

Abstract
Cardiac arrhythmia is a common and often lethal manifestation of many forms of heart disease. Gap junction remodeling has been postulated to contribute to the increased propensity for arrhythmogenesis in diseased myocardium, although a causative role in vivo remains speculative. By generating mice with cardiac-restricted knockout of connexin43 (Cx43), we have circumvented the perinatal lethal developmental defect associated with germline inactivation of this gap junction channel gene and uncovered an essential role for Cx43 in the maintenance of electrical stability. Mice with cardiac-specific loss of Cx43 have normal heart structure and contractile function, and yet they uniformly (28 of 28 conditional Cx43 knockout mice observed) develop sudden cardiac death from spontaneous ventricular arrhythmias by 2 months of age. Optical mapping of the epicardial electrical activation pattern in Cx43 conditional knockout mice revealed that ventricular conduction velocity was significantly slowed by up to 55% in the transverse direction and 42% in the longitudinal direction, resulting in an increase in anisotropic ratio compared with control littermates (2.1±0.13 versus 1.66±0.06; P<0.01). This novel genetic murine model of primary sudden cardiac death defines gap junctional abnormalities as a key molecular feature of the arrhythmogenic substrate.
Keywords: gap junction, connexin43, arrhythmia, conduction
Gap junction channels, composed of highly homologous proteins known as connexins in vertebrate species, have been implicated in diverse processes, including the electrotonic coupling of excitable tissues, such as cardiac muscle.1,2 Theoretical modeling3–5 and studies of connexin (Cx) expression in diseased human and experimental animal hearts suggest that altered gap junction expression or function may underlie the high incidence of arrhythmias associated with many forms of heart disease, including hypertrophic, ischemic, and dilated cardiomyopathies.6–14 Specifically, in diseased myocardium, Cx43, the major constituent of ventricular gap junctions, is often diminished in abundance and fails to preferentially localize to the intercalated disk. This pathologic process, referred to as gap junction remodeling, is hypothesized to be an essential component of the substrate promoting some atrial as well as lethal ventricular tachyarrhythmias.
Efforts to genetically define the role of gap junction channels in cardiac conduction and arrhythmogenesis in vivo have been limited by the perinatal lethal developmental phenotype observed in Cx43 knockout (KO) mice.15 Data on conduction properties in Cx43 heterozygous KO mice, which develop normally and have normal lifespans, have been contradictory, likely reflecting differing methodologies. Some investigators have reported conduction slowing,16 whereas others find no statistically significant electrophysiological differences compared with wild-type hearts.17 Regardless, hearts from Cx43 heterozygous mice seem to be abnormally susceptible to the development of ventricular tachycardia in response to acute ischemia, providing evidence, at least in isolated heart preparations, that altered gap junction expression may be proarrhythmic.11
To test whether Cx43 is essential for cardiac electrical stability and whether altered gap junction expression is arrhythmogenic in vivo, we conditionally inactivated the Cx43 gene exclusively in cardiomyocytes. Cardiac morphogenesis in conditional KO (CKO) mice is normal, demonstrating no intrinsic cardiomyocyte cell–autonomous requirement for Cx43 during heart development. Despite normal cardiac structure and contractile performance, Cx43 CKO mice show profound conduction defects and uniformly develop spontaneous ventricular arrhythmias and sudden cardiac death by 2 months of age. Thus, in contrast to all existing murine models of altered cardiac electrophysiology, primary derangements in Cx43 expression lead to formation of a highly arrhythmogenic substrate culminating in uniform sudden cardiac death.
Materials and Methods
Gene Targeting and Production of Cx43 CKO Mice
Embryonic stem (ES) cell manipulation and electroporation were performed according to established techniques.18 Blastocyst injection for the production of Cx43+/flox mice was performed in the Mouse Genetics Shared Resource Facility of the Mount Sinai School of Medicine.
Southern Blot Analysis and Polymerase Chain Reaction
Southern blotting and polymerase chain reaction (PCR) were performed according to established techniques. Southern blots were probed with a 700-bp 3′ flanking probe from a region outside of the targeting vector.
Western Blot Analysis
Western blot analyses were performed with polyclonal antibodies to Cx4319 and Cx40 (Alpha Diagnostic). Blotting for Cx45 was performed with a monoclonal antibody (directed against amino acids 354–367; Chemicon) and embryonic mouse-heart lysate from embryonic day (E) 13.5 was used as a positive control. For normalization of signals, blotting was also performed with anti–β-tubulin (Zymed Laboratories) or antisarcomeric myosin heavy chain (MHC) (Developmental Studies Hybridoma Bank) monoclonal antibodies, followed by blotting with horseradish peroxidase–conjugated secondary antibody, chemiluminescent processing (ECL, Amersham Pharmacia Biotech), and autoradiography.
Histology, Immunofluorescence, and Confocal Analysis
Hearts were removed from experimental mice at predetermined times and flash-frozen in liquid nitrogen. Hearts were later thawed and refrozen in O.C.T. freezing medium before sectioning. Embryos at E12.5 were frozen directly in O.C.T. medium before sectioning. Frozen sections were fixed in acetone followed by immunostaining. Double staining of myocardial tissue with anti-Cx43 antisera and wheat germ agglutinin to visualize myocyte borders was performed as previously described.20 Cx43 was detected using a polyclonal antibody (custom manufactured by Research Genetics) directed against the same epitope used by Yamamoto et al.19 Sections stained by immunofluorescence were visualized by confocal microscopy. For the evaluation of myocardial fibrosis, tissue was fixed in 4% paraformaldehyde and embedded in paraffin, followed by staining with modified Mason’s trichrome stain.
Telemetry Monitoring
For ambulatory electrocardiographic monitoring, miniature telemetry transmitter devices (Data Sciences International) were implanted as previously described.21 The animals were allowed 48 hours to recover from the surgery before telemetry recordings were acquired. Recordings from CKO mice were continuous beginning 48 hours after implantation until the time of death.
Echocardiography
Echocardiography was performed under light anesthesia with avertin using an Acuson Sequoia echocardiography machine and a 15-MHz linear probe. Measurements were performed online in a blinded fashion. Fractional shortening (FS), left ventricular volumes, and ejection fraction (EF) were calculated according to a standard formula, as applied previously in rodents.22,23
Optical Mapping of Ventricular Activation
Mice were heparinized (heparin sodium 0.5 U/g intraperitoneally) and killed by cervical dislocation, and hearts were removed and perfused as previously described.17,24 After 15 minutes of equilibration, the hearts were stained with a voltage-sensitive dye (di-4-ANEPPS, Molecular Probes). Epicardial pacing was performed with a unipolar electrode (stimuli at 1.5 × diastolic threshold) at a cycle length of 120 ms (control and MHC-CKO). High-resolution optical mapping of voltage-dependent fluorescence was performed on an upright Olympus microscope (BX50WI) with a reflected light fluorescence attachment (BX-FLA). Excitation light from a 100-watt mercury arc lamp (Olympus) was filtered (480 to 550; dichroic mirror 570 nm), and the emitted fluorescent light (>590 nm) was projected onto a CCD camera (Dalsa). Images were acquired at 912 frames per second with 12-bit resolution from a 64×64-pixel array, which provided a spatial resolution of 40 μm (4× objective, NA 0.28).
To obtain a representative sequence of activation during epicardial pacing, 10 to 15 beats were averaged. No pharmacological or mechanical manipulations were used to limit motion. Because contraction begins during repolarization, it is possible to identify action potential upstrokes using dF/dtmax. Local fluorescence maxima and minima were determined within an 8-ms window centered on the dF/dtmax. A pixel was considered activated when its fluorescent signal exceeded 50% of this local fluorescence range.
Conduction Velocity
Conduction velocities were calculated as described previously.17 Briefly, local conduction velocities were calculated from the gradient of activation times. Vectors near the stimulating electrode and at a distance from the electrode were excluded to remove stimulus artifacts and the effects of 3-dimensional propagation. In addition, vectors that deviated >60 degrees from their neighboring sites were excluded from analysis to remove the effect of wavefront collisions.
Statistics
Western blot densitometry, echocardiographic and conduction velocity (CV) data are expressed as mean±SEM. Comparisons between groups were performed with a 2-tailed t test using Microsoft Excel software. For analysis of CVmax, CVmin, and anisotropic ratio, because only 2 of these parameters can be considered independent, we chose to test significance for CVmin and anisotropic ratio. Kaplan-Meier survival curves for the CKO mice were constructed and compared (with the logrank test) using StatView software. P<0.05 was considered statistically significant.
Results
Conditional Inactivation of Cx43 in the Heart
In an attempt to circumvent the lethal developmental phenotype associated with germline deletion of Cx43 and explore the role of this gap junction channel protein in cardiac arrhythmogenesis in vivo, we used the Cre/loxP system to inactivate Cx43 expression exclusively in cardiomyocytes.25 We generated a targeting vector in which a floxed neomycin (Neo) resistance cassette was inserted into the single Cx43 intron and a third loxP site was inserted into exon 2 of the Cx43 gene, just downstream of the open reading frame (Figure 1A). Genomic DNA from Neo-resistant ES cell clones was first screened for homologous recombination by Southern blotting (Figure 1B) and secondarily screened by PCR for the presence of the third loxP site (Figure 1C). Of a total of 64 clones screened, 5 (7.8%) had undergone homologous recombination and had the downstream loxP site. The targeting event did not affect endogenous Cx43 gene expression,26 because wild-type and targeted ES cells expressed equivalent amounts of Cx43 protein (Figure 1D). The floxed allele was successfully transmitted through the germline and used to produce Cx43flox/flox mice.
Figure 1.

Generation and characterization of Cx43+/flox ES cells. A, Cx43 targeting vector and strategy for generating homologous recombination. Shaded triangles represent loxP sequences in the targeting vector. IVS indicates intervening sequence; ORF, open reading frame; and Neo, neomycin-resistance cassette. B, Southern blotting of wild-type (Cx43+/+) and targeted Cx43+/flox ES cell– clone genomic DNA digested with the indicated restriction enzymes. The blot was probed with a 700-bp 3′flanking probe from a region outside of the targeting vector. C, PCR screening for the presence of the 3′-most loxP site in a targeted Cx43+/flox ES cell clone. Lane 1, Wild-type ES cell genomic DNA as a template; lane 2, genomic DNA from a targeted ES cell clone is used as a template; lane 3, the targeting vector (0.1 ng) is used as a template. PCR primers flank the sequence where the 3′-most loxP site was inserted. DNA sequence containing the loxP site is ≈50 bp larger than the corresponding wild-type sequence. D, Western blot depicting Cx43 protein expression in wild-type (lane 1) and targeted Cx43+/flox (lane 2) ES cell clones. Blotting for β-tubulin was performed on the same membrane to indicate relative loading. Similar results were seen in 3 independently targeted ES cell lines.
Cx43 CKO Mice Develop Normally
Homozygous Cx43flox/flox mice were crossed with strains of mice expressing Cre recombinase (Cre) exclusively in cardiomyocytes. These included a transgenic strain in which Cre was driven by regulatory elements from the α-MHC gene27 and a second strain in which Cre has been knocked in to the myosin light chain 2v (MLC2v) locus.28 Both the α-MHC–Cre:Cx43flox/flox CKO (MHC-CKO) and the MLC2v-Cre:Cx43flox/flox CKO (MLC-CKO) mice were born with the expected mendelian frequency and were grossly indistinguishable from their non-KO littermates.
Efficient inactivation of Cx43 expression in the hearts of CKO mice was examined by immunofluorescence. At both E12.5 (Figures 2A through 2C) and at 4 weeks postpartum (Figures 2D through 2F), a marked reduction of Cx43 expression was observed in CKO hearts, with extensive areas that were completely devoid of Cx43 immunoreactivity.
Figure 2.

Immunofluorescent staining for Cx43 in wild-type and MHC-CKO hearts. Sections from E12.5 (A through C) and 4-week-old (D through F) hearts were stained for Cx43 (green fluorescent stain), counter-stained using wheat germ agglutinin (red fluorescent stain), and imaged with confocal microscopy. Abundant Cx43 staining in control littermate hearts is evident at both embryonic (A) and postnatal (D) stages. In contrast, extensive areas of the myocardium in embryonic (B and C) and post-natal (E and F) CKO mice are devoid of Cx43 staining, although some residual expression is detectable. Bar=50 μm.
To quantify the reduction in Cx43 expression, Western blot analysis was performed. At 4 weeks of age, compared with control littermates, Cx43 expression was reduced by 95% in the MHC-CKO mice (P=0.027; n=3 MHC-CKO mice and 4 controls) and by 86% in MLC-CKO mice (P<0.01; n=4 MLC-CKO and 4 controls), consistent with the immunofluorescence data (Figure 3A). There was no apparent compensatory increase in the expression of other connexins known to be expressed in myocytes. By Western blot analysis, Cx40 levels in both the MHC-CKO and MLC-CKO strains were unchanged compared with littermate controls (Figure 3B). Cx45 expression was below the limits of detection in both CKO and control hearts, in agreement with previous studies of normal murine heart.29 In addition, no expression of Cx40 or Cx45 was detected by immunofluorescence in working ventricular myocardium (not shown).
Figure 3.

Connexin expression in ventricular lysates from CKO mice. A, Western blot analysis of Cx43 expression in mice with the indicated genotypes. B, Western blot analysis of Cx40 expression in mice with the indicated genotypes. Blots were probed with a monoclonal antibody directed against sarcomeric MHC to confirm uniform loading. Representative samples are shown.
Histological examination of CKO hearts at several time points revealed no evidence of the right ventricular outflow tract (RVOT) obstruction phenotype associated with germline ablation of Cx43. Both at 1 week after birth (n=2 CKO and 3 Cre− littermate hearts) and in older mice at 8 weeks of age (n=3 CKO and 3 Cre− littermate hearts), the ventricular muscle appeared entirely normal, without fibrosis, hypertrophy, or myofibrillar disarray (not shown).
Contractile performance, left ventricular chamber sizes, and wall thicknesses, as assessed by echocardiography in 1-month-old MHC-CKO mice, were no different than in non-KO littermates (Table). Thus, Cre recombinase, driven by regulatory elements from either the α-MHC or MLC2v genes, efficiently and specifically inactivated Cx43 expression in the myocyte compartment of the heart, with no discernible compensatory effect on the abundance of other cardiac connexins. Moreover, there appeared to be no intrinsic cardiomyocyte cell–autonomous requirement for Cx43 during heart development. This conclusion is consistent with evidence suggesting a primary neural crest origin of the Cx43 KO phenotype.30
Table 1.
Echocardiographic Studies of Cx43 Conditional Knockout (MHC-CKO) Mice
| Cre−, flox/flox (n=4) | MHC-CKO (n=6) | |
|---|---|---|
| IDD, mm | 3±0.2 | 3±0.1 |
| IDS, mm | 1.6±0.2 | 1.7±0.1 |
| FS, % | 46.3±4 | 45±2.9 |
| LV diastolic volume, μL | 28±4 | 28±3 |
| LV systolic volume, μL | 5±2 | 5±1 |
| EF, % | 83.7±4.8 | 82.6±2.6 |
| AWTs, mm | 1.5±0.1 | 1.4±0.1 |
| AWTd, mm | 0.9±0.1 | 0.9±0.1 |
| PWTs, mm | 1.2±0.1 | 1.2±0.1 |
| PWTd, mm | 0.9±0.1 | 0.9±0.1 |
| HR, bpm | 525±9 | 462±37 |
| Age, wk | 5 | 5 |
| Weight, g | 20.3±0.6 | 21.0±0.4 |
P=NS for all of the above comparisons. IDD indicates intraventricular dimension in diastole; IDS, intraventricular dimension in systole; FS, fractional shortening; LV, left ventricle; AWTs, anterior wall thickness in systole; AWTd, anterior wall thickness in diastole; PWTs, posterior wall thickness in systole; PWTd, posterior wall thickness in diastole; and HR, heart rate.
Sudden Cardiac Death in Cx43 CKO Mice
Although the Cx43 CKO mice appeared morphologically and functionally normal, they began to die suddenly at 2 to 3 weeks of age (Figure 4). Within the first 2 months of life, 13 of 13 MLC-CKO mice and 15 of 15 MHC-CKO mice died suddenly, all without previous signs of illness. In contrast, there was no mortality among the Cre−:Cx43flox/flox littermates (n=25) during the first 6 months of life.
Figure 4.

Kaplan-Meier survival curves for CKO mice. Kaplan-Meier survival curves for MHC-CKO and MLC-CKO mice (n=15 MHC-CKO mice, n=13 MLC-CKO mice; P<0.01 for a comparison of the 2 curves). There have been no deaths in Cre−:Cx43flox/flox littermates during more than 6 months of observation.
We implanted miniaturized telemetry transmitter devices in four 5-week-old MHC-CKO mice to record the cardiac rhythm during episodes of sudden death.21 During the continuous recording period, all 4 MHC-CKO mice were in normal sinus rhythm, with no evidence of ventricular ectopy. In 3 of the mice, we successfully captured the abrupt onset of spontaneous ventricular tachyarrhythmias, confirming that the deaths were arrhythmic in nature (Figure 5). In the fourth mouse, the recording device was inadvertently inactive at the time of death.
Figure 5.

Telemetry recordings of lethal arrhythmias in MHC-CKO mice. Continuous electrocardiographic recordings from miniaturized implanted transmitter devices in 3 MHC-CKO mice at 5 (top), 8 (middle), and 7 (bottom) weeks of age. Telemetry devices were implanted for a total of 4 (top), 27 (middle), and 15 (bottom) days before the death events. The recordings show normal sinus rhythm initially, followed by ventricular tachycardia. The arrhythmias quickly degenerated into ventricular fibrillation. The recording in the top panel was made at 6 PM, that in the middle panel at 8 AM, and that in the bottom panel at 5 AM.
Abnormal Conduction Properties and Arrhythmias in MHC-CKO Hearts
To directly examine the consequences of loss of Cx43 on ventricular conduction, we optically mapped electrical activity in the hearts of MHC-CKO mice and littermate controls using a voltage-sensitive dye. In hearts from 5- to 8-week-old control mice (n=6; Figure 6B), the normal anisotropic pattern of electrical activation was observed,17,21 without evidence of conduction abnormalities or spontaneous arrhythmias. In contrast, hearts from 5- to 8-week-old MHC-CKO mice demonstrated markedly abnormal conduction parameters (Figure 6C). Six of the 10 MHC-CKO mouse hearts tested developed spontaneous polymorphic ventricular tachyarrhythmias that could not be terminated by high-frequency pacing. The remaining MHC-CKO hearts initially were in normal sinus rhythm and could be paced. However, conduction parameters in these hearts were markedly abnormal. Compared with control hearts, CV in these 4 MHC-CKO hearts was substantially reduced in all directions, as visualized by optical mapping. CV was reduced most drastically in the transverse (CVmin) direction (control CVmin=0.38±0.02 m/s; MHC-CKO CVmin=0.17±0.02 m/s; P<0.001; Figure 6D), presumably reflecting the lessening of rotational anisotropy attributable to extreme uncoupling between epicardial and deeper cell layers.31,32 CVmax decreased from 0.62±0.02 m/s in controls to 0.36±0.05 m/s in MHC-CKO mice (significance was not tested). As a result, the anisotropic ratio (CVmax/CVmin) was significantly increased in the CKO hearts (1.66±0.06 in control hearts versus 2.1±0.13 in MHC-CKO hearts; P<0.01; Figure 6D).
Figure 6.
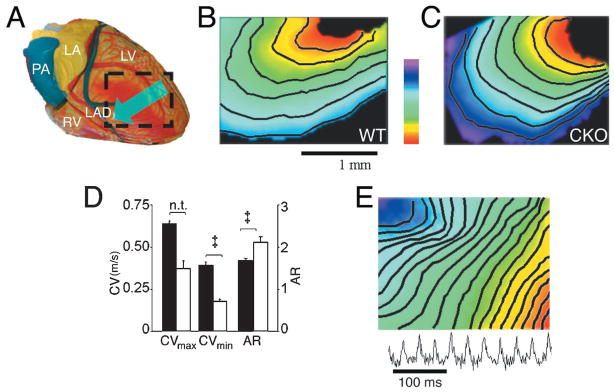
Optical-mapping studies in MHC-CKO mice. A, Schematic of the anterior surface of the heart showing the approximate area imaged and used to calculate conduction velocities. Hearts in this study were paced at the left ventricular lateral wall, and the epicardial activation pattern was recorded as it spread across the anterior wall toward the ventricular septum, as indicated by the arrow. B, Anterior view of a control littermate heart showing the expected smooth epicardial activation pattern from the site of pacing at the lateral wall and spreading toward the ventricular septum. CVmax in this control heart is 0.58 m/s, and CVmin is 0.32 m/s. C, Representative epicardial activation pattern of an age-matched MHC-CKO mouse heart paced in the same fashion as the control heart. CVmax in this MHC-CKO heart is 0.42 m/s, and CVmin is 0.15 m/s. D, CVmax, CVmin, and anisotropic ratio (AR) for the control (n=6) and MHC-CKO (n=4) mice that were successfully paced. See text for details of measurements. E, Epicardial activation pattern in an MHC-CKO mouse with incessant ventricular tachyarrhythmia. The bottom trace shows a pseudo-ECG that summarizes the activity recorded during the episode. Color scale bar in panels B and C indicates 0 (red) to 4 ms (purple); in panel E from 0 to 10 ms.
The MHC-CKO hearts were highly susceptible to spontaneous ventricular arrhythmias, which occurred before or during the standard pacing protocol (Figure 6E). Interestingly, within the group of MHC-CKO hearts, spontaneous arrhythmias at explantation that prevented pacing were more common in mice older than 7 weeks of age (3 of 3 MHC-CKO hearts) than in those younger than 7 weeks (3 of 7 MHC-CKO hearts). There were no age-dependent differences in conduction parameters in the control group. In total, 8 of 10 MHC-CKO mice studied had spontaneous ventricular arrhythmias, compared with 0 of 6 controls.
Discussion
In the past decade, several animal and human studies have described alterations in the abundance or subcellular localization of connexin proteins in arrhythmogenic cardiac conditions, a process referred to as gap junction remodeling.7–10,12 However, determining the unique contribution of gap junctional remodeling to the substrate for cardiac arrhythmias has been difficult, inasmuch as the diseased heart invariably shows a multitude of structural and functional perturbations. In the present study, by specifically ablating the expression of Cx43 in the myocardium, we sought to circumvent the lethal developmental defect caused by germline deletion of Cx43 and address this question directly.
The first novel finding in this study is the demonstration that there is no intrinsic cardiomyocyte cell–autonomous requirement for Cx43 during heart development. A similar conclusion was made for the RXR gene when inactivated by crossing with the same MLC2v-Cre mouse used in this study.28 Whether recombination was catalyzed in the Cx43 floxed mice with Cre recombinase expressed under the transcriptional control of the MLC2v or αMHC regulatory elements, where Cx43 expression is inactivated no later than E12.5, heart development proceeded normally, without the RVOT phenotype observed with germline knockout of Cx43.15 Thus, the lethal developmental defect resulting from global inactivation of Cx43 seems to result from a nonmyocyte lineage, consistent with previous experimental evidence suggesting a primary neural-crest origin of this phenotype.30,33 Lineage-restricted inactivation of Cx43 using strains of mice expressing Cre recombinase preferentially in the cardiac neural crest, such as Wnt-134 or Pax3,35 may unequivocally answer this question. In addition, targeted ablation of Cx43 in additional lineages will likely provide additional insight into the tissue-specific functions of this widely expressed gap junction channel protein.
By circumventing the lethal RVOT phenotype caused by germline deletion of Cx43 with the conditional strategy, we were able to examine the consequences of loss of Cx43 exclusively in the myocardium. Despite normal heart structure and contractile performance, Cx43 CKO mice uniformly developed sudden cardiac death, apparently from spontaneous ventricular tachycardia. These data support the critical role of the gap junction channel in maintaining cardiac electrical stability. Indeed, it seems that loss of Cx43 function, even in the absence of other identifiable structural cardiac abnormalities, can lead to a substrate in which spontaneous, lethal cardiac arrhythmias are inevitable.
Although CV is markedly slowed in the Cx43 CKO mice, successful impulse propagation is maintained, at least until the abrupt onset of lethal ventricular tachyarrhythmias. This observation is consistent with modeling suggesting that the safety factor for conduction is paradoxically increased with reduced gap junctional coupling.5 Because only scattered myocytes express Cx43 in the CKO mice, our data suggest that the myocardium must be coupled by low-level expression of gap junction channels formed from other connexin isoforms.
The electrophysiological mechanisms leading to the lethal ventricular arrhythmias and the factors accounting for the timing of sudden cardiac death in the CKO mice are uncertain. Cellular uncoupling resulting from loss of Cx43 gap junction channels might unmask ectopic foci or trigger arrhythmias by enhancing the generation of early afterdepolarizations (EADs).36 Computer modeling studies indicate that moderate decreases in junctional coupling predispose to the generation of EADs, whereas higher levels of junctional resistance limit their propagation.37 Thus, the residual level of junctional coupling in the CKOs may allow for the generation and propagation of EADs. Indeed, a pause-dependent EAD may account for the initiation of the arrhythmia shown in Figure 5, top.
By enhancing dispersion of action potential duration (APD) and repolarization, reduced cell coupling also predisposes to the formation of unidirectional block and reentry.36,38 Modeling of a multicellular theoretical fiber suggests that decreased gap junction coupling, although causing a paradoxical increase in the safety factor as CV decreases, also may facilitate microreentry.5 Furthermore, on the basis of in vitro studies of dissociated myocytes,39 it has been theorized that extreme conduction slowing in combination with geometrical factors may permit reentrant excitation to occur in extremely small areas of cardiac tissue. These factors in combination with the progressive growth of individual myocytes after birth, which is predicted to increase the discontinuous nature of propagation, may also play a role in the time course of the arrhythmic phenotype.3
In recent years, increasing attention has focused on the complex interactions between passive and active membrane properties. The Cx43 CKO mice provide a novel experimental system to examine these relationships in a multicellular preparation, particularly with respect to formation of a substrate that clearly enhances the propensity for spontaneous arrhythmias. For example, theoretical simulations by Rudy and colleagues5,36,38 predict that reduced cellular coupling markedly increases APD dispersion and also renders action potential propagation increasingly dependent on the L-type calcium current. Moreover, Laurita et al40 have presented evidence that APD restitution is influenced by cell-to-cell coupling. While recognizing the significant differences in the shape and ionic components of the action potential in various species, additional analyses of impulse propagation in intact hearts or multicellular fibers from wild-type and homozygous Cx43 CKO mice may yield experimental data that can be directly compared with predictions based on theoretical modeling. In addition, with the availability of the Cx43 CKO mice, one can envision increasingly complex genetic strategies in which both the extent of cellular coupling and the magnitude of specific sarcolemmal currents are simultaneously modified, thereby modeling the complex electrophysiological derangements typically observed in diseased myocardium.
In summary, the results of the present study provide strong evidence that loss of Cx43 expression may serve as a critical event in the formation of the arrhythmogenic substrate. Indeed, altered gap junction channel expression in the heart seems sufficient to induce spontaneous ventricular tachycardia with complete penetrance. Moreover, in contrast to other murine models associated with premature cardiovascular mortality, Cx43 CKO mice develop uniform sudden cardiac death in the absence of cardiac dysfunction and morphological abnormalities. Because gap junction remodeling has been described in many forms of human cardiac disease, restoration of normal intercellular coupling in the myopathic heart may well serve as a novel target in the treatment of patients at risk for lethal ventricular arrhythmias.
Acknowledgments
This study was supported in part by grants HL04222 (to D.E.G.), HL39707 (to G.E.M.), and HL38449, HL30557, and HL64757 (to G.I.F.) from the National Institutes of Health and an Established Investigator Award from the American Heart Association (to G.I.F.). Confocal laser scanning microscopy was performed at the Mount Sinai School of Medicine–Confocal Laser Scanning Microscopy core facility, supported with funding from National Institutes of Health shared instrumentation grant (1 SS10 RR0 9145-01) and National Science Foundation Major Research Instrumentation grant (DBI-9724504). The authors acknowledge the kind gift of mouse Cx43 genomic DNA from Dr Janet Rossant (Samuel Lunenfeld Research Institute, Toronto, Ontario) and anti-Cx43 antisera from Dr Elliot L. Hertzberg (Albert Einstein College of Medicine, Bronx, NY). We would like to thank Dr John T. Fallon, Dr Kevin Kelley (Mount Sinai Mouse Genetics Shared Resource Facility), Dr Scott Henderson (the Mount Sinai School of Medicine–Confocal Laser Scanning Microscopy core facility), Fang-Yu Liu, Jie Zhang, Veronica Gulle, Samantha Buckley, and Maya Srinivas for their help on this project.
References
- 1.Kumar NM, Gilula NB. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 2.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 3.Spach M, Heidlage J, Dolber P, Barr R. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–311. doi: 10.1161/01.res.86.3.302. [DOI] [PubMed] [Google Scholar]
- 4.Quan W, Rudy Y. Unidirectional block and reentry of cardiac excitation: a model study. Circ Res. 1990;66:367–382. doi: 10.1161/01.res.66.2.367. [DOI] [PubMed] [Google Scholar]
- 5.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue: roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 6.Luke RA, Saffitz JE. Remodeling of ventricular conduction pathways in healed canine infarct border zones. J Clin Invest. 1991;87:1594–1602. doi: 10.1172/JCI115173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–875. doi: 10.1161/01.cir.88.3.864. [DOI] [PubMed] [Google Scholar]
- 8.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease: an immunohisto-chemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 9.Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. 1997;95:988–996. doi: 10.1161/01.cir.95.4.988. [DOI] [PubMed] [Google Scholar]
- 10.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, Pennell DJ, Fox K, Pepper J, Poole-Wilson PA, Severs NJ. Downregulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation. 1998;97:651–660. doi: 10.1161/01.cir.97.7.651. [DOI] [PubMed] [Google Scholar]
- 11.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 12.Matsushita T, Oyamada M, Fujimoto K, Yasuda Y, Masuda S, Wada Y, Oka T, Takamatsu T. Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res. 1999;85:1046–1055. doi: 10.1161/01.res.85.11.1046. [DOI] [PubMed] [Google Scholar]
- 13.Peters NS, Wit AL. Myocardial architecture and ventricular arrhythmogenesis. Circulation. 1998;97:1746–1754. doi: 10.1161/01.cir.97.17.1746. [DOI] [PubMed] [Google Scholar]
- 14.Costeas C, Peters NS, Waldecker B, Ciaccio EJ, Wit AL, Coromilas J. Mechanisms causing sustained ventricular tachycardia with multiple QRS morphologies: results of mapping studies in the infarcted canine heart. Circulation. 1997;96:3721–3731. doi: 10.1161/01.cir.96.10.3721. [DOI] [PubMed] [Google Scholar]
- 15.Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- 16.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991–1998. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–1375. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 18.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto T, Ochalski A, Hertzberg EL, Nagy JI. LM and EM immunolocalization of the gap junctional protein connexin 43 in rat brain. Brain Res. 1990;508:313–319. doi: 10.1016/0006-8993(90)90415-8. [DOI] [PubMed] [Google Scholar]
- 20.Dolber PC, Beyer EC, Junker JL, Spach MS. Distribution of gap junctions in dog and rat ventricle studied with a double-label technique. J Mol Cell Cardiol. 1992;24:1443–1457. doi: 10.1016/0022-2828(92)91085-j. [DOI] [PubMed] [Google Scholar]
- 21.Lee P, Morley G, Huang Q, Fischer A, Seiler S, Horner JW, Factor S, Vaidya D, Jalife J, Fishman GI. Conditional lineage ablation to model human diseases. Proc Natl Acad Sci U S A. 1998;15:11371–11376. doi: 10.1073/pnas.95.19.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazquez de Prada JA, Jiang L, Handschumacher MD, Xie SW, Rivera JM, Schwammenthal E, Guerrero JL, Weyman AE, Levine RA, Picard MH. Quantification of pericardial effusions by three-dimensional echocardiography. J Am Coll Cardiol. 1994;24:254–259. doi: 10.1016/0735-1097(94)90571-1. [DOI] [PubMed] [Google Scholar]
- 23.Cittadini A, Stromer H, Katz SE, Clark R, Moses AC, Morgan JP, Douglas PS. Differential cardiac effects of growth hormone and insulin-like growth factor-1 in the rat: a combined in vivo and in vitro evaluation. Circulation. 1996;93:800–809. doi: 10.1161/01.cir.93.4.800. [DOI] [PubMed] [Google Scholar]
- 24.Vaidya D, Morley GE, Samie FH, Jalife J. Reentry and fibrillation in the mouse heart: a challenge to the critical mass hypothesis. Circ Res. 1999;85:174–181. doi: 10.1161/01.res.85.2.174. [DOI] [PubMed] [Google Scholar]
- 25.Baubonis W, Sauer B. Genomic targeting with purified Cre recombinase. Nucleic Acids Res. 1993;21:2025–2029. doi: 10.1093/nar/21.9.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targeting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 27.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells: targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Kubalak SW, Chien KR. Ventricular muscle-restricted targeting of the RXRα gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development. 1998;125:1943–1949. doi: 10.1242/dev.125.10.1943. [DOI] [PubMed] [Google Scholar]
- 29.Coppen SR, Dupont E, Rothery S, Severs NJ. Connexin45 expression is preferentially associated with the ventricular conduction system in mouse and rat heart. Circ Res. 1998;82:232–243. doi: 10.1161/01.res.82.2.232. [DOI] [PubMed] [Google Scholar]
- 30.Ewart JL, Cohen MF, Meyer RA, Huang GY, Wessels A, Gourdie RG, Chin AJ, Park SMJ, Lazatin BO, Villabon S, Lo CW. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development. 1997;124:1281–1292. doi: 10.1242/dev.124.7.1281. [DOI] [PubMed] [Google Scholar]
- 31.Muzikant AL, Henriquez CS. Paced activation mapping reveals organization of myocardial fibers: a simulation study. J Cardiovasc Electrophysiol. 1997;8:281–294. doi: 10.1111/j.1540-8167.1997.tb00791.x. [DOI] [PubMed] [Google Scholar]
- 32.Berenfeld O, Pertsov AM. Dynamics of intramural scroll waves in three-dimensional continuous myocardium with rotational anisotropy. J Theor Biol. 1999;199:383–394. doi: 10.1006/jtbi.1999.0965. [DOI] [PubMed] [Google Scholar]
- 33.Lo CW, Cohen MF, Huang GY, Lazatin BO, Patel N, Sullivan R, Pauken C, Park SM. Cx43 gap junction gene expression and gap junctional communication in mouse neural crest cells. Dev Genet. 1997;20:119–132. doi: 10.1002/(SICI)1520-6408(1997)20:2<119::AID-DVG5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 34.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Liu KC, Jin F, Lu MM, Epstein JA. Transgenic rescue of congenital heart disease and spina bifida in Splotch mice. Development. 1999;126:2495–2503. doi: 10.1242/dev.126.11.2495. [DOI] [PubMed] [Google Scholar]
- 36.Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 37.Saiz J, Ferrero JMJ, Monserrat M, Ferrero JM, Thakor NV. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Trans Biomed Eng. 1999;46:138–147. doi: 10.1109/10.740876. [DOI] [PubMed] [Google Scholar]
- 38.Viswanathan PC, Rudy Y. Cellular arrhythmogenic effects of congenital and acquired long-QT syndrome in the heterogeneous myocardium. Circulation. 2000;101:1192–1198. doi: 10.1161/01.cir.101.10.1192. [DOI] [PubMed] [Google Scholar]
- 39.Rohr S, Kucera JP, Kleber AG. Slow conduction in cardiac tissue, I: effects of a reduction of excitability versus a reduction of electrical coupling on microconduction. Circ Res. 1998;83:781–794. doi: 10.1161/01.res.83.8.781. [DOI] [PubMed] [Google Scholar]
- 40.Laurita KR, Girouard SD, Rudy Y, Rosenbaum DS. Role of passive electrical properties during action potential restitution in intact heart. Am J Physiol. 1997;273:H1205–H1214. doi: 10.1152/ajpheart.1997.273.3.H1205. [DOI] [PubMed] [Google Scholar]