

Abstract
Previous experiments from our laboratories have identified peptides derived from the human astrovirus coat protein (CP) that bind C1q and mannose binding lectin (MBL) inhibiting activation of the classical and lectin pathways of complement, respectively. The purpose of this study was to evaluate the function of these coat protein peptides (CPPs) in an in vitro model of complement-mediated disease (ABO incompatibility), preliminarily assess their in vivo complement suppression profile and develop more highly potent derivatives of these molecules. E23A, a 30 amino acid CPP derivative previously demonstrated to inhibit classical pathway activation was able to dose-dependently inhibit lysis of AB erythrocytes treated with mismatched human O serum. Additionally, when injected into rats, E23A inhibited the animals’ serum from lysing antibody-sensitized erythrocytes, providing preliminary in vivo functional evidence that this CPP can cross the species barrier to inhibit serum complement activity in rodents. A rational drug design approach was implemented to identify more potent CPP derivatives, resulting in the identification and characterization of a 15 residue peptide (Polar Assortant (PA)), which demonstrated both superior inhibition of classical complement pathway activation and robust binding to C1q collagen-like tails. PA also inhibited ABO incompatibility in vitro and demonstrated in vivo complement suppression up to 24 hours post-injection. CPP’s ability to inhibit ABO incompatibility in vitro, proof of concept in vivo inhibitory activity in rats and the development of the highly potent PA derivative set the stage for preclinical testing of this molecule in small animal models of complement-mediated disease.
Keywords: coat protein, peptides, complement, C1q, MBL, ABO incompatibility
1. Introduction
Innate immunity is a critical component of host defense and consists of multiple systems to prevent invasion of pathogens. The complement cascade plays a fundamental role in innate immunity by contributing to opsonization of microbial surfaces with C3b and C4b, neutrophil and phagocyte recruitment through C3a and C5a anaphylatoxin generation, and cell lysis via membrane attack complex assembly (reviewed in Köhl, 2006). Inappropriate activation or dysregulated control of complement activation plays a central role in the pathogenesis of many human diseases (Ricklin and Lambris, 2007), including antibody-initiated, complement-mediated acute intravascular hemolytic disease processes such as acute ABO incompatible blood transfusion reactions (reviewed by Strobel, 2008) or hemolytic disease of the newborn (Roberts, 2008). In these disease processes, preexisting antibodies, typically natural antibodies, bind to erythrocyte surface antigens initiating classical complement pathway activation via C1 (Schreiber, 1998). Subsequent terminal complement cascade activation leads to membrane attack complex formation on the erythrocyte surface and intravascular hemolysis. Due to the amplification cascade of complement, acute intravascular hemolysis is a very rapid process frequently resulting in death, acute renal failure due to hemoglobinemia, or fetal hydrops (Pruss et al., 2003). To date, exchange blood transfusion remains the primary therapy for hemolytic disease of the newborn (Thayyil and Milligan, 2006) with no pharmacological interventions to treat these disease processes.
Our group has previously demonstrated that the coat protein (CP) of human astrovirus type 1 inhibits activation of the classical and lectin complement pathways and generation of subsequent downstream complement effectors (Bonaparte et al., 2008; Hair et al., 2010). CP was found to bind C1q, disrupting its interaction with the cognate serine protease complex (C1r-C1s-C1s-C1r), and preventing C1s cleavage (Hair et al., 2010). Additionally, CP was shown to bind MBL and presumably exerts its effect on the lectin pathway by interaction with the MASP2 binding site on MBL (Hair et al, 2010). The discovery that a region of the astrovirus CP shares homology with human neutrophil defensin peptide 1 (HNP-1) (Groeneveld et al., 2007) led to the development of coat protein peptides (CPPs) that also bound C1q and inhibited classical pathway activation, but were only 15 – 30 amino acids in length (Gronemus et al., 2010). Two CPPs derived from the native CP sequence were identified that demonstrated superior inhibitory activity: E23A (PAICQRATATLGTVGSNTSGTTEIAACILL, 30 aa) and d8-22 (PAICQRAEIEACILL, 15aa) (Gronemus et al, 2010).
The ability to halt classical complement pathway activation presents an opportunity to develop a novel therapeutic to treat antibody-initiated complement-mediated diseases such as ABO incompatibility. As a prerequisite to the development of a viable therapeutic to inhibit classical pathway activation, we tested whether CPP could inhibit ABO incompatible hemolysis in vitro as well as perform preliminary test-of-concept in vivo injections and measure complement activity in serum from subsequent blood draws. CPP E23A demonstrated inhibition of ABO incompatibility in in vitro and in vivo suppressed complement activation in rats. In addition, further rational drug design manipulations of the CPPs were conducted and a highly potent CPP derivative, Polar Assortant (PA), was identified that showed superior functional activity over other CPP analogs in vitro, efficacy in the in vitro ABO incompatibility assay and in vivo complement suppression. Collectively, these findings suggest that CPPs are viable candidates for development of a novel therapeutic inhibitor for disease processes in which the classical and lectin pathways of complement are implicated.
2. Materials and Methods
2.1. Materials
Sheep erythrocytes were purchased from MP Biomedicals, LLC. Anti-sheep-erythrocyte-IgM was purchased from Rockland Incorporated. Antibody-sensitized sheep erythrocytes (EA) were prepared as previously described (Bonaparte et al., 2008). Standard complement buffers were prepared for hemolytic assays: GVBS++ (Veronal-buffered saline, 0.1% gelatin, 0.15 mM CaCl2, and 1.0 mM MgCl2) and GVBS− − (Veronal buffered saline, 0.1% gelatin, 0.01 M EDTA). Coat Protein Peptides (CPP) were synthesized by GenScript and NEP. Lyophilized peptides were reconstituted to 10 mM in DMSO. Pooled normal human serum (NHS) was derived from the blood of healthy human volunteers in accordance with an Institutional Review Board approved protocol (IRB 02-06-EX-0216, Eastern Virginia Medical School) and prepared as previously described (Cunnion et al., 2001). Factor B-depleted serum and purified C1q were purchased from CompTech. Purified globular head region (GHR) and collagen-like region (CLR) of C1q were prepared as previously described (Thielens et al., 1993). Male Wistar rats were purchased from Harlan and used in accordance with an EVMS IACUC approved protocol (#09-001).
2.2. ABO incompatibility hemolytic assays
Whole blood was obtained from a healthy human volunteer with blood type AB. Erythrocytes were purified, washed with GVBS− −, and then re-suspended in GVBS++. Serum was prepared from a healthy human volunteer with blood type O. Increasing amounts of CPP E23A or AcPA were then incubated with 60 μL of O serum on ice in a total volume of 275 μL GVBS++ for 1 h. Equal volumes of either 100% DMSO for E23A or 20% DMSO, 0.1 M Na2HPO4, 0.9% NaCl for AcPA were also incubated with serum as vehicle controls. Following this, 100 μL of the erythrocytes were added to the reaction, and incubated for 1 h at 37° C. Reactions were stopped with 2 mL GVBS− − and centrifuged. To quantify hemolysis, the free hemoglobin in the supernatant was measured by spectrophotometry at 412 nm.
2.3. Mannan C3 deposition assay
Functional activity of the lectin pathway was assessed by ELISA using immobilized mannan as a ligand. Mannan was obtained from Sigma (from Saccharomyces cerevisiae; M7504), dissolved in phosphate buffered saline (PBS) (10 mg/ml) and stored at −20 °C. Maxisorb plates (Nunc) were coated with mannan (10 μg/ml; 100 μl per well) in coating buffer (100mM Na2CO3/NaHCO3, pH 9.6), for 16 h at room temperature or for 2 h at 37 °C. After each step, plates were washed three times with PBS containing 0.05% Tween-20. All incubation volumes were 100 μl. Residual binding sites were blocked by incubation with PBS containing 1% BSA for 1 h at 37 °C. Serum samples were treated with increasing amounts of E23A or DMSO vehicle control and diluted in GVB++ (VBS containing 0.5 mM MgCl2, 2 mM CaCl2, 0.05% Tween-20, and 0.1% gelatin; pH 7.5). The plates were then sequentially incubated for 1 h at 37 °C, followed by washing. C3 deposition was detected using monoclonal anti-human C3 (mAb RFK22) conjugated to digoxygenin (dig). Binding of mAb was detected using dig-conjugated sheep anti-mouse antibodies (Fab fragments) followed by HRP-conjugated sheep anti-dig antibodies (Fab fragments, both from Boehringer Mannheim). Enzyme activity of HRP was detected following incubation with 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (from Sigma; 2.5 mg/ml in 0.1M Citrate/Na2HPO4 buffer, pH 4.2) in the presence of 0.01% H2O2, for 30–60 min at room temperature. The OD at 415 nm was measured.
2.4. CPP injection into rats
Peptide E23A was dissolved in DMSO at a concentration of 20 mg/mL. 20 mg of E23A was injected into the peritoneum of three male Wistar rats (approximately 250 grams each) on separate occasions. Three male Wistar rats (approximately 250 grams each) underwent intra-peritoneal injection with 1 mL of DMSO as a vehicle control. Phlebotomy was performed just prior to injection with E23A and again 1 h after injection. Serum was extracted from the whole blood for use in hemolytic assays.
AcPA was dissolved in 20% DMSO, 0.1M Na2HPO4, 0.9% NaCl at a concentration of 20 mg/mL. 20 mg of AcPA was then injected via intrajugular catheter into three male Wistar rats (approximately 250 grams each). One 250 gram rat underwent intrajugular injection of an equal volume of 20% DMSO, 0.1M Na2HPO4, 0.9% NaCl as a vehicle control. Blood draws were performed from the catheter just prior to AcPA injection and again at 30 minutes, 4 hours and 24 hours after injection. Serum was extracted from the whole blood as above.
2.5. Hemolytic assays with Factor B-depleted serum, NHS, and rat serum
Thirty microliters of Factor B-depleted serum or normal human serum (NHS) were incubated both with and without 2.5 μL of 10 mM of CPPs at 37 °C for 1 h in 1.7 mL microfuge tubes. The final concentration of peptide in serum was 0.77 mM. Serum was also incubated with 2.5 μL of DMSO as a control. Following incubation, the treated serum was diluted to 800 μL total volume in GVBS++. 250 μL of the diluted serum was then transferred to a glass tube containing 400 μL of additional GVBS++. 100 μL of EA were then added and incubated for 1 h at 37 °C. 100 μL of EA were incubated in 650 μL GVBS++ without serum as a control. Immediately after the incubation, 4 mL of ice-cold GVBS− − was added to the glass tubes, which were then centrifuged at 413 × g for 5 minutes. Hemolysis was quantified as described above. The percent lysis of each sample was standardized to that of the untreated serum control.
Rat serum was used to lyse 100 μL of EA in 750 μL total volume of GVBS++ at 37 °C for 1 h. The amount of serum for each assay was optimized to find the dynamic range of hemolysis, and equal volumes of pre- and post-treatment serum were then used. Reactions were quenched with 4 mL GVBS− − and centrifuged. Hemolysis was quantified as described above.
2.6. Surface plasmon resonance (SPR)
Binding of selected peptides to C1q was performed at room temperature using a BI-3000 SPR instrument (Biosensing Instrument, Tempe, AZ) and a Fusion 100 syringe pump from Chemyx Inc. Activation of the dextran coated sensor chips (Biosensing Instrument, Tempe, AZ) was achieved by injection of 80 mg 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 10 mg N-hydroxysuccinimide (NHS) at a flow rate of 40 μl/min. Peptides were diluted in 5 mM sodium acetate (pH 3.8) and immobilized onto the sensor chips following the completion of activation. Unused immobilization sites were blocked with 1 M ethanolamine-hydrochloride (pH 8.5). Binding of C1q was measured in PBS-Tween 20 (0.01%) at a flow rate of 20 μl/min. Addition of 0.01% Tween 20 blocked non-specific binding of C1q to the sensor chip. Surface regeneration after C1q injection was accomplished by injecting 10 mM sodium hydroxide. SPR data analysis was conducted using Data Analysis (version 2.4.2) and Kinetic Analysis (version 2.0.0.3) software from Biosensing Instrument. The association and dissociation constants, kon and koff, of the binding were determined by globally fitting the obtained SPR sensorgrams with the 1:1 model taking into account the mass transport effect (Myszka, 1997). The corresponding equilibrium dissociation constant is KD=koff/kon. Data were derived from two or more experiments and reported as mean ± S.E. In all cases, C1q was used as the soluble analyte.
2.7. PA-biotin binding to C1q, GHR and CLR
Microtiter plates (Nunc) were coated with equimolar amounts of C1q, the globular head region (GHR) and the collagen-like region (CLR) of C1q at 25nM in 50 μl of carbonate buffer; gelatin was coated at 30 μg/mL as a control. Plates were incubated at 4°C overnight then washed to remove unbound protein with PBS-T (phosphate buffered saline with 0.05% Tween) followed by blocking with 0.5% gelatin/PBS-T for 2 h at room temperature. Wells were washed and block buffer containing 0.4 M NaCl and various amounts of a C-terminally biotinylated PA (New England Peptide) was added and incubated for 1 h at room temperature. After washing, the binding of PA-biotin was assessed using avidin-HRP (1:7500, BD Pharmingen) in block buffer. Plates were developed using TMB substrate (Thermo Scientific), stopped with 1N H2SO4, and read at 450 nm. Absorbance values from wells not incubated with peptide were subtracted as background.
2.8. Statistical analysis
Means and standard error of the means (SEM) were calculated from independent replicate experiments. Statistical comparisons were made using Student’s t-test. The significance of the trend in the ABO inhibition assay was measured by ANOVA. Statistical analysis was performed using Graphpad InStat 3 software.
3. Results
3.1. In vitro inhibition of ABO incompatible hemolysis with E23A peptide
Previous work from our laboratories has identified two coat protein peptides (CPPs) derived from the human astrovirus type 1 coat protein (CP) sequence, E23A (PAICQRATATLGTVGSNTSGTTEIAACILL, 30 aa) and Δ8-22 (PAICQRAEIEACILL, 15aa), that demonstrated potent suppression of complement activity in CH50-type hemolytic assays (Gronemus et al., 2010). Given the superior activity of E23A, we tested whether this peptide was able to inhibit complement-mediated ABO incompatibility in an in vitro assay. ABO incompatibility is a potentially life threatening medical condition where natural antibodies bind to erythrocytes expressing major antigens A, B, or both, leading to rapid complement-mediated intravascular hemolysis. We performed an in vitro hemolytic assay with human O serum as the source of complement and human AB erythrocytes as the target. As demonstrated in Fig. 1A, untreated O serum lysed AB erythrocytes as expected. E23A significantly inhibited ABO incompatible hemolysis in a dose dependent manner (p<0.0001) compared to the DMSO buffer control (Fig. 1A), suggesting the potential utility of CPPs in treating this disease process.
Fig. 1.

E23A inhibits complement activity both in vitro and in vivo. (A) Human O serum was incubated with increasing concentrations of E23A in DMSO and then added to human AB erythrocytes. Values are the means of three independent experiments. Error bars represent the SEM. (B) NHS was incubated with increasing concentrations of E23A in DMSO and then added to mannan coated plates. C3 deposition was measured by ELISA. (C) Three male Wistar rats received IP injection of either 20 mg E23A or vehicle control (DMSO). Blood was collected before and one hour after injection, from which serum was isolated and tested in hemolytic assays with erythrocytes. Values are the means of three independent experiments. Error bars represent the SEM.
3.2. In vitro inhibition of lectin pathway activity with E23A peptide
We have previously shown that full-length CP will inhibit lectin pathway activation, however this property has not been previously tested using CPPs. We therefore performed an ELISA assay measuring C3-fragment binding in mannan-coated wells. NHS was incubated with increasing amounts of E23A and then added to the wells. As expected, E23A inhibited C3 deposition in a dose-dependent manner compared to DMSO control (Fig. 1B). This result demonstrates that E23A has the ability to inhibit both the lectin and classical complement pathways.
3.3. In vivo inhibition of complement activity in rats by E23A peptide
A prerequisite to development of a novel compound for potential human use is demonstration of in vivo activity. In order to test whether astrovirus derived CPPs could inhibit complement activation in vivo, we performed limited proof-of-concept injections of E23A into rats. Three male Wistar rats underwent intra-peritoneal injections of the peptide and three received vehicle control (DMSO) alone. Serum from blood drawn one hour after injection showed a 51.3–60.5% (p=0.023) decrease of complement activation in hemolytic assays compared with the hemolytic activity of pre-injection serum (Fig. 1C). The animals suffered no overt adverse effects during this time nor did they have any signs of overt toxicity upon necropsy. These limited experiments demonstrate that CPPs cross the species barrier and inhibit complement activation in vivo.
3.4. Optimization of Δ8-22 and derivative peptides
The efficacy of E23A in the in vitro model of ABO compatibility as well as its ability to inhibit complement activation in vivo provides the rationale for further development of more potent analogs of CPPs for eventual testing in small animal models of complement-mediated disease. While E23A showed convincing activity in vitro and in vivo, its relative insolubility made further characterization challenging (data not shown). To this end, we exploited the Δ8-22 peptide (Fig. 2A) for additional characterization as it displays similar activity to E23A in hemolytic assays (Gronemus et al., 2010) and is 15 residues in length compared to E23A which is 30 residues long. To evaluate the contribution of the individual residues of Δ8-22 to the inhibition of complement activation, derivative peptides were synthesized with alanine substituted at each residue (Fig. 2A). Two additional peptides 15 residues in length (internal control segments 1 and 2 [ICS 1 and 2]), were synthesized as negative controls. Previous experiments indicated these sequences were not likely to have functional activity (Gronemus et al., 2010). Hemolytic assays were performed in Factor B-depleted serum. Factor B is a component of the alternative pathway convertase, so serum depleted of Factor B is incapable of alternative pathway activation. As predicted, ICS1 and 2 demonstrated no inhibition of complement activation as seen with Factor B-depleted serum alone and the DMSO vehicle control (Fig. 2B). Many of the alanine substitutions had little or no inhibitory effect compared to E23A or the parental molecule Δ8-22. Substituting alanine for glutamic acid at residue 8 or 10 demonstrated a trend towards improved inhibition compared to Δ8-22 but was not statistically significant (p=0.10 each). In an attempt to improve the solubility properties of Δ8-22, we rearranged the peptide sequence such that most of the hydrophobic residues were placed at the N terminus and the hydrophilic residues were placed at the C terminus (Fig. 2A). This rearranged peptide derived from Δ8-22 was designated polar assortant (PA). PA demonstrated a robust and significant inhibition of classical pathway activity (Figure 2B), decreasing classical pathway activation to 63% compared with untreated serum (p=0.0002).
Fig. 2.

Δ8-22 derivative activity in the hemolytic assay. (A) Peptide sequences. (B) Hemolytic assays performed with Δ8-22, alanine substitution peptides, internal control peptides sequences (ICS 1 and 2), and polar assortant (PA). The final concentration of peptide in serum was 0.77 mM. Values are the means of three independent experiments. Error bars represent the SEM. Experiments were performed with Factor B-depleted serum.
3.5. Analysis of CPP binding kinetics to C1q by SPR
Earlier studies from our laboratory have demonstrated that human astrovirus CP as well as CPP derivatives bind C1q and inhibit classical pathway activation (Bonaparte et al., 2008; Hair et al., 2010; Gronemus et al., 2010). We also showed that CP appears to displace the serine proteases associated with collagen-like region (CLR) of C1q, C1s-C1r-C1r-C1s (Hair et al., 2010). To evaluate the kinetics of CPP interactions with C1q, we utilized SPR to measure the affinity of immobilized peptides E23A, Δ8-22, and polar assortant (PA) with human C1q. Figure 3 depicted the SPR sensorgrams, which were fitted by the 1:1 model with mass transport. The mean equilibrium dissociation constants (KD) were 6.08 nM for E23A, 5.43 nM for Δ8-22, and 33.3 nM for PA (Table 1), respectively. These dissociation constants show that the CPPs bind C1q with high affinity.
Fig. 3.
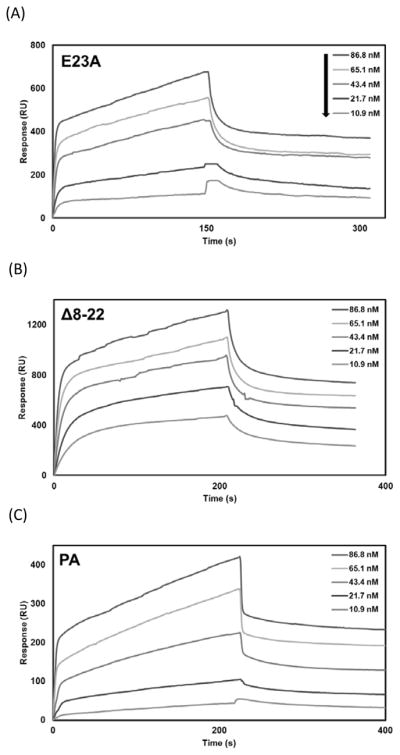
Interactions between CPP and C1q measured by surface plasmon resonance (SPR). CPP were immobilized onto the surface of a dextran sensor chip and C1q was injected at 86.8, 65.1, 43.4, 21.7 and 10.9 nM. SPR sensorgrams for C1q interaction with immobilized E23A (A), Δ8-22 (B) and PA (C). Data analysis consisted of globally fitting the SPR sensorgrams for different C1q concentrations with a 1:1 model taking into account mass transport.
Table 1.
Kinetic properties of CPP binding to C1q
| Immobilized Protein | Soluble analyte | kon (×104) M−1s−1 | koff (×10−4) s−1 | KDa (nM) |
|---|---|---|---|---|
| E23A | C1q | 11.1 ± 2.34 | 6.52 ± 0.351 | 6.08 ± 0.966 |
| Δ8-22 | C1q | 19.8 ± 0.395 | 10.6 ± 3.94 | 5.43 ± 2.10 |
| PA | C1q | 2.58 ± 0.596 | 8.08 ± 0.206 | 33.3 ± 8.49 |
KD values were calculated from kon/koff for each experiment and were averaged from at least two separate experiments.
3.6. Characterization of the polar assortant (PA)
Given the strong inhibitory activity of the PA peptide, a dose response experiment was performed in Factor B-depleted serum to measure the potency of PA. Inhibition of classical pathway mediated hemolysis was clearly demonstrated at concentrations of PA ≥ 0.193 mM (p<0.001 by ANOVA) with an S-shaped dose response curve (Fig. 4A). While PA demonstrated high affinity binding to C1q, we sought to better understand the relative binding affinities of PA for intact C1q as well as the C1q globular head region (GHR) and C1q collagen-like region (CLR) in a plate assay using gelatin as a control protein. A C-terminally biotinylated PA peptide (PA-biotin) was found to bind immobilized C1q and the CLR in a dose-dependent manner (Fig. 4B). Additionally, significantly more PA-biotin bound to whole C1q and the CLR compared to the GHR or gelatin for all tested concentrations of PA-biotin greater than 0.156 μM (p < 0.05). Half-maximal values were calculated as 0.59 μM and 0.66 μM of PA-biotin, for C1q and the CLR, respectively. These data strongly suggest that the binding of PA-biotin to C1q is specific for the CLR of C1q.
Fig. 4.

Polar assortant (PA) inhibits classical pathway hemolysis and binds C1q CLR. (A) Hemolytic assays performed with different concentrations of PA in Factor B-depleted serum. Factor B-depleted serum was incubated with 0.77 mM of peptide inhibitors in DMSO and then added to sensitized erythrocytes. Values are the means of three independent experiments. Error bars represent the SEM. (B) C1q, GHR, CLR or gelatin (negative control) were absorbed to a microtiter plate and incubated with various amounts of biotinylated PA which was then detected with a streptavidin HRP conjugate. Data are the means from 3 independent experiments. Error bars denote SEM.
In order to confirm the classical pathway inhibitory activity of PA, the PA peptide was synthesized by a second manufacturer. PA peptide synthesized by both manufacturers (GenScript [GS] and NEP) demonstrated comparable activity and robustly inhibited classical pathway activation in Factor B-depleted serum (Fig. 5B), achieving 75–85% inhibition compared with Factor B-depleted serum alone or DMSO (p<0.006). To determine the minimal sequence requirements for the inhibitory activity of PA, PA peptides serially deleted of between 1 to 3 residues at the amino and carboxy termini were synthesized (Fig. 5A). Deleting any residue from either the N or C terminus abolished the inhibitory activity of PA (Fig. 5B), suggesting that the terminal residues are critical for function and that 15 amino acids may be the minimal size requirement for maximal activity.
Fig. 5.

Effect of PA deletions on hemolytic activity. (A) PA sequences with N- and C-terminal deletions. (B) Hemolytic assays performed with PA synthesized by different vendors (GenScript (GS) or NEP), and PA peptides with amino and carboxy terminus deletions in Factor B-depleted serum. Factor B-depleted serum was incubated with 0.77 mM of peptide inhibitors in DMSO and then added to sensitized erythrocytes. Values are the means of three independent experiments. Error bars represent the SEM.
In order to evaluate which residues of PA were critical for functionality, PA peptides were synthesized with alanine substituted at each residue (Fig. 6A) and analyzed in the hemolytic assay using Factor B-depleted serum. Substitution of the leucine residues at position 3 and 5, the isoleucine residue at position 4, or the proline residue at position 7 reduced the ability of the PA to inhibit hemolysis (Fig. 6B). Alanine substitutions at other residues did not affect activity. These results suggest that residues 3, 4 and 5 play an important role in the function of PA. Not surprisingly, the proline, which could potentially produce a 60° angle in the middle of the molecule, also appears to have an important impact on function. To determine if acetylation of the amino terminus of PA would affect its functionality, an amino-acetylated PA peptide was synthesized (Ac-PA). The acetylated PA peptide demonstrated equivalent activity to the parent compound demonstrating that neutralization of the positive charge at the amino terminus of the peptide has no effect on its activity (Fig. 6B). A subsequent dose-response curve comparing PA to the acetylated version of the peptide showed no significant difference in activity over a range of concentrations (data not shown).
Fig. 6.

Effect of PA alanine substitutions on hemolytic activity. (A) PA sequences with alanine substitutions. (B) Hemolytic assays performed with PA, PA alanine substitution peptides, and acetylated PA in Factor B-depleted serum. The final concentration of peptide in serum was 0.77 mM. Values are the means of three independent experiments. Error bars represent the SEM.
3.7. In vitro inhibition of ABO incompatible hemolysis with AcPA peptide
In order to evaluate if AcPA could inhibit ABO incompatible hemolysis in a manner similar to E23A, we performed an in vitro hemolytic assay with human O serum as the source of complement and human AB erythrocytes as the target as previously described. Untreated O serum lysed AB erythrocytes as expected, while AcPA significantly inhibited ABO incompatible hemolysis in a dose-dependent manner (p<0.0001) compared to the 20% DMSO buffer control (Fig. 7A).
Fig. 7. AcPA inhibition of complement activity in vitro and in vivo.

E23A inhibits complement activity both in vitro and in vivo. (A) Human O serum was incubated with increasing concentrations of AcPA in 20% DMSO and then added to human AB erythrocytes. Values are the means of three independent experiments. Error bars represent the SEM. (B) Three male Wistar rats received intravenous injection of 20 mg AcPA and one rat received vehicle control ( 20% DMSO). Blood was collected before injection and at 0.5, 4, and 24 hours after injection, from which serum was isolated and tested in hemolytic assays with erythrocytes.
3.8 In vivo inhibition of complement activity in rats by AcPA peptide
In order to evaluate if AcPA could inhibit in vivo complement activation in a manner similar to E23A, we performed intravenous injections in commercially available catheterized Wistar rats (Harlan). As demonstrated in Fig. 7B, the 3 rats treated with AcPA showed sustained inhibition of classical pathway activity (61.7–71.3%) from 30 minutes post injection through 24 hours. The amount of inhibition resulting from AcPA intravenous injection is similar to the level of inhibition seen for E23A injected intraperitoneally. The single rat treated with 20% DMSO vehicle control demonstrated some transient inhibition of classical pathway mediated hemolysis that probably resulted from intravenous DMSO administration.
4. Discussion
Previously we demonstrated that human astrovirus CP binds C1q (Bonaparte et al., 2008) and MBL (Hair et al., 2010), inhibiting classical and lectin pathway activation, respectively. A critical 30 amino acid region of the CP molecule was subsequently identified that bound C1q and inhibited classical pathway activation. A derivative of this construct named E23A was found to have enhanced ability to inhibit classical complement activation. In order to test the ability of this compound to inhibit an important clinical disease process, we utilized an in vitro model of ABO incompatibility using mismatched human erythrocytes and serum. ABO incompatibility, which can occur due to inadvertent incompatible transfusions or passive transplacental transfer of incompatible maternal antibody to the fetus, results in life threatening rapid intravascular complement-mediated hemolysis of critical importance in neonatal and transfusion medicine (Jantapour et al., 2008). E23A and AcPA potently inhibited human ABO incompatibility (Fig. 1A and 7A) demonstrating the potential therapeutic value of CPPs in clinical disease. We have also performed proof-of-concept in vivo testing of E23A and AcPA (Fig. 1C and 7B) showing that injection into rats inhibited serum lysis of antibody-sensitized erythrocytes. Thus, these compounds appear to demonstrate similar functionality in vivo as has been demonstrated in vitro, such that future determination of pharmacodynamics and pharmacokinetics in this rat model is feasible and warranted.
A fifteen amino acid derivative of the 30 residue coat protein peptide, Δ8-22, retained functional activity and was chosen for further manipulation. Residues were rearranged to yield a construct with the proline in the middle of the sequence and the majority of the charged and polar residues at the carboxy terminus, termed polar assortant (PA) (Fig. 2A). As predicted, aqueous solubility of the compound was significantly improved. Interestingly, the ability of this construct to inhibit classical complement activation was increased over Δ8-22 (Fig. 2B). Deletion analysis of the PA revealed that removal of one residue at the N or C terminus resulted in loss of inhibitory activity in the hemolytic assay (Fig. 5B) suggesting that 15 residues is the minimal length of the CPP necessary to inhibit complement activation. Altering the sequence appeared to alter function as shown by the polar assortant (PA). Altering the composition also appeared to affect function as seen with the alanine scan and truncation peptides. We speculate that this improved functionality could be due to having increased the polarity of the molecule, or having a proline-induced angular ‘kink’ in the molecule, or both.
Our previous work has shown that CP can displace the cognate serine proteases C1s-C1r-C1r-C1s from C1q CLR and that substitution of a lysine critical for MASP-2 binding to MBL abolished CP binding to MBL (Hair et al., 2010). In order to disrupt the interaction of the pattern recognition molecules, C1q and MBL, with their cognate serine proteases, CPPs would be predicted to have similar binding affinities. It has been previously shown by SPR that immobilized C1q binds to its cognate serine proteases (C1s-C1r-C1r-C1s) with a KD of 2.72 nM (Phillips et al., 2009). The binding of E23A, Δ8-22, and PA to C1q with similar affinity for C1s-C1r-C1r-C1s (i.e., nM range) (Fig. 3 and Table 1) is consistent with our hypothesis that these peptides may function by competitively displacing, at least partially, C1s-C1r-C1r-C1s from the CLR of C1q. Binding of PA to the CLR of C1q was also verified in a binding assay (Fig. 4B). It is of interest to note that while PA displays superior inhibitory activity compared to all the other CPPs, its binding affinity to C1q as assessed by SPR is lower than that of E23A and Δ8-22 (33.3 nM versus 6.08 and 5.43, respectively). It had been previously demonstrated by ELISA that E23A and Δ8-22 bound C1q with lower affinity to other CPPs even though they showed increased inhibitory activity against classical pathway activation (Gronemus et al., 2010). Thus, tighter binding does not necessarily correlate with inhibitory activity. That is, the overall strength of binding does not inform one about the orientation of the peptide and it may be the case that a lower affinity orientation has a greater effect on preventing the conformational changes necessary to elicit C1s-C1r-C1r-C1s activation. In addition to these SPR results, CPP E23A inhibits lectin pathway activation (Fig. 1B) suggesting that as with the parental CP molecule, CPPs can also bind MBL to inhibit lectin pathway activation. Studies to analyze the precise molecular details as to how the CPPs bind C1q and MBL to inhibit classical and lectin pathway activation are currently underway.
One of the ultimate goals of this research is to develop the CPP into a therapeutic molecule for use in disease processes induced by dysregulated classical and lectin pathway activation. To our knowledge there has been only one other report of a peptidic inhibitor of classical pathway activation that functions by binding to C1q. This 15 residue cyclical peptide termed 2J (CEGPFGPRHDLTFCW), initially identified through panning a phage-display library against C1q binding, bound to the globular head domain of C1q and inhibited classical pathway activation in serum isolated from humans, non-human primates, rats and mice in vitro (Roos et al., 2001). While promising, no further work on this molecule has been reported including testing of its efficacy in vivo. In contrast to peptide 2J, CPPs are postulated to bind to the CLRs of C1q and MBL (Hair et al., 2010 and Fig. 4B) inhibiting both the classical and lectin pathways respectively and retaining their activity in vivo. Another very well characterized peptidic complement inhibitor is Compstatin, a 13 residue molecule that binds C3 preventing its activation. Compstatin is currently in clinical trials for age-related macular degeneration and is likely to have other indications as well (reviewed in Ricklin and Lambris, 2008). The success of Compstatin clearly demonstrates that peptidic inhibitors of specific complement factors have genuine potential as therapeutics for complement-mediated disease processes.
In conclusion, CPPs show proof-of-concept that these peptidic molecules are reasonable candidates as potential therapeutic agents for complement-mediated diseases such as ABO incompatibility and that these compounds appear to be functional in vivo. The polar assortant (PA) construct appears to be superior to prior compounds in terms of aqueous solubility and potency for inhibiting classical complement pathway activation. These studies pave the way for in vivo testing to determine the pharmacological profile of PA as a prerequisite for testing this compound in animal models of disease caused by dysregulated activation of the classical and lectin pathways.
References
- Bonaparte RS, Hair PS, Banthia D, Marshall DM, Cunnion KM, Krishna NK. Human astrovirus coat protein inhibits serum complement activation via C1, the first component of the classical pathway. J Virol. 2008;82:817–827. doi: 10.1128/JVI.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnion KM, Lee JC, Frank MM. Capsule production and growth phase influence binding of complement to Staphylococcus aureus. Infect Immun. 2001;69:6796–6803. doi: 10.1128/IAI.69.11.6796-6803.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneveld TWL, Ramwadhdoebé TH, Trouw LA, van den Ham DL, van der Borden V, Drijfhout JW, Hiemstra PS, Daha MR, Roos A. Human neutrophil peptide-1 inhibits both the classical and the lectin pathway of complement activation. Mol Immunol. 2007;44:3608–3614. doi: 10.1016/j.molimm.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Gronemus JQ, Hair PS, Crawford KB, Nyalwidhe JO, Cunnion KM, Krishna NK. Potent inhibition of the classical pathway of complement by a novel C1q-binding peptide derived from the human astrovirus coat protein. Mol Immunol. 2010;48:305–313. doi: 10.1016/j.molimm.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Hair PS, Gronemus JQ, Crawford KB, Salvi VP, Cunnion KM, Thielens NM, Arlaud GJ, Rawal N, Krishna NK. Human astrovirus coat protein binds C1q and MBL and inhibits the classical and lectin pathways of complement inhibition. Mol Immunol. 2010;47:792–798. doi: 10.1016/j.molimm.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Janatpour KA, Kalmin ND, Jensen HM, Holland PV. Clinical outcomes of ABO-incompatible RBC transfusions. Am J Clin Pathol. 2008;129:276–281. doi: 10.1309/VXY1ULAFUY6E6JT3. [DOI] [PubMed] [Google Scholar]
- Köhl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol. 2006;586:71–94. doi: 10.1007/0-387-34134-X_6. [DOI] [PubMed] [Google Scholar]
- Phillips AE, Toth J, Dodds AW, Girija UV, Furze CM, Pala E, Sim RB, Reid KBM, Schwaeble WJ, Schimd R, Keeble AH, Wallis R. Analogous interactions in initiating complexes of the classical and lectin pathways of complement. J Immunol. 2009;182:7708–7717. doi: 10.4049/jimmunol.0900666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotech. 1997;8:50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- Pruss A, Salama A, Ahrens N, Hansen A, Kiesewetter H, Koscielny J, Dörner T. Immune hemolysis-serological and clinical aspects. Clin Exp Med. 2003;3:55–64. doi: 10.1007/s10238-003-0009-4. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Compstatin: a complement inhibitor on its way to clinical application. Adv Exp Med Biol. 2008;632:273–292. doi: 10.1007/978-0-387-78952-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts IA. The changing face of haemolytic disease of the newborn. Early Hum Dev. 2008;84:515–523. doi: 10.1016/j.earlhumdev.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Roos A, Nauta AJ, Broers D, Faber-Krol MC, Trouw LA, Drijfhout JW, Daha MR. Specific inhibition of the classical complement pathway by C1q-binding peptides. J Immunol. 2001;93:942–951. doi: 10.4049/jimmunol.167.12.7052. [DOI] [PubMed] [Google Scholar]
- Schreiber AD. Autoimmune hemolytic anemia. In: Volanakis JE, Frank MM, editors. The Human Complement System in Health and Disease. 1. Marcel Dekker, Inc; New York, NY: 1998. pp. 547–569. [Google Scholar]
- Strobel E. Hemolytic Transfusion Reactions. Transfus Med Hemother. 2008;35:346–353. doi: 10.1159/000154811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayyil S, Milligan DW. Single versus double volume exchange transfusion in jaundiced newborn infants. Cochrane Database Syst Rev. 2006;4:CD004592. doi: 10.1002/14651858.CD004592.pub2. [DOI] [PubMed] [Google Scholar]
- Thielens NM, Bally IM, Ebenbichler CF, Dierich MP, Arlaud GJ. Further characterization of the interaction between the C1q subcomponent of C1 and the transmembrane envelope glycoprotein gp41 of HIV-1. J Immunol. 1993;151:6583–6592. [PubMed] [Google Scholar]