

Abstract
Triple-negative breast cancer (TNBC), as defined by the absence of estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 expression, is a challenging disease with the poorest prognosis of all breast cancer subtypes. Importantly, there are currently no known molecular targets for this subgroup of patients. Recent advances in genomics and gene expression profiling have shed new light on the molecule heterogeneity of TNBC. We present an overview of the scientific evidence suggesting that clinical outcome in TNBC is affected by tumor-infiltrating immune cells. We also describe tumor-associated antigens recently identified in TNBC. Finally, we review the current literature on promising immunotherapies for TNBC, including tumor vaccine approaches, immune-checkpoint inhibitors, antagonists of immunosuppressive molecules and adoptive cell therapies. It is our contention that selected patients with TNBC with lymphocytic tumor infiltrates at diagnosis may benefit from immune-based therapies and that these immunotherapies will be most beneficial in combination with cytotoxic drugs that potentiate adaptive anti-tumor immunity.
Keywords: basal like, breast cancer, immunotherapy, T cell
Defining triple-negative breast cancer: the need for international standards
Breast cancer is a heterogeneous disease composed of different subtypes. Clinically, the classification of breast cancer depends on the expression of three biomarkers: estrogen receptors (ERs), progesterone receptors (PRs) and human epidermal growth factor receptor 2 (HER2). Breast cancers that are negative for ER, PR and HER2 are considered as triple-negative breast cancer (TNBC) and represent 15–20% of newly diagnosed cases. While HER2 assessment follows standardized guidelines [Wolff et al. 2006], hormone receptor assessment can vary across countries. We believe this lack of uniformity in hormone receptor assessment is a major obstacle to the worldwide research effort on TNBC. Reproducible assessment of triple-negative status would enable better definition of patients with TNBC and greatly improve the quality of research. According to the American Society of Clinical Oncology and the College of American Pathologists, ER and PR status should be considered positive when at least 1% of tumor cells are positive by immunohistochemistry (IHC) [Hammond et al. 2010]. Compliance with these recommendations and guidelines should be the gold standard for identifying patients with TNBC.
Clinical characteristics of TNBC
Compared with other forms of breast cancer, TNBC is associated with a younger age at diagnosis [Bauer et al. 2007; Dent et al. 2007; Anders et al. 2011], African American ethnicity [Bauer et al. 2007; Lund et al. 2009], advanced stage at diagnosis, increased risk of visceral metastasis and poorer outcome [Perou et al. 2000; Nielsen et al. 2004; Carey, 2006; Liedtke et al. 2008; Dent et al. 2009; Cancello et al. 2010; Metzger et al. 2010]. Because TNBC lacks a therapeutic target, patients do not benefit from endocrine therapy or HER2-targeted therapy. Accordingly, TNBC remains an important challenge in today’s clinical practice.
Cytotoxic chemotherapy is the only systemic treatment option for patients with TNBC. Although as a group TNBC is associated with a poor prognosis, some patients respond well to anthracycline-based chemotherapy, suggesting a significant degree of molecular heterogeneity. Unfortunately, the molecular mechanisms underlying this heterogeneity and its relationship to treatment response are still poorly understood.
Heterogeneity of TNBC
Significant research effort has recently been deployed to better understand the molecular heterogeneity of TNBC with the objective of identifying potential therapeutic targets. One of the first molecular insights into TNBC was the observation that it often presents a basal-like profile. However, not all TNBCs are basal-like [Perou et al. 2000]. It has been suggested that approximately 70% of TNBCs present a basal-like gene expression profile [Rakha and Ellis, 2009]. At the histopathological level, TNBC (defined by IHC) and basal-like breast cancers (defined by gene expression) share several characteristics, such as high histologic grade, pushing borders of invasion and stromal lymphocytic infiltrates [Fulford et al. 2006; Livasy et al. 2007]. Also, both TNBC and basal-like breast cancers show links with BRCA1-mutated breast cancers and the BRCA1 pathway in general. Accordingly, up to 90% of BRCA1-associated tumors are TNBC by IHC [Foulkes et al. 2003; Arnes et al. 2005] and mice deficient in Brca1 and p53 in mammary epithelial cells develop tumors that are both triple negative and basal like [Molyneux et al. 2010].
Although the majority of TNBCs present a basal-like gene expression profile, some cases lack expression of basal markers, such as epidermal growth factor receptor (EGFR) and cytokeratin (CK) 5/6. Notably, these non-basal TNBCs are associated with better outcomes than core basal-like breast cancers [Cheang et al. 2008]. Various studies have proposed the use of surrogate markers detected by IHC to identify core basal-like breast cancers. These include basal CKs (i.e. CK5/6, CK14, and CK17), EGFR and C-kit [Nielsen et al. 2004]. Expression of these surrogate markers in TNBC is associated with shorter survival compared with the remaining patients with TNBC [Cheang et al. 2008]. However, before using these surrogates as prognostic tools, standardized cutoff levels must be defined.
Gene expression subtypes in TNBC
Recent advances in genomics offer great potential to better understand the heterogeneity of TNBC. Using gene expression profiling from over 3000 cases, Lehmann and colleagues recently described six different TNBC subtypes [Lehmann et al. 2011]. These TNBC molecular subtypes included two basal-like, two mesenchymal, a luminal androgen-receptor positive and an immunomodulatory subtype. The two basal-like subtypes were characterized by high levels of genes involved in cell proliferation and DNA damage response. The two mesenchymal subtypes shared common features with previously identified claudin-low breast cancers [Prat et al. 2010]. These mesenchymal subtypes showed increased expression of genes involved in cell motility (e.g. Rho pathway), cellular differentiation, and growth pathways [e.g. ALK, transforming growth factor β (TGFβ), EGFR, platelet-derived growth factor, G-protein coupled receptor, ERK1/2 signaling and Wnt/β-catenin]. The androgen-receptor-positive TNBC subtype was associated with genes involved in steroid synthesis, porphyrin metabolism, and androgen/estrogen metabolism. Notably, this subtype is reminiscent of previously described apocrine breast cancers [Farmer et al. 2005]. Finally, the immunomodulatory subtype was shown to be characterized by elevated expression of genes involved in T-cell function, immune transcription, interferon (IFN) response and antigen processing. Notably, the immunomodulatory subtype showed overlap with medullary breast cancers, a rare form of TNBC with prominent lymphocytic reaction that is associated with a favorable prognosis [Bertucci et al. 2006].
Immune infiltrates and prognosis in breast cancer
The identification of a TNBC subtype characterized by elevated expression of immune genes suggests that some patients may benefit from immune-based therapies. Over recent years, overwhelming data have revealed the importance of tumor-infiltrating lymphocytes (TILs) in controlling the clinical progression of various epithelial cancers [Fridman et al. 2012]. In colorectal cancers, for instance, immune-scoring determined by IHC of intratumoral CD8+ T cells has shown superior prognostic power than standard tumor staging (AJCC/UICC-TNM classification) methods [Mlecnik et al. 2011]. In breast cancer, two large series, both in newly diagnosed or early stage breast cancer, support a correlation between immune gene signature and better clinical outcomes [Denkert et al. 2010; Mahmoud et al. 2011]. Unsupervised gene expression profiling of breast cancer-associated stroma has also revealed a gene signature predictive of good prognosis (>98% 5-year survival) that was enriched for cytotoxic CD8+ T cell genes and natural killer cell activity [Finak et al. 2008].
While CD8+ T-cell infiltrates are generally associated with better prognosis, CD4+ T cells, which include T-regulatory cells, and tumor-associated macrophages (TAMs) have been associated with worse outcomes. A recent study assessed whether intratumoral T cells and macrophages correlated with clinical outcome in patients with breast cancer. IHC analysis of tissue microarrays derived from 179 treatment-naïve breast tumors revealed that high levels of macrophages and CD4+ T cells correlated with reduced overall survival (OS), while high levels of CD8+ T cells combined with low levels of macrophages and CD4+ T cells correlated with increased OS [DeNardo et al. 2011]. Thus, CD8+ T cells can control human breast cancer, but the presence of immunosuppressive cells, that is, CD4+ T-regulatory cells and macrophages, abrogates this action.
Intratumoral B cells have also been associated with favorable prognosis in breast cancer. In 200 consecutive lymph node negative cases, a B-cell metagene primarily formed by immunoglobulin heavy- and light-chain genes was associated with metastasis-free survival in highly proliferating breast tumors [Schmidt et al. 2008]. Interestingly, immunoglobulin κ C as a single marker has recently been shown to have similar predictive and prognostic value in breast cancer as the entire B-cell metagene [Schmidt et al. 2012].
Immune signature in TNBC
One of the most important conclusions of recent meta-analyses in breast cancer is that the correlation between lymphocytic infiltrates and clinical outcomes varies across subtypes. Historically, traditional assessments of lymphocytic infiltrates (by haematoxylin and eosin stain) have highlighted some uncertainty. However, recent advances in gene expression profiling now reveals that several immune signatures are associated with favorable outcomes. DeNardo and colleagues performed a meta-analysis of CD68 and CD8 gene expression in 4000 breast cancer cases, and reported that a CD68high/CD8low immune gene signature correlated with reduced OS for basal or HER2+ breast cancer subtypes, but not for luminal breast cancers [DeNardo et al. 2011]. Similarly, a metagene of STAT1 signaling, a surrogate of IFN response, was associated with better outcomes in TNBC and HER2+ breast cancers, but not in luminal cases [Desmedt et al. 2008; Ignatiadis et al. 2012]. Another independent group identified an immune response prognostic gene module in ER– but not ER+ breast cancers [Teschendorff et al. 2007]. Consistent with these findings, a recent study investigated the relationship between TIL at diagnosis and clinical outcome after anthracycline-based chemotherapy and reported significant prognostic association only for ER–/HER2+ and TNBC, but not for luminal breast cancers. Patients with TNBC with at least 50% TILs (10.5% of patients) had a 5-year disease-free survival of 89% compared with 62% (p = 0.018, hazard ratio 0.29, 95% confidence interval 0.11–0.81).
Amongst ER– breast cancers, accumulating evidence suggests that basal TNBC may be the most regulated by intratumoral T cells, and thus the most responsive to immunotherapies. Liu and colleagues recently performed a large-scale IHC analysis of CD8 staining on 3400 breast cancer cases representing different subtypes [Liu et al. 2012]. Multivariate analysis indicated that amongst all breast cancer subtypes, only core basal TNBC demonstrated a significant correlation between intratumoral CD8 staining and favorable prognosis. The B-cell metagene has also been associated with good outcome in TNBC. A gene expression analysis of 579 cases of TNBC revealed that a ratio of high B-cell and low interleukin (IL)-8 metagenes identified 32% of patients with TNBC with good prognosis [Rody et al. 2011]. Taken together, these studies suggest that clinical outcomes in ER– breast cancers, especially TNBC, are particularly influenced by tumor immune responses.
The intrinsic nature of TNBC might help explain its increased propensity to inflammatory responses. For instance, signaling pathways that downregulate hormone receptor and HER2 expression may in return stimulate proinflammatory activity. A recent study identified lactoferrin as a repressor of ER, PR, and HER2 expression in breast cancer cells and a direct stimulator of endothelin 1 [Ha et al. 2011]. Alarmins like lactoferrin are proinflammatory molecules that play important intracellular roles and are released upon tissue damage. When released, lactoferrin induces the production cytokines and chemokines, such as IL-10, IL-4, macrophage inflammatory protein (MIP)-1α and MIP-2 [Yang et al. 2009]. Lactoferrin may thus contribute to a triple-negative phenotype in breast cancer and could represent a novel target to stimulate antitumor immune responses.
Immune responses improves chemotherapy of TNBC
Accumulating data now suggest that certain chemotherapeutic drugs, such as anthracyclines, mediate their anticancer activity not only by direct cytotoxic effects but also through activation of CD8+ T-cell responses. Thus, the correlation between intratumoral immune responses and clinical outcomes in TNBC is potentially related to the role of immune cells in the activity of cytotoxic chemotherapeutics. In mice, chemotherapy with anthracyclines requires priming of IFNγ-producing CD8+ T cells [Ghiringhelli et al. 2009]. In humans, correlative studies have reported that high intratumoral levels of IFNγ and CD8+ T cells [Mattarollo et al. 2011], or TILs, are associated with better clinical responses to anthracycline-based chemotherapy [Denkert et al. 2010]. Consistent with these studies, West and colleagues recently reported that in ER– breast cancer, high lymphocyte gene expression is associated with a remarkably high rate (74%) of pathological complete response to neoadjuvant anthracycline-based chemotherapy [West et al. 2011].
The immune-stimulating property of anthracyclines is a three-step process: preapoptotic translocation of calreticulin (CRT) on the tumor cell surface; postapoptotic release of the chromatin-binding protein high-mobility group B1 (HMGB1); and extracellular release of adenosine triphosphate (ATP). CRT, HMGB1, and ATP act in concert to promote tumor antigen presentation by dendritic cells via activation of CD91, Toll-like receptor 4 (TLR), and purinergic P2X7 receptors respectively. Notably, chemotherapy-induced autophagy is essential for the release of ATP and subsequent anticancer immunity. This suggests that patients with TNBC with autophagy-deficient tumor cells might benefit from therapeutic strategies designed to compensate this process.
Because of the suppressive effects of TAMs on antitumor CD8+ T cells, chemosensitivity might also be regulated by the ratio of TAMs to CD8+ T cells in human breast tumors. In support of this, the CD68/CD8A ratio was found to be a predictive biomarker for therapeutic response to neoadjuvant chemotherapy in women with breast cancer. Interestingly, chemotherapy with paclitaxel was shown to increase TAM infiltration in breast tumors [DeNardo et al. 2011]. Indeed, it was shown that breast tumors receiving neoadjuvant chemotherapy have higher levels of TMAs compared with those undergoing primary surgery without preoperative treatment. Using a transgenic mouse model of breast cancer, the authors further demonstrated that TMA depletion significantly improves chemotherapy by enhancing antitumor CD8+ T-cell responses.
Tumor-associated antigens in TNBC
In order to kill tumor cells, CD8+ T cells must recognize specific antigens on tumor cells presented by major histocompatibility complex class I molecules. Hundreds of tumor-associated antigens (TAAs) and respective T-cell epitopes have been identified (see the Cancer Immunity Peptide Database: http://cancerimmunity.org/peptide/). TAAs have been classified into five major groups on the basis of their expression pattern: tissue-differentiation antigens that are shared with the tissue of origin; mutation antigens that arise from point mutations in genes expressed ubiquitously; viral antigens that are associated with transforming viruses; antigens derived from nontransformed cells, such as blood vessel cells and fibroblasts; and antigens preferentially expressed in tumors as a result of epigenetic changes, such as cancer-testis (CT) antigens.
Because CT antigens are not shared with normal somatic cells, they represent attractive candidates for cancer immunotherapy. Over 150 CT antigens have been described thus far, and of these, 83 are encoded on the X-chromosome (also known as CT-X antigens). Grigoriadis and colleagues recently reported the analysis of CT-X antigen expression in human breast cancer samples [Grigoriadis et al. 2009]. Interestingly, the study revealed higher expression of MAGE-A and NY-ESO-1 in ER– versus ER+ breast cancers, and possible coexpression of these CT-X antigens with basal cell markers. Similar findings were subsequently reported by Curigliano and colleagues [Curigliano et al. 2011]. Consistent with these finding, IHC analysis of eight CT antigens, including MAGE-A and NY-ESO-1, also demonstrated significantly more frequent expression in ER– versus ER+ human breast cancers [Chen et al. 2011]. By IHC, CT antigens were found expressed in 12–24% of ER– tumors versus 2–6% in ER+ tumors. In another recent study, Karn and colleagues reported genes differently expressed in human TNBC [Karn et al. 2012]. From a total of 133 bimodally expressed genes, 69 correlated with known molecular subtypes (described above). Interestingly, amongst the 64 remaining genes that showed no correlation with known subtypes of TNBC, the most prominent group of genes encoded for CT antigens. Taken together, these studies strongly suggest that CT antigen expression, including MAGE-A and NY-ESO-1, is a common feature of TNBC.
NY-ESO-1 as an immunotherapeutic target in TNBC
The prognostic implications of CT antigen expression in TNBC remain unknown. Nevertheless, CT antigens represent attractive candidates for cancer immunotherapy. Amongst the different CT antigens, NY-ESO-1 is believed to be one of the most immunogenic. NY-ESO-1 expression has been associated with spontaneous antitumor immune responses in patients with cancer [Valmori et al. 2000; Theurillat et al. 2007]. Ademuyiwa and colleagues recently investigated whether NY-ESO-1 expression in TNBC was also associated with antitumor immune responses [Ademuyiwa et al. 2012]. Consistent with previous studies, NY-ESO-1 expression could be detected by IHC in 16% of TNBC compared with only 2% in ER+ tumors. Remarkably, 8 out of 11 (72.7%) patients with NY-ESO-1+ TNBC had detectable antibody responses to NY-ESO-1. Furthermore, patients with NY-ESO-1+ TNBC had higher levels of CD8+ T-cell infiltrates compared with those with NY-ESO-1– TNBC. As highlighted by the authors, ‘these results suggest that the subset of patients with TNBC whose tumors express NY-ESO-1 have particularly high inherent immunogenicity’, and may represent a population most likely to benefit from immunotherapy.
Immunotherapies for TNBC
The genetic and epigenetic alterations in TNBC provide a set of tumor-associated antigens that the immune system can in theory use to distinguish tumor cells from normal cells [Shah et al. 2012; Stephens et al. 2012]. While evidence suggests that antitumor immunity can control TNBC progression and patient outcome, tumors persist despite being infiltrated with tumor-specific CD8+ T cells. This apparent paradox is at least partly due to the exhausted nature of tumor-infiltrating T cells and the presence of immunosuppressive factors in the tumor microenvironment. We hereafter present an overview of the most promising immunotherapies that may benefit patients with TNBC (Figure 1). These include vaccination strategies to bolster tumor antigens recognized by immune cells, immunotherapies aimed at overcoming immune exhaustion and therapeutic strategies to block tumor-mediated immunosuppression.
Figure 1.
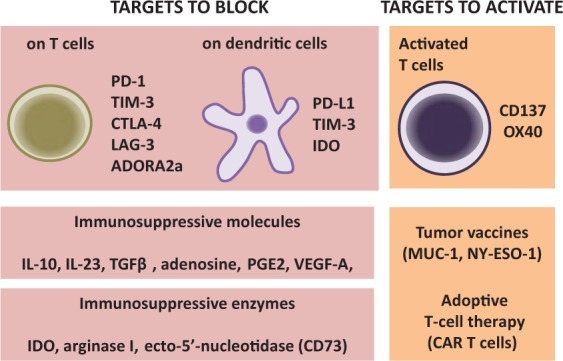
Immunotherapeutic opportunities for triple negative breast cancer (TNBC). Compared with other subtypes, the outcome of TNBC has been shown to be particularly influenced by tumor-infiltrating lymphocytes. Therefore, different immunotherapeutic strategies, aiming at blocking or activating specific targets, are currently envisaged. One of the most promising approaches consists of the use of antagonists against inhibitory receptors that become upregulated on antitumor T cells, such as programmed cell death protein 1 (PD-1), T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), cytotoxic T-lymphocyte antigen 4 (CTLA-4), and lymphocyte-activation gene 3 (LAG-3). Antitumor T cells also express the adenosine receptor A2A (ADORA2a), and antagonists of ADORA2a have been shown to enhance antitumor immunity. PD-L1 and IDO (indoleamine 2,3-dioxygenase), expressed on dendritic cells and tumor cells, as well as immunosuppressive cytokines [such as interleukin (IL)-10, IL-23, transforming growth factor β (TGFβ), adenosine, prostaglandin E2 (PGE2), vascular endothelial growth factor A (VEGF-A)] or immunosuppressive enzymes (such as CD73 and arginase I), could also be targeted to stimulate antitumor immune responses. Alternatively, some approaches aim to activate specific immune targets, such as the coreceptors CD137 (41BB) and OX40, expressed on T cells. Finally, recent evidence suggests that specific tumor antigens, such as MUC-1 and NY-ESO-1, may constitute targetable vaccine antigens. The adoptive transfer of chimeric antigen receptor (CAR)-expressing T cells specific for these tumor antigens is also envisaged.
MAGE-A3 and MUC-1 tumor vaccines
Therapeutic tumor vaccines targeting the CT-X antigen MAGE-A3, expressed in breast cancer, lung cancer, and melanoma, are currently being tested in clinical studies. In a phase II trial, GlaxoSmithKline (GSK) reported that patients with non-small cell lung cancer (NSCLC) whose tumors had been removed by surgery experienced 25% fewer recurrences following vaccination against MAGE-A3. GSK now aims to enrol around 2300 patients with NSCLC positive for MAGE-A3 antigen in a phase III trial [Tyagi and Mirakhur, 2009]. The outcome of this phase III clinical trial could be decisive in the development of tumor vaccines targeting MAGE-A3 or other CT-X antigens expressed in TNBC.
Another candidate target of an immunotherapeutic vaccine is the MUC-1 antigen, which is expressed in the vast majority of breast cancers, including TNBC. While both normal and cancerous breast cells express MUC-1, cancer cells express an aberrantly glycosylated form of MUC-1 due to a lack of core 1,3-galactosyltransferase (T-synthase) [Ju and Cummings, 2002]. The presence of circulating antibodies against MUC-1 at the time of cancer diagnosis has been correlated with a favorable disease outcome in patients with breast cancer [Von Mensdorff-Pouilly et al. 2000]. Furthermore, CD8+ T cells isolated from patients with breast cancer can recognize epitopes present on MUC-1 [Brossart et al. 1999]. Unfortunately, early MUC-1 vaccines have failed to elicit effective antitumor immune responses. A recent preclinical study, however, demonstrated that a glycosylated MUC-1-derived glycopeptide covalently linked to a TLR agonist can elicit potent humoral and cellular immune responses and generate a therapeutic response [Lakshminarayanan et al. 2012]. This study highlighted the importance of maintaining conformational elements of aberrant glycosylation of MUC-1 in order to achieve successful vaccination.
Immune checkpoint blockade
In humans with cancer, antitumor immunity is often ineffective due to the tight regulation associated with the maintenance of immune homeostasis. One of the major limitations is a process known as ‘T-cell exhaustion’, which results from chronic exposure to antigens and is characterized by the upregulation of inhibitory receptors. These inhibitory receptors serve as immune checkpoints in order to prevent uncontrolled immune reactions. Blocking of one or several of these immune checkpoints with monoclonal antibodies (mAbs) has been shown to rescue otherwise exhausted antitumor T cells, and most importantly, has been associated with objective clinical responses in cancer patients. The first immune-checkpoint inhibitor to be tested in a clinical trial was ipilimumab (Yervoy, Bristol-Myers Squibb), an anti-cytotoxic T-lymphocyte antigen 4 (CTLA-4) mAb. CTLA-4 belongs to the immunoglobulin superfamily of receptors, which also includes programmed cell death protein 1 (PD-1), B and T lymphocyte attenuator, T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), and V-domain immunoglobulin suppressor of T cell activation. In 2011, the US Food and Drug Administration approved the use of ipilimumab in patients with metastatic melanoma, either as initial therapy or after relapse.
Anti-CTLA-4 mAb therapy enhances the antitumor function of CD8+ T cells, increases the ratio of CD8+ T cells to Foxp3+ T regulatory cells, and inhibits the suppressive function of T regulatory cells [Quezada et al. 2006]. CTLA-4 blockade has also been shown to expand a subpopulation of tumor-infiltrating CD4+ T cells that express high levels of inducible T-cell costimulator (ICOS) and secrete IFNγ [Chen et al. 2009]. These CD4+ ICOS+ T cells might play a role in the therapeutic activity of anti-CTLA-4 mAb therapy, as their frequency correlates with survival in treated patients with melanoma. The major drawback to anti-CTLA-4 mAb therapy is the generation of autoimmune toxicities due to on-target effects. It has been reported that up to 25% of patients treated with ipilimumab developed serious grade 3–4 adverse events [Kähler and Hauschild, 2011], reflecting the importance of CTLA-4 in maintaining immune homeostasis. Unfortunately, toxicity is not always associated with therapeutic benefit. Thus, a major challenge in the use of anti-CTLA-4 mAb is to define favorable clinical settings that strike an optimum balance between tumor immunity and autoimmunity.
PD-1 is another inhibitory coreceptor expressed on activated and exhausted T cells. Its ligand, PD-L1, is often found overexpressed in various types of cancer. Administration of blocking anti-PD-1/anti-PD-L1 mAbs enhance adaptive antitumor immune responses by preventing T-cell exhaustion [Hirano et al. 2005]. Anti-PD-1 mAb blocks interactions between PD-1 and its ligands, PD-L1 and PD-L2, whereas anti-PD-L1 mAb blocks interactions between PD-L1 and both PD-1 and CD80. PD-1 is expressed by activated CD4+ and CD8+ T cells, B cells, monocytes and natural killer T cells. It has two ligands, PD-L1 and PD-L2, with distinct expression profiles. Expression of PD-L1 has been shown to be associated with poor prognosis in melanoma and hepatocellular carcinoma [Gao et al. 2009; Gadiot et al. 2011]. Notably, cytotoxic chemotherapeutics such as paclitaxel, etoposide, and 5-fluorouracil have been shown to upregulate PD-L1 expression on breast cancer cells [Zhang et al. 2008].
Two phase I trials recently reported clinical responses with anti-PD-1 or anti-PD-L1 mAb in pretreated patients with diverse tumor types [Brahmer et al. 2012; Topalian et al. 2012]. Anti-PD-1 mAb therapy was associated with objective responses in 18% of patients with NSCLC (14 of 76 patients), 28% of patients with melanoma (26 of 94 patients), and 27% of patients with renal-cell cancer (9 of 33 patients) [Topalian et al. 2012]. Anti-PD-L1 mAb therapy was associated with objective responses in 17% of patients with melanoma (9 of 52) 12% of patients with renal-cell cancer (2 of 17), 10% of patients with NSCLC (5 of 49), and 6% of patients with ovarian cancer (1 of 17) [Brahmer et al. 2012]. Notably, anti-PD-1 and anti-PD-L1 mAb therapy caused drug-related grade 3 or 4 adverse events in 14% and 9% of patients respectively. Strikingly, in the context of anti-PD-1 mAb therapy, objective responses occurred only in PD-L1+ tumors (36% response rate) compared with no clinical responses in PD-L1– tumors.
Combination of checkpoint inhibitors
While inhibition of a single immune checkpoint can prolong the survival of patients with cancer, an important question that remains is whether combinatorial checkpoint blockade can by synergistic in promoting anticancer activity. The first combination of immune checkpoint inhibitors to be tested in mice was the combination of anti-CTLA-4 and anti-PD-1 mAbs. Curran and colleagues demonstrated that blockade of CTLA-4 and PD-1 in mice allows CD8+ and CD4+ T cells to survive in the tumor microenvironment, proliferate, and carry out effector function [Curran et al. 2010].
More recently, TIM-3 has been identified as another important inhibitory receptor expressed by exhausted CD8+ T cells. In mouse models of cancer, it was shown that the most dysfunctional tumor-infiltrating CD8+ T cells actually coexpress PD-1 and TIM-3 [Sakuishi et al. 2010]. Based on these findings, a direct comparison of the therapeutic activity of anti-CTLA-4, anti-PD-1 and anti-TIM-3 mAbs was made in various mouse models of cancer [Ngiow et al. 2011]. It was observed that the combination of anti-PD-1 and anti-TIM-3 mAbs had the most potent anticancer effect against well established experimental and carcinogen-induced tumors. From a molecular point of view, a recent paper by Rangachari and colleagues identified Bat3 as a key regulator of TIM-3 activity on T lymphocytes [Rangachari et al. 2012]. Bat3, through its binding to the intracellular tail of TIM-3, prevents TIM-3 mediated cell death or exhaustion in T lymphocytes. Interestingly, the authors demonstrated that Bat3 is highly downregulated in TIM3+ or PD1+ TILs and that this downmodulation is associated with a decreased cytotoxic potential as revealed by a reduced secretion of IFNγ and tumor necrosis factor α (TNFα).
In addition to its inhibitory role on CD8 T cells, TIM-3 has also been reported as a key regulator of nucleic acid mediated antitumor immunity. In a very recent paper, TIM-3 was shown to be upregulated on tumor-associated dendritic cells (TADCs) extracted from both mouse and human tumors [Chiba et al. 2012]. The authors identified IL-10, vascular endothelial growth factor A, and arginase I as the main tumor-released immunosuppressive factors responsible for TIM-3 upregulation on TADCs. TIM-3 expression in TADCs was linked to an impaired nucleic acid mediated innate immune response as revealed by a reduced secretion of cytokines such as IFNβ or IL-12. Accordingly, it was proven that anti-TIM-3 mAb therapy greatly enhanced the antitumor efficacy of nucleic-acid based adjuvants in a B16F10 mouse melanoma model, and that this synergistic activity depended on IFNβ and IL-12 secretion. More importantly, using a CD11c DTR mouse strain (in which CD11c can be depleted upon diphtheria toxin administration), it was demonstrated that TIM-3 expression on TADCs (and not on CD8 T cells) was the main limit to the triggering of a nucleic acid mediated antitumor immune response. From a mechanistic point of view, TIM-3 limited dendritic cell innate immune response in a HMGB1-dependent fashion, restraining the HMGB1-mediated transport of nucleic acid into endosome and thus limiting the activation of cytosolic sensors responsible for nucleic-acid-mediated immune response. Finally, the authors extend the relevance of their study showing that TIM-3 mAb therapy strongly synergizes with standard chemotherapy in a subcutaneous colon tumor model, which reinforces the rational for combining ‘immunogenic cell death’ inducing chemotherapeutic agents with immune checkpoint inhibitors for cancer therapy.
Lymphocyte-activation gene 3 (LAG-3) is another recently identified inhibitory receptor that acts to limit effector T-cell function and augment the suppressive activity of T regulatory cells. Woo and colleagues recently revealed that PD-1 and LAG-3 are extensively coexpressed by tumor-infiltrating T cells in mice, and that combined blockade of PD-1 and LAG-3 provokes potent synergistic antitumor immune responses against mouse models of cancer [Woo et al. 2012]. These studies suggest that combined blockade of immune checkpoint inhibitors may represent a promising strategy for cancer immunotherapy.
Agonistic of tumor necrosis factor receptor superfamily
Members of the TNF receptor superfamily also play an important role as regulators of T-cell function [Croft et al. 2012]. Activation of these costimulatory receptors may further enhance the generation of tumor-reactive T cells in the context of cancer therapy. Costimulatory receptors of the TNF receptor family are composed of OX40 (CD134), 4-1BB (CD137), CD27, CD30, and herpes virus entry mediator. When activated, each of these receptors can enhance cytokine production and T-cell proliferation in response to T-cell receptor signaling. OX40 and CD137 activation are particularly effective at allowing activated T cells to survive and proliferate in the late phase of immune responses. The administration of agonistic mAbs against OX40 or CD137 has been shown to enhance tumor immunity and induce regression of established mouse models of cancer [Piconese et al. 2008; Narazaki et al. 2010; Palazón et al. 2011]. Taken together, the use of agonists to costimulatory receptors or antagonists to inhibitory receptors may provide efficient means to rescue or enhance the activity of tumor-reactive T cells.
Blocking the immunosuppressors
Targeting immunosuppression by soluble mediators is another attractive approach for cancer immunotherapy. A plethora of immunosuppressive factors has been associated with tumorigenesis, including TGFβ, indoleamine 2,3-dioxygenase (IDO), arginase, prostaglandin E2, and extracellular adenosine [Stagg et al. 2007; Stagg and Smyth, 2010]. Determining which immunosuppressive factors are minimally required for maintaining tumor tolerance in a given patient population remains a great challenge. Recent studies in mouse models of cancer and clinical correlative studies suggest that IL-23 may be a key cytokine governing the balance between protumorigenic and antitumorigenic immune responses [Langowski et al. 2006; Teng et al. 2011; Gangemi et al. 2012]. In support of this model, mice genetically deficient in IL-23 are significantly protected against a wide range of malignancies and mice treated with a blocking antibody against IL-23 have a decreased risk of tumor formation and a faster elimination of transplanted tumor cells [Teng et al. 2011].
Enzymes that metabolize L-arginine (such as arginase I), the tryptophan-catabolizing enzyme IDO, as well as enzymes that regulate extracellular adenosine levels (such as the ecto-nucleotidases CD39 and CD73) also significantly contribute to the inhibition of anticancer immune responses [Stagg et al. 2010; Singer et al. 2011]. CD73 is at a critical checkpoint in the conversion of immune-activating ATP into immunosuppressive adenosine, making it a potential therapeutic target. Tumors often overexpress CD73 as a consequence of tissue hypoxia or, in the case of breast cancer, consequent to the loss of ER expression. Proof of principle studies have revealed that anti-CD73 mAb therapy can reduce tumor burden and prevent metastasis in mice [Stagg et al. 2010, 2011, 2012; Wang et al. 2011]. While tumor-derived CD73 is a significant contributor to the generation of adenosine, host CD73 also exacerbates tumorigenesis, highlighted by the reduced susceptibility of CD73-deficient mice against a number of transplantable and spontaneous tumors. Given the promising results of anti-CD73 targeted therapy in mice, future studies aimed at translating this approach into the clinic are warranted. A combination of these agents with immunogenic cytotoxic agents could also be a reasonable approach to enhance tumor antigen recognition by the host.
Adoptive T-cell therapy
Another promising immunotherapy consists of the adoptive transfer of tumor-specific T cells. Work pioneered by the Rosenberg lab established that autologous TILs can be isolated from primary tumors, expanded ex vivo, and adoptively transferred back to patients following lymphodepletion. For patients with metastatic melanoma capable of withstanding treatment, TIL therapy combined with IL-2 is the best available option, with responses rates ranging from 49% to 72% [Rosenberg et al. 2011]. TIL therapy, however, has not proven to be readily applicable in most clinical settings, and to date, only patients with melanoma have consistently demonstrated favorable outcomes.
Building up from the successes of TIL therapy in patients with melanoma, the past decade has seen the emergence of a novel form of adoptive cell therapy based on the transfer of genetically engineered T cells expressing a single-chain antibody structure fused to an intracellular T-cell receptor signaling domain called chimeric antigen receptor (CAR) [Jena et al. 2010]. The inclusion of CD28 and CD137 costimulatory signaling in the intracellular domain of CAR has substantially improved their therapeutic efficacy. The therapeutic potential of CAR-expressing T cells for treatment of chronic lymphoid leukemia was recently revealed in a clinical study in which infusion of autologous T cells modified to express a CD19-specific CAR induced complete regression of a patient with refractory disease [Porter et al. 2011]. In a breast cancer clinical trial, however, T cells genetically engineered to target HER2+ tumors resulted in the death of the patient, presumably due to cross reactivity of CAR T cells with normal cells expressing low levels of HER2 [Morgan et al. 2010].
A search for tumor antigens associated with TNBC recently identified mesothelin as a novel immunotherapy target [Tchou et al. 2012]. Mesothelin is a cell-surface glycoprotein normally present on mesothelial cells that has previously shown promise as an immunotherapeutic target in ovarian cancer and mesothelioma. Tchou and colleagues screened 99 primary breast cancer samples by IHC for mesothelin expression [Tchou et al. 2012]. While mesothelin was rarely expressed in ER+ or HER2+ breast cancers, 67% (29 out of 43) of patients with TNBC exhibited mesothelin expression in at least 5% of tumor cells, with 19% of patients with TNBC expressing mesothelin in over 50% of tumor cells. In proof-of-concept experiments, the authors also demonstrated specific cytotoxicity of primary mesothelin-positive breast cancer cells by genetically modified T cells expressing a CAR specific for mesothelin. While further validation in larger cohorts is needed, this study suggests that mesothelin might represent a valid and previously unrecognized target for adoptive T-cell therapy of TNBC.
Conclusion
Unlike other molecular subtypes of breast cancers, triple-negative disease does not benefit from targeted therapies and is associated with worse clinical outcomes. As such, TNBC remains a challenge in medical practice. Among the new therapeutic options for TNBC, cellular or molecular immunotherapies appear to be very promising approaches. Their rational has been supported by several studies showing that TNBC outcome is correlated with tumor-immune infiltrate. Building on these results, a variety of different immunotherapeutic strategies are currently being tested in preclinical and clinical studies, with some showing promising therapeutic activity. Among them, the use of immune-checkpoint blocking antibodies, such as anti-PD-1 or anti-PD-L1, is particularly attractive. Importantly, immune-stimulating therapies might act synergistically when combined with chemotherapeutic drugs currently used in the clinic. As we have discussed, other immune-based strategies that might be promising for TNBC include the adoptive transfer of CAR-engineered T cells or the development of tumor vaccines against CT antigens, which appear highly expressed in TNBC.
In conclusion, in our endeavor towards new therapeutic approaches, one should keep in mind that TNBC itself is a highly heterogeneous disease, implying that not all patients will benefit from given immunotherapeutic strategies. The next challenge will be to find the optimal clinical setting for each new treatment.
Footnotes
Funding: J. Stagg is supported by the Famille Jean-Guy Sabourin Pharmaceutical Research Chair of University of Montreal.
Conflict of interest statement: The authors declare no conflicts of interest in preparing this article.
Contributor Information
John Stagg, Centre de Recherche du Centre Hospitalier de l’Université de Montréal, Faculté de Pharmacie et Institut du Cancer de Montréal, Montréal, Québec, Canada H2L 4M1.
Bertrand Allard, Centre de Recherche du Centre Hospitalier de l’Université de Montréal, Faculté de Pharmacie et Institut du Cancer de Montréal, Montréal, Québec, Canada.
References
- Ademuyiwa F., Bshara W., Attwood K., Morrison C., Edge S., Ambrosone C., et al. (2012) NY-ESO-1 cancer testis antigen demonstrates high immunogenicity in triple negative breast cancer. PLoS ONE 7: e38783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C., Deal A., Miller C., Khorram C., Meng H., Burrows E., et al. (2011) The prognostic contribution of clinical breast cancer subtype, age, and race among patients with breast cancer brain metastases. Cancer 117: 1602–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnes J., Brunet J., Stefansson I., Bégin L., Wong N., Chappuis P., et al. (2005) Placental cadherin and the basal epithelial phenotype of BRCA1-related breast cancer. Clin Cancer Res 11: 4003–4011 [DOI] [PubMed] [Google Scholar]
- Bauer K., Brown M., Cress R., Parise C., Caggiano V. (2007) Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 109: 1721–1728 [DOI] [PubMed] [Google Scholar]
- Bertucci F., Finetti P., Cervera N., Charafe-Jauffret E., Mamessier E., Adélaïde J., et al. (2006) Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res 66: 4636–4644 [DOI] [PubMed] [Google Scholar]
- Brahmer J., Tykodi S., Chow L., Hwu W., Topalian S., Hwu P., et al. (2012) Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N Engl J Med 366: 2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossart P., Heinrich K., Stuhler G., Behnke L., Reichardt V., Stevanovic S., et al. (1999) Identification of HLA-A2–restricted T-cell epitopes derived from the MUC1 tumor antigen for broadly applicable vaccine therapies. Blood 93: 4309–4317 [PubMed] [Google Scholar]
- Cancello G., Maisonneuve P., Rotmensz N., Viale G., Mastropasqua M., Pruneri G., et al. (2010) Prognosis and adjuvant treatment effects in selected breast cancer subtypes of very young women (<35 years) with operable breast cancer. Ann Oncol 21:1974–1981 [DOI] [PubMed] [Google Scholar]
- Carey L. (2006) Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 295: 2492–2502 [DOI] [PubMed] [Google Scholar]
- Cheang M., Voduc D., Bajdik C., Leung S., McKinney S., Chia S., et al. (2008) Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 14: 1368–1376 [DOI] [PubMed] [Google Scholar]
- Chen H., Liakou C., Kamat A., Pettaway C., Ward J., Tang D., et al. (2009) Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-γ levels in both nonmalignant and malignant prostate tissues. Proc Natl Acad Sci 106: 2729–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Ross D., Chiu R., Zhou X., Chen Y., Lee P., et al. (2011) Multiple cancer/testis antigens are preferentially expressed in hormone-receptor negative and high-grade breast cancers. PLoS ONE 6: e17876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S., Baghdadi M., Akiba H., Yoshiyama H., Kinoshita I., Dosaka-Akita H., et al. (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 29 July 2012. (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M., Duan W., Choi H., Eun S., Madireddi S., Mehta A. (2012) TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol 33: 144–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curigliano G., Viale G., Ghioni M., Jungbluth A., Bagnardi V., Spagnoli G., et al. (2011) Cancer–testis antigen expression in triple-negative breast cancer. Ann Oncol 22: 98–103 [DOI] [PubMed] [Google Scholar]
- Curran M., Montalvo W., Yagita H., Allison J. (2010) PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci 107: 4275–4280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo D., Brennan D., Rexhepaj E., Ruffell B., Shiao S., Madden S., et al. (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery 1: 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C., Loibl S., Noske A., Roller M., Müller B., Komor M., et al. (2010) Tumor-Associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28: 105–113 [DOI] [PubMed] [Google Scholar]
- Dent R., Hanna W., Trudeau M., Rawlinson E., Sun P., Narod S. (2009) Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res Treat 115: 423–428 [DOI] [PubMed] [Google Scholar]
- Dent R., Trudeau M., Pritchard K., Hanna W., Kahn H., Sawka C., et al. (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13: 4429–4434 [DOI] [PubMed] [Google Scholar]
- Desmedt C., Haibe-Kains B., Wirapati P., Buyse M., Larsimont D., Bontempi G., et al. (2008) Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 14: 5158–5165 [DOI] [PubMed] [Google Scholar]
- Farmer P., Bonnefoi H., Becette V., Tubiana-Hulin M., Fumoleau P., Larsimont D., et al. (2005) Identification of molecular apocrine breast tumours by microarray analysis. Breast Cancer Res 7: 1–1 [DOI] [PubMed] [Google Scholar]
- Finak G., Bertos N., Pepin F., Sadekova S., Souleimanova M., Zhao H., et al. (2008) Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14: 518–527 [DOI] [PubMed] [Google Scholar]
- Foulkes W., Stefansson I., Chappuis P., Bégin L., Goffin J., Wong N., et al. (2003) Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J Natl Cancer Inst 95: 1482–1485 [DOI] [PubMed] [Google Scholar]
- Fridman W., Pagès F., Sautès-Fridman C., Galon J. (2012) The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12: 298–306 [DOI] [PubMed] [Google Scholar]
- Fulford L., Easton D., Reis-Filho J., Sofronis A., Gillett C., Lakhani S., et al. (2006) Specific morphological features predictive for the basal phenotype in grade 3 invasive ductal carcinoma of breast. Histopathology 49: 22–34 [DOI] [PubMed] [Google Scholar]
- Gadiot J., Hooijkaas A., Kaiser A., van Tinteren H., van Boven H., Blank C. (2011) Overall survival and PD-L1 expression in metastasized malignant melanoma. Cancer 117: 2192–2201 [DOI] [PubMed] [Google Scholar]
- Gangemi S., Minciullo P., Adamo B., Franchina T., Ricciardi G., Ferraro M., et al. (2012) Clinical significance of circulating interleukin-23 as a prognostic factor in breast cancer patients. J Cell Biochem 113: 2122–2125 [DOI] [PubMed] [Google Scholar]
- Gao Q., Wang X., Qiu S., Yamato I., Sho M., Nakajima Y., et al. (2009) Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res 15: 971–979 [DOI] [PubMed] [Google Scholar]
- Ghiringhelli F., Apetoh L., Tesniere A., Aymeric L., Ma Y., Ortiz C., et al. (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med 15: 1170–1178 [DOI] [PubMed] [Google Scholar]
- Grigoriadis A., Caballero O., Hoek K., da Silva L., Chen Y., Shin S., et al. (2009) CT-X antigen expression in human breast cancer. Proc Natl Acad Sci 106: 13493–13498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha N., Nair V., Reddy D., Mudvari P., Ohshiro K., Ghanta K., et al. (2011) Lactoferrin–endothelin-1 axis contributes to the development and invasiveness of triple-negative breast cancer phenotypes. Cancer Res 71: 7259–7269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond M., Hayes D., Dowsett M., Allred D., Hagerty K., Badve S., et al. (2010) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 28: 2784–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano F., Kaneko K., Tamura H., Dong H., Wang S., Ichikawa M., et al. (2005) Blockade of B7–H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 65: 1089–1096 [PubMed] [Google Scholar]
- Ignatiadis M., Singhal S., Desmedt C., Haibe-Kains B., Criscitiello C., Andre F., et al. (2012) Gene modules and response to neoadjuvant chemotherapy in breast cancer subtypes: a pooled analysis. J Clin Oncol 30: 1996–2004 [DOI] [PubMed] [Google Scholar]
- Jena B., Dotti G., Cooper L. (2010) Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood 116: 1035–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju T., Cummings R. (2002) A unique molecular chaperone Cosmc required for activity of the mammalian core 1 B3-galactosyltransferase. Proc Natl Acad Sci 99: 16613–16618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kähler K., Hauschild A. (2011) Treatment and side effect management of CTLA-4 antibody therapy in metastatic melanoma. J Dtsch Dermatol Ges 9: 277–286 [DOI] [PubMed] [Google Scholar]
- Karn T., Pusztai L., Ruckhäberle E., Liedtke C., Müller V., Schmidt M., et al. (2012) Melanoma antigen family A identified by the bimodality index defines a subset of triple negative breast cancers as candidates for immune response augmentation. Eur J Cancer 48: 12–23 [DOI] [PubMed] [Google Scholar]
- Lakshminarayanan V., Thompson P., Wolfert M., Buskas T., Bradley J., Pathangey L., et al. (2012) Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc Natl Acad Sci 109: 261–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langowski J., Zhang X., Wu L., Mattson J., Chen T., Smith K., et al. (2006) IL-23 promotes tumour incidence and growth. Nature 442: 461–465 [DOI] [PubMed] [Google Scholar]
- Lehmann B., Bauer J., Chen X., Sanders M., Chakravarthy A., Shyr Y., et al. (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121: 2750–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C., Mazouni C., Hess K., André F., Tordai A., Mejia J., et al. (2008) Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26: 1275–1281 [DOI] [PubMed] [Google Scholar]
- Liu S., Lachapelle J., Leung S., Gao D., Foulkes W., Nielsen T. (2012) CD8+ lymphocyte infiltration is an independent favorable prognostic indicator in basal-like breast cancer. Breast Cancer Res 14: R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livasy C., Perou C., Karaca G., Cowan D., Maia D., Jackson S., et al. (2007) Identification of a basal-like subtype of breast ductal carcinoma in situ. Hum Pathol 38: 197–204 [DOI] [PubMed] [Google Scholar]
- Lund M., Trivers K., Porter P., Coates R., Leyland-Jones B., Brawley O., et al. (2009) Race and triple negative threats to breast cancer survival: a population-based study in Atlanta, GA. Breast Cancer Res Treat 113: 357–370 [DOI] [PubMed] [Google Scholar]
- Mahmoud S., Paish E., Powe D., Macmillan R., Grainge M., Lee A., et al. (2011) Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 29: 1949–1955 [DOI] [PubMed] [Google Scholar]
- Mattarollo S., Loi S., Duret H., Ma Y., Zitvogel L., Smyth M. (2011) Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 71: 4809–4820 [DOI] [PubMed] [Google Scholar]
- Metzger O., de Azambuja E., Quinaux E., Francis P., Buyse M., Crown J., et al. (2010) 11 Lymph node ratio is an independent risk classifier in node positive breast cancer patients: results of the phase III BIG 02–98 trial. Eur J Cancer Suppl 8: 61 [Google Scholar]
- Mlecnik B., Tosolini M., Kirilovsky A., Berger A., Bindea G., Meatchi T., et al. (2011) Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol 29: 610–618 [DOI] [PubMed] [Google Scholar]
- Molyneux G., Geyer F., Magnay F., McCarthy A., Kendrick H., Natrajan R., et al. (2010) BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7: 403–417 [DOI] [PubMed] [Google Scholar]
- Morgan R., Yang J., Kitano M., Dudley M., Laurencot C., Rosenberg S. (2010) Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18: 843–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narazaki H., Zhu Y., Luo L., Zhu G., Chen L. (2010) CD137 agonist antibody prevents cancer recurrence: contribution of CD137 on Both hematopoietic and nonhematopoietic cells. Blood 115: 1941–1948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngiow S., von Scheidt B., Akiba H., Yagita H., Teng M., Smyth M. (2011) Anti-TIM3 antibody promotes T Ccell IFN-γ–mediated antitumor immunity and suppresses established tumors. Cancer Res 71: 3540–3551 [DOI] [PubMed] [Google Scholar]
- Nielsen T., Hsu F., Jensen K., Cheang M., Karaca G., Hu Z., et al. (2004) Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res 10: 5367–5374 [DOI] [PubMed] [Google Scholar]
- Palazón A., Teijeira A., Martínez-Forero I., Hervás-Stubbs S., Roncal C., Peñuelas I., et al. (2011) Agonist ati-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Res 71: 801–811 [DOI] [PubMed] [Google Scholar]
- Perou C., Sørlie T., Eisen M., van de, Rijn M., Jeffrey S., Rees C., et al. (2000) Molecular portraits of human breast tumours. Nature 406: 747–752 [DOI] [PubMed] [Google Scholar]
- Piconese S., Valzasina B., Colombo M. (2008) OX40 Triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med 205: 825–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter D., Levine B., Kalos M., Bagg A., June C. (2011) Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A., Parker J., Karginova O., Fan C., Livasy C., Herschkowitz J., et al. (2010) Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 12: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezada S., Peggs K., Curran M., Allison J. (2006) CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest 116: 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakha E., Ellis I. (2009) Triple-negative/basal-like breast cancer. Pathology 41: 40–47 [DOI] [PubMed] [Google Scholar]
- Rangachari M., Zhu C., Sakuishi K., Xiao S., Karman J., Chen A., et al. (2012) Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med 18: 1394–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A., Karn T., Liedtke C., Pusztai L., Ruckhaeberle E., Hanker L., et al. (2011) A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res 13: R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg S., Yang J., Sherry R., Kammula U., Hughes M., Phan G., et al. (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17: 4550–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K., Apetoh L., Sullivan J., Blazar B., Kuchroo V., Anderson A. (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207: 2187–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M., Böhm D., von Törne C., Steiner E., Puhl A., Pilch H., et al. (2008) The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res 68: 5405–5413 [DOI] [PubMed] [Google Scholar]
- Schmidt M., Hellwig B., Hammad S., Othman A., Lohr M., Chen Z., et al. (2012) A comprehensive analysis of human gene expression profiles identifies stromal immunoglobulin kappa c as a compatible prognostic marker in human solid tumors. Clin Cancer Res 18: 2695–2703 [DOI] [PubMed] [Google Scholar]
- Shah S., Roth A., Goya R., Oloumi A., Ha G., Zhao Y., et al. (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486: 395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer K., Gottfried E., Kreutz M., Mackensen A.(2011) Suppression of T-cell responses by tumor metabolites. Cancer Immunol Immunother 60: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J., Beavis P., Divisekera U., Liu M., Möller A., Darcy P., et al. (2012) CD73-deficient mice are resistant to carcinogenesis. Cancer Res 72: 2190–2196 [DOI] [PubMed] [Google Scholar]
- Stagg J., Divisekera U., Duret H., Sparwasser T., Teng M., Darcy P., et al. (2011) CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71: 2892–2900 [DOI] [PubMed] [Google Scholar]
- Stagg J., Divisekera U., McLaughlin N., Sharkey J., Pommey S., Denoyer D., et al. (2010) Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci 107: 1547–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg J., Johnstone R., Smyth M. (2007) From cancer immunosurveillance to cancer immunotherapy. Immunol Rev 220: 82–101 [DOI] [PubMed] [Google Scholar]
- Stagg J., Smyth M. (2010) Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29: 5346–5358 [DOI] [PubMed] [Google Scholar]
- Stephens P., Tarpey P., Davies H., Loo P., Greenman C., Wedge D., et al. (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchou J., Wang L., Selven B., Zhang H., Conejo-Garcia J., Borghaei H., et al. (2012) Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat 133: 799–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng M., von Scheidt B., Duret H., Towne J., Smyth M. (2011) Anti-IL-23 monoclonal antibody synergizes in combination with targeted therapies or IL-2 to suppress tumor growth and metastases. Cancer Res 71: 2077–2086 [DOI] [PubMed] [Google Scholar]
- Teschendorff A., Miremadi A., Pinder S., Ellis I., Caldas C. (2007) An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol 8: R157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theurillat J., Ingold F., Frei C., Zippelius A., Varga Z., Seifert B., et al. (2007) NY-ESO-1 protein expression in primary breast carcinoma and metastases—correlation with CD8+ T-cell and CD79a+ plasmacytic/B-cell Infiltration. Int J Cancer 120: 2411–2417 [DOI] [PubMed] [Google Scholar]
- Topalian S., Hodi F., Brahmer J., Gettinger S., Smith D., McDermott D., et al. (2012) Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med 366: 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi P., Mirakhur B. (2009) MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin Lung Cancer 10: 371–374 [DOI] [PubMed] [Google Scholar]
- Valmori D., Dutoit V., Liénard D., Rimoldi D., Pittet M., Champagne P., et al. (2000) Naturally occurring human lymphocyte antigen-A2 restricted CD8+ T-cell response to the cancer testis antigen NY-ESO-1 in melanoma patients. Cancer Res 60: 4499–4506 [PubMed] [Google Scholar]
- Von Mensdorff-Pouilly S., Verstraeten A., Kenemans P., Snijdewint F., Kok A., Kamp G., et al. (2000) Survival in early breast cancer patients is favorably influenced by a natural humoral immune response to polymorphic epithelial mucin. J Clin Oncol 18: 574–574 [DOI] [PubMed] [Google Scholar]
- Wang L., Fan J., Thompson L., Zhang Y., Shin T., Curiel T., et al. (2011) CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 121: 2371–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West N., Milne K., Truong P., Macpherson N., Nelson B., Watson P. (2011) Tumor-infiltrating lymphocytes predict response to anthracycline-based chemotherapy in estrogen receptor-negative breast cancer. Breast Cancer Res 13: R126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff A., Hammond M., Schwartz J., Hagerty K., Allred D., Cote R., et al. (2006) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol 25: 118–145 [DOI] [PubMed] [Google Scholar]
- Woo S., Turnis M., Goldberg M., Bankoti J., Selby M., Nirschl C., et al. (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72: 917–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., de la Rosa G., Tewary P., Oppenheim J. (2009) Alarmins link neutrophils and dendritic cells. Trends Immunol 30: 531–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Su D., Liang M., Fu J. (2008) Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol Immunol 45: 1470–1476 [DOI] [PubMed] [Google Scholar]