-2958. [DOI] [PubMed] [Google Scholar]
- 27.Kucherlapati M, Nguyen A, Kuraguchi M, Yang K, Fan K, Bronson R, et al. Tumor progression in Apc(1638N) mice with Exo1 and Fen1 deficiencies. Oncogene. 2007;26:6297–306. doi: 10.1038/sj.onc.1210453. [DOI] [PubMed] [Google Scholar]
- 28.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, et al. Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res. 1999;59:1301–7. [PubMed] [Google Scholar]
- 29.Nakayama T, Yamazumi K, Uemura T, Yoshizaki A, Yakata Y, Matsuu-Matsuyama M, et al. X radiation up-regulates the occurrence and the multiplicity of invasive carcinomas in the intestinal tract of Apc(min/+) mice. Radiat Res. 2007;168:433–9. doi: 10.1667/RR0869.1. [DOI] [PubMed] [Google Scholar]
- 30.Reichling T, Goss KH, Carson DJ, Holdcraft RW, Ley-Ebert C, Witte D, et al. Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005;65:166–76. [PubMed] [Google Scholar]
- 31.Green RA, Kaplan KB. Chromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APC. J Cell Biol. 2003;163:949–61. doi: 10.1083/jcb.200307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pollard P, Deheragoda M, Segditsas S, Lewis A, Rowan A, Howarth K, et al. The Apc 1322T mouse develops severe polyposis associated with submaximal nuclear beta-catenin expression. Gastroenterology. 2009;136:2204–13. e1–13. doi: 10.1053/j.gastro.2009.02.058. [DOI] [PubMed] [Google Scholar]
- 33.Lamlum H, Ilyas M, Rowan A, Clark S, Johnson V, Bell J, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson’s ‘two-hit’ hypothesis. Nature medicine. 1999;5:1071–5. doi: 10.1038/12511. [DOI] [PubMed] [Google Scholar]
- 34.Cheung AF, Carter AM, Kostova KK, Woodruff JF, Crowley D, Bronson RT, et al. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010;29:1857–64. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis A, Segditsas S, Deheragoda M, Pollard P, Jeffery R, Nye E, et al. Severe polyposis in Apc(1322T) mice is associated with submaximal Wnt signalling and increased expression of the stem cell marker Lgr5. Gut. 2010;59:1680–6. doi: 10.1136/gut.2009.193680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishikawa TO, Tamai Y, Li Q, Oshima M, Taketo MM. Requirement for tumor suppressor Apc in the morphogenesis of anterior and ventral mouse embryo. Dev Biol. 2003;253:230–46. doi: 10.1016/s0012-1606(02)00020-9. [DOI] [PubMed] [Google Scholar]
- 37.Li Q, Ishikawa TO, Oshima M, Taketo MM. The threshold level of adenomatous polyposis coli protein for mouse intestinal tumorigenesis. Cancer Res. 2005;65:8622–7. doi: 10.1158/0008-5472.CAN-05-2145. [DOI] [PubMed] [Google Scholar]
- 38.Gaspar C, Franken P, Molenaar L, Breukel C, van der Valk M, Smits R, et al. A targeted constitutive mutation in the APC tumor suppressor gene underlies mammary but not intestinal tumorigenesis. PLoS genetics. 2009;5:e1000547. doi: 10.1371/journal.pgen.1000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeineldin M, Cunningham J, McGuinness W, Alltizer P, Cowley B, Blanchat B, et al. A knock-in mouse model reveals roles for nuclear Apc in cell proliferation, Wnt signal inhibition and tumor suppression. Oncogene. 2011 doi: 10.1038/onc.2011.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oshima H, Oshima M, Kobayashi M, Tsutsumi M, Taketo MM. Morphological and molecular processes of polyp formation in Apc(delta716) knockout mice. Cancer Res. 1997;57:1644–9. [PubMed] [Google Scholar]
- 41.Anderson CB, Neufeld KL, White RL. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc Natl Acad Sci U S A. 2002;99:8683–8. doi: 10.1073/pnas.122235399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blaker H, Scholten M, Sutter C, Otto HF, Penzel R. Somatic mutations in familial adenomatous polyps. Nuclear translocation of beta-catenin requires more than biallelic APC inactivation. American journal of clinical pathology. 2003;120:418–23. doi: 10.1309/4E4W-G3AY-GJNC-D11P. [DOI] [PubMed] [Google Scholar]
- 43.Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, et al. Colonic tumorigenesis in BubR1+/−ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–70. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aoki K, Tamai Y, Horiike S, Oshima M, Taketo MM. Colonic polyposis caused by mTOR-mediated chromosomal instability in Apc+/Delta716 Cdx2+/− compound mutant mice. Nat Genet. 2003;35:323–30. doi: 10.1038/ng1265. [DOI] [PubMed] [Google Scholar]
- 45.Qian Z, Chen L, Fernald AA, Williams BO, Le Beau MM. A critical role for Apc in hematopoietic stem and progenitor cell survival. J Exp Med. 2008;205:2163–75. doi: 10.1084/jem.20080578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watkins-Chow DE, Pavan WJ. Genomic copy number and expression variation within the C57BL/6J inbred mouse strain. Genome Res. 2008;18:60–6. doi: 10.1101/gr.6927808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silverman KA, Koratkar R, Siracusa LD, Buchberg AM. Identification of the modifier of Min 2 (Mom2) locus, a new mutation that influences Apc-induced intestinal neoplasia. Genome Res. 2002;12:88–97. doi: 10.1101/gr.206002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, Buchberg AM. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell. 1995;81:957–66. doi: 10.1016/0092-8674(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 49.Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P, Mulherkar R, et al. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet. 1997;17:88–91. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- 50.Fijneman RJ, Bade LK, Peham JR, van de Wiel MA, van Hinsbergh VW, Meijer GA, et al. Pla2g2a attenuates colon tumorigenesis in azoxymethane-treated C57BL/6 mice; expression studies reveal Pla2g2a target genes and pathways. Cell Oncol. 2009;31:345–56. doi: 10.3233/CLO-2009-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baran AA, Silverman KA, Zeskand J, Koratkar R, Palmer A, McCullen K, et al. The modifier of Min 2 (Mom2) locus: embryonic lethality of a mutation in the Atp5a1 gene suggests a novel mechanism of polyp suppression. Genome Res. 2007;17:566–76. doi: 10.1101/gr.6089707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kwong LN, Shedlovsky A, Biehl BS, Clipson L, Pasch CA, Dove WF. Identification of Mom7, a novel modifier of Apc(Min/+) on mouse chromosome 18. Genetics. 2007;176:1237–44. doi: 10.1534/genetics.107.071217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suraweera N, Haines J, McCart A, Rogers P, Latchford A, Coster M, et al. Genetic determinants modulate susceptibility to pregnancy-associated tumourigenesis in a recombinant line of Min mice. Hum Mol Genet. 2006;15:3429–35. doi: 10.1093/hmg/ddl419. [DOI] [PubMed] [Google Scholar]
- 54.Crist RC, Roth JJ, Lisanti MP, Siracusa LD, Buchberg AM. Identification of Mom12 and Mom13, two novel modifier loci of Apc (Min) -mediated intestinal tumorigenesis. Cell Cycle. 2011;10:1092–9. doi: 10.4161/cc.10.7.15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crist RC, Roth JJ, Baran AA, McEntee BJ, Siracusa LD, Buchberg AM. The armadillo repeat domain of Apc suppresses intestinal tumorigenesis. Mamm Genome. 2010;21:450–7. doi: 10.1007/s00335-010-9288-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dove WF, Cormier RT, Gould KA, Halberg RB, Merritt AJ, Newton MA, et al. The intestinal epithelium and its neoplasms: genetic, cellular and tissue interactions. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 1998;353:915–23. doi: 10.1098/rstb.1998.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dove WF, Clipson L, Gould KA, Luongo C, Marshall DJ, Moser AR, et al. Intestinal neoplasia in the ApcMin mouse: independence from the microbial and natural killer (beige locus) status. Cancer Res. 1997;57:812–4. [PubMed] [Google Scholar]
- 58.Fox JG, Dangler CA, Whary MT, Edelman W, Kucherlapati R, Wang TC. Mice carrying a truncated Apc gene have diminished gastric epithelial proliferation, gastric inflammation, and humoral immunity in response to Helicobacter felis infection. Cancer Res. 1997;57:3972–8. [PubMed] [Google Scholar]
- 59.Zell JA, Ignatenko NA, Yerushalmi HF, Ziogas A, Besselsen DG, Gerner EW, et al. Risk and risk reduction involving arginine intake and meat consumption in colorectal tumorigenesis and survival. Int J Cancer. 2007;120:459–68. doi: 10.1002/ijc.22311. [DOI] [PubMed] [Google Scholar]
- 60.Song J, Sohn KJ, Medline A, Ash C, Gallinger S, Kim YI. Chemopreventive effects of dietary folate on intestinal polyps in Apc+/−Msh2−/− mice. Cancer Res. 2000;60:3191–9. [PubMed] [Google Scholar]
- 61.Mollersen L, Paulsen JE, Olstorn HB, Knutsen HK, Alexander J. Dietary retinoic acid supplementation stimulates intestinal tumour formation and growth in multiple intestinal neoplasia (Min)/+ mice. Carcinogenesis. 2004;25:149–53. doi: 10.1093/carcin/bgg176. [DOI] [PubMed] [Google Scholar]
- 62.Smith KJ, Levy DB, Maupin P, Pollard TD, Vogelstein B, Kinzler KW. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–5. [PubMed] [Google Scholar]
- 63.Munemitsu S, Souza B, Muller O, Albert I, Rubinfeld B, Polakis P. The APC gene product associates with microtubules in vivo and promotes their assembly in vitro. Cancer Res. 1994;54:3676–81. [PubMed] [Google Scholar]
- 64.Marshall TW, Lloyd IE, Delalande JM, Nathke I, Rosenblatt J. The tumor suppressor adenomatous polyposis coli controls the direction in which a cell extrudes from an epithelium. Mol Biol Cell. 2011;22:3962–70. doi: 10.1091/mbc.E11-05-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lewis A, Davis H, Deheragoda M, Pollard P, Nye E, Jeffery R, et al. The C-terminus of Apc does not influence intestinal adenoma development or progression. J Pathol. 2012;226:73–83. doi: 10.1002/path.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wodarz A, Nathke I. Cell polarity in development and cancer. Nat Cell Biol. 2007;9:1016–24. doi: 10.1038/ncb433. [DOI] [PubMed] [Google Scholar]
- 67.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 68.Yoshimi K, Tanaka T, Takizawa A, Kato M, Hirabayashi M, Mashimo T, et al. Enhanced colitis-associated colon carcinogenesis in a novel Apc mutant rat. Cancer Sci. 2009;100:2022–7. doi: 10.1111/j.1349-7006.2009.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smits R, Kielman MF, Breukel C, Zurcher C, Neufeld K, Jagmohan-Changur S, et al. Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev. 1999;13:1309–21. doi: 10.1101/gad.13.10.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Quesada CF, Kimata H, Mori M, Nishimura M, Tsuneyoshi T, Baba S. Piroxicam and acarbose as chemopreventive agents for spontaneous intestinal adenomas in APC gene 1309 knockout mice. Japanese journal of cancer research: Gann. 1998;89:392–6. doi: 10.1111/j.1349-7006.1998.tb00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91:8969–73. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smits R, van der Houven van Oordt W, Luz A, Zurcher C, Jagmohan-Changur S, Breukel C, et al. Apc1638N: a mouse model for familial adenomatous polyposis-associated desmoid tumors and cutaneous cysts. Gastroenterology. 1998;114:275–83. doi: 10.1016/s0016-5085(98)70478-0. [DOI] [PubMed] [Google Scholar]
- 73.Sasai H, Masaki M, Wakitani K. Suppression of polypogenesis in a new mouse strain with a truncated Apc(Delta474) by a novel COX-2 inhibitor, JTE-522. Carcinogenesis. 2000;21:953–8. doi: 10.1093/carcin/21.5.953. [DOI] [PubMed] [Google Scholar]
- 74.Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645–56. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 75.Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, Lund JJ, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS genetics. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robanus-Maandag EC, Koelink PJ, Breukel C, Salvatori DC, Jagmohan-Changur SC, Bosch CA, et al. A new conditional Apc-mutant mouse model for colorectal cancer. Carcinogenesis. 2010;31:946–52. doi: 10.1093/carcin/bgq046. [DOI] [PubMed] [Google Scholar]
- 77.Liu J, Xing Y, Hinds TR, Zheng J, Xu W. The third 20 amino acid repeat is the tightest binding site of APC for beta-catenin. J Mol Biol. 2006;360:133–44. doi: 10.1016/j.jmb.2006.04.064. [DOI] [PubMed] [Google Scholar]
- 78.Taketo MM. Mouse models of gastrointestinal tumors. Cancer Science. 2006;97:355–61. doi: 10.1111/j.1349-7006.2006.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116:1517–26. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Flisikowska T, Merkl C, Landmann M, Eser S, Rezaei N, Cui X, et al. A Porcine Model of Familial Adenomatous Polyposis. Gastroenterology. 2012 doi: 10.1053/j.gastro.2012.07.110. [DOI] [PubMed] [Google Scholar]
- 81.Sodir NM, Chen X, Park R, Nickel AE, Conti PS, Moats R, et al. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+ mice. Cancer Res. 2006;66:8430–8. doi: 10.1158/0008-5472.CAN-06-1437. [DOI] [PubMed] [Google Scholar]
- 82.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, et al. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nature medicine. 1998;4:1053–7. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 83.Saez E, Tontonoz P, Nelson MC, Alvarez JG, Ming UT, Baird SM, et al. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nature medicine. 1998;4:1058–61. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 84.Leedham SJ, Rodenas-Cuadrado P, Howarth K, Lewis A, Mallappa S, Segditsas S, et al. A basal gradient of Wnt and stem-cell number influences regional tumour distribution in human and mouse intestinal tracts. Gut. 2012 doi: 10.1136/gutjnl-2011-301601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cooper HS, Chang WC, Coudry R, Gary MA, Everley L, Spittle CS, et al. Generation of a unique strain of multiple intestinal neoplasia (Apc(+/Min-FCCC)) mice with significantly increased numbers of colorectal adenomas. Molecular carcinogenesis. 2005;44:31–41. doi: 10.1002/mc.20114. [DOI] [PubMed] [Google Scholar]
- 86.Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Le Plenier S, Houbron C, et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Laboratory investigation; a journal of technical methods and pathology. 2004;84:1619–30. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- 87.Boardman LA. Heritable colorectal cancer syndromes: recognition and preventive management. Gastroenterology clinics of North America. 2002;31:1107–31. doi: 10.1016/s0889-8553(02)00049-3. [DOI] [PubMed] [Google Scholar]
- 88.Furuuchi K, Tada M, Yamada H, Kataoka A, Furuuchi N, Hamada J, et al. Somatic mutations of the APC gene in primary breast cancers. Am J Pathol. 2000;156:1997–2005. doi: 10.1016/s0002-9440(10)65072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin Z, Tamura G, Tsuchiya T, Sakata K, Kashiwaba M, Osakabe M, et al. Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br J Cancer. 2001;85:69–73. doi: 10.1054/bjoc.2001.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarrio D, Moreno-Bueno G, Hardisson D, Sanchez-Estevez C, Guo M, Herman JG, et al. Epigenetic and genetic alterations of APC and CDH1 genes in lobular breast cancer: relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int J Cancer. 2003;106:208–15. doi: 10.1002/ijc.11197. [DOI] [PubMed] [Google Scholar]
- 91.Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN. ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc Natl Acad Sci U S A. 1993;90:8977–81. doi: 10.1073/pnas.90.19.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuraguchi M, Ohene-Baah NY, Sonkin D, Bronson RT, Kucherlapati R. Genetic mechanisms in Apc-mediated mammary tumorigenesis. PLoS genetics. 2009;5:e1000367. doi: 10.1371/journal.pgen.1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shibata H, Takano H, Ito M, Shioya H, Hirota M, Matsumoto H, et al. Alpha-catenin is essential in intestinal adenoma formation. Proc Natl Acad Sci U S A. 2007;104:18199–204. doi: 10.1073/pnas.0705730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Irving AA, Halberg RB, Albrecht DM, Plum LA, Krentz KJ, Clipson L, et al. Supplementation by vitamin D compounds does not affect colonic tumor development in vitamin D sufficient murine models. Archives of biochemistry and biophysics. 2011;515:64–71. doi: 10.1016/j.abb.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Niho N, Mutoh M, Komiya M, Ohta T, Sugimura T, Wakabayashi K. Improvement of hyperlipidemia by indomethacin in Min mice. Int J Cancer. 2007;121:1665–9. doi: 10.1002/ijc.22872. [DOI] [PubMed] [Google Scholar]
- 96.Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003;63:6090–5. [PubMed] [Google Scholar]
- 97.Yokoyama A, Nomura R, Kurosumi M, Shimomura A, Onouchi T, Iizuka-Kogo A, et al. The C-terminal domain of the adenomatous polyposis coli (Apc) protein is involved in thyroid morphogenesis and function. Medical molecular morphology. 2011;44:207–12. doi: 10.1007/s00795-010-0529-9. [DOI] [PubMed] [Google Scholar]
- 98.Oshima M, Oshima H, Kobayashi M, Tsutsumi M, Taketo MM. Evidence against dominant negative mechanisms of intestinal polyp formation by Apc gene mutations. Cancer Res. 1995;55:2719–22. [PubMed] [Google Scholar]