

Abstract
c-Ski is a transcriptional corepressor that interacts strongly with Smad2, Smad3, and Smad4 but only weakly with Smad1 and Smad5. Through binding to Smad proteins, c-Ski suppresses signaling of transforming growth factor-β (TGF-β) as well as bone morphogenetic proteins (BMPs). In the present study, we found that a mutant of c-Ski, termed c-Ski (ARPG) inhibited TGF-β/activin signaling but not BMP signaling. Selectivity was confirmed in luciferase reporter assays and by determination of cellular responses in mammalian cells (BMP-induced osteoblastic differentiation of C2C12 cells and TGF-β–induced epithelial-to-mesenchymal transdifferentiation of NMuMG cells) and Xenopus embryos. The ARPG mutant recruited histone deacetylases 1 (HDAC1) to the Smad3-Smad4 complex but not to the Smad1/5-Smad4 complex. c-Ski (ARPG) was unable to interact with Smad4, and the selective loss of suppression of BMP signaling by c-Ski (ARPG) was attributed to the lack of Smad4 binding. We also found that c-Ski interacted with Smad3 or Smad4 without disrupting Smad3-Smad4 heteromer formation. c-Ski (ARPG) would be useful for selectively suppressing TGF-β/activin signaling.
INTRODUCTION
Cytokines of the transforming growth factor-β (TGF-β) superfamily are multifunctional proteins that regulate growth, differentiation, apoptosis, and morphogenesis of a wide variety of cell types. TGF-β and related factors bind to two different types of serine/threonine kinase receptors, termed type I and type II (Derynck et al., 1998; Attisano and Wrana, 2000; Shi and Massagué, 2003). Type I receptor is activated by type II receptor upon ligand binding and transduces signals into cytoplasm through phosphorylation of receptor-regulated Smads (R-Smads). Phosphorylated R-Smads interact with Co-Smad (Smad4) and translocate into the nucleus. Nuclear Smad complexes regulate transcription of target genes in cooperation with transcriptional activators/repressors as well as with coactivators/corepressors (Miyazawa et al., 2002). Of the eight different mammalian Smad proteins, Smad1, Smad5, and Smad8 are R-Smads activated by BMP family members, whereas Smad2 and Smad3 are R-Smads activated by TGF-β and activin.
Transcription of target genes by TGF-β is upregulated by binding of Smads to transcriptional coactivators, including p300 and CBP, which induce acetylation of histones and loosen chromatin structure (Massagué and Chen, 2000; Miyazono et al., 2001). In contrast, transcriptional corepressors, including TGIF, c-Ski, and SnoN, physically interact with Smads, and repress TGF-β signaling through recruitment of histone deacetylases (HDACs) (Massagué and Chen, 2000; Liu et al., 2001).
c-Ski was originally identified as a cellular counterpart of a retroviral oncogene product, v-Ski (Li et al., 1986). c-Ski physically interacts with Smad2, Smad3, and Smad4, thus antagonizing signal transduction in the TGF-β pathway (Akiyoshi et al., 1999; Luo et al., 1999; Sun et al., 1999; Xu et al., 2000). Overexpression of c-Ski abolished TGF-β–induced growth inhibition (Luo et al., 1999; Sun et al., 1999; Xu et al., 2000). High levels of expression of c-Ski were reported in several tumor cell lines (Nomura et al., 1989; Fumagalli et al., 1993) and in melanomas in vivo (Reed et al., 2001). Inhibition of TGF-β signaling is thus regarded as a part of the mechanism of oncogenesis by Ski proteins (He et al., 2003). c-Ski recruits HDACs via N-CoR as well as mSin3A and represses transcriptional activities of Smads. In addition, c-Ski has been shown to compete with p300 for binding to Smad3, which may also play an important role in transcriptional repression by c-Ski (Akiyoshi et al., 1999). Recently, a novel model of the interaction of c-Ski with Smad-complexes was proposed (Wu et al., 2002). In the model, c-Ski binds to L3 loop of Smad4, a loop that is responsible for interaction with R-Smads; thus, c-Ski inhibits formation of active Smad complex between R-Smads and Co-Smad. Although BMP-specific R-Smads bind to c-Ski only weakly (Akiyoshi et al., 1999; Mizuide et al., 2003), it has been shown that c-Ski inhibits BMP signaling in mammals and in Xenopus (Amaravadi et al., 1997; Wang et al., 2000).
The ARPG mutant of Ski protein was first described in v-Ski as a transformation-defective mutant (Colmenares et al., 1991). The ARPG mutation is an insertional mutation of four amino acids (Ala-Arg-Pro-Gly), replacing Asp in a potential amphipathic helix region (residue 181 in c-Ski and residue 145 in v-Ski), which is supposed to disrupt helix structure. v-Ski–infected cells exhibit morphological transformation, anchorage-independent growth, and increased growth rate, whereas v-Ski (ARPG)-infected cells do not. v-Ski induces muscle differentiation in quail embryo cells as judged by induction of MyoD, myogenin, and myotube formation (Colmenares and Stavnezer, 1989), whereas v-Ski (ARPG) induced only myotube formation (Colmenares et al., 1991). v-Ski (ARPG) thus appeared to have signal-regulating characteristics different from those of c-Ski and v-Ski. Subsequently, Nomura et al. (1999) found that ARPG mutation causes loss of interaction with NCoR.
In the present study, we found that c-Ski (ARPG) selectively loses inhibitory activity on BMP signaling. Selectivity of inhibition was confirmed in luciferase reporter assays and by determination of cellular responses in mammalian cells and Xenopus embryos. We also found that c-Ski (ARPG) recruited HDAC1 to the Smad3-Smad4 complex, but not to the Smad1/5-Smad4 complex, explaining the observed selectivity. The difference in recruitment of HDAC1 to the two types of Smad complexes was attributed to loss of Smad4 binding by c-Ski (ARPG). In addition, we examined the effect of c-Ski on Smad3-Smad4 heteromer formation and found that c-Ski can interact with Smad3 or Smad4 without disrupting Smad3-Smad4 heteromer. The molecular bases of binding of c-Ski to Smads in cells stimulated or not with TGF-β or BMP will be discussed.
MATERIALS AND METHODS
Cell Culture
COS-7 and HepG2 cells were maintained in DMEM (Sigma, St. Louis, MO) containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. NMuMG cells were cultured in DMEM (Sigma) containing 10% FBS and 5 μg/ml insulin. C2C12 cells were maintained in DMEM (GIBCO, Rockville, MD) containing 20% FBS.
Plasmids and Ligands
The construction of plasmids containing c-Ski was described previously (Akiyoshi et al., 1999). c-Ski (ARPG) was provided by Dr. Nomura. c-Ski W274E and c-Ski ΔS2/3 were constructed as described by Wu et al. (2002). Other mutants of c-Ski were constructed by a PCR-based approach. To increase levels of expression, inserts were subcloned into an expression vector, pcDEF3 (Goldman et al., 1996). All of the PCR products were sequenced. For expression in Xenopus embryos, Flag-tagged human c-Ski and c-Ski (ARPG)-coding sequences were subcloned into pCS2+ vector using EcoRI and XhoI restriction sites. An expression plasmid containing activin (pSP64T-activin βB) has been described (Cho et al., 1991). Recombinant TGF-β3, BMP-6, and BMP-7 were from R&D Systems (Minneapolis, MN).
Transfection, Immunoprecipitation, and Immunoblotting
COS-7 cells were transiently transfected using FuGENE6 (Boehringer Mannheim, Indianapolis, IN). Twenty-four hours after transfection, cells were lysed with lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100). Immunoprecipitation and immunoblotting were performed as described (Kawabata et al., 1998). Anti-Flag M2 (Sigma) and anti-Myc 9E10 (PharMingen, San Diego, CA) were used for immunoprecipitation and immunoblotting of tagged proteins. Anti–phospho-Smad3 (pSmad3) was described previously (Ota et al., 2002).
Luciferase Assay
Reporter plasmids used were (CAGA)9-MLP-Luc (Dennler et al., 1998) and 3GC2-Lux (Ishida et al., 2000). HepG2 cells were transiently transfected with an appropriate combination of reporter constructs, expression plasmids, and pcDNA3. Smad5 was cotransfected when we used 3GC2-Lux. Total amounts of transfected DNAs were the same in each experiment. Twenty-four hours after transfection, cells were stimulated with ligands (TGF-β, 2.5 ng/ml; BMP-7, 500 ng/ml) and cultured for another 24 h. Cell lysates were then prepared and luciferase activities in the lysates were measured by the Dual-Luciferase Reporter System (Promega, Madison, WI). Renilla luciferase activity under the control of thymidine kinase promoter was used for normalization. In some experiments, expression level of recombinant proteins in lysates was determined by immunoblotting.
Construction of Recombinant Adenoviruses
Recombinant adenoviruses carrying LacZ, Smad6, and Smad7 cDNAs were described previously (Fujii et al., 1999). cDNAs encoding Flag-tagged c-Ski or c-Ski (ARPG) mutant were subcloned into the SwaI site of the pAxCAwt cassette cosmid. Each cosmid carrying the expression unit and adenovirus DNA-terminal protein complex were cotransfected into E1 transcomplemental cell line 293. The recombinant adenoviruses generated by homologous recombination were isolated. High-titered stocks of recombinant viruses were grown in 293 cells and purified by ViraPrep kit (Virapur, Carlsbad, CA) according to the manufacturer's instructions.
Assay for Alkaline Phosphatase Activity
C2C12 cells were seeded at a density of 4 × 105 cells/well on six-well plates. The next day, cells were infected with adenoviruses carrying various cDNAs at a m.o.i. of 100. Twenty-four hours after infection, C2C12 cells were stimulated with BMP-6 (200 ng/ml) for 96 h in DMEM containing 0.5% FBS, with a change of medium at 48 h after stimulation. Cells were then washed, and cellular proteins were extracted with a lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 1% Triton X-100). Alkaline phosphatase activity was determined using p-nitrophenyl phosphate (Sigma) as a substrate (Asahina et al., 1993). Protein concentration of extracts was determined by BCA protein assay kit (Pierce, Rockford, IL) using bovine serum albumin as a standard.
Assay for Epithelial-to-Mesenchymal Transdifferentiation
NMuMG cells were seeded at a density of 2 × 105 cells/well on six-well plates. The next day, cells were infected with adenoviruses carrying various cDNAs at a m.o.i. of 300. Twenty-four hours later, cells were stimulated with TGF-β3 (5 ng/ml). Another 24 h later, cell morphology was observed under microscopy.
Xenopus Embryo Manipulation and Microinjection
Embryo manipulations and microinjections were conducted as described previously (Cho et al., 1991). RNAs were injected into the animal pole at the four-cell stage or into the marginal zone of a ventral blastomere at the four-cell stage. Capped synthetic RNA was generated by in vitro transcription of linearized templates using a Megascript kit (Ambion, Austin, TX).
Reverse Transcription-Polymerase Chain Reaction
RNA was isolated from pooled animal caps (at least 15) and reverse transcription-polymerase chain reaction (RT-PCR) analysis was performed as described (Nakayama et al., 1998) using the following PCR conditions: 94°C for 5 min, followed by a variable number of cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min. Muscle actin, neural cell adhesion molecule (NCAM), and histone H4 primers have been described previously (Nakayama et al., 1998). PCR products were visualized on ethidium-bromide–stained agarose gels.
RESULTS
c-Ski (ARPG) Inhibits TGF-β Signaling But Does Not Inhibit BMP Signaling in Mammalian Cells
Wang et al. (2000) reported that c-Ski inhibited BMP-induced transactivation of 15× GCCG-Luc reporter and osteoblastic differentiation of W20–17 cells as well as endogenous BMP signaling in Xenopus embryos. We also observed inhibition of BMP signaling by c-Ski, v-Ski, and SnoN using a BMP-responsive reporter, 3GC2-Lux (unpublished data).
ARPG mutation of Ski proteins has been reported to modulate effects of c-Ski or v-Ski on target cells (Colmenares et al., 1989; Nomura et al., 1999). We examined how c-Ski (ARPG) affects TGF-β signaling and BMP signaling using luciferase reporter assays (Figure 1). Interestingly, c-Ski (ARPG) suppressed TGF-β signaling but not BMP signaling. To confirm this selectivity, we next examined the effect of c-Ski and c-Ski (ARPG) on cell responses induced by TGF-β and by BMP.
Figure 1.

c-Ski (ARPG) selectively suppressed TGF-β signaling. Luciferase reporter assay was conducted using (CAGA)9-MLP-Luc (A, top) and 3GC2-Lux (B, bottom). Cells were stimulated with TGF-β (2.5 ng/ml) (A) or BMP-7 (500 ng/ml) (B). □, unstimulated samples; ▪, ligand-stimulated samples. Expression of recombinant proteins was determined by immunoblotting analysis of the cell lysates using anti-Flag M2 antibody (bottom).
C2C12 cells are undifferentiated mesenchymal cells that differentiate into osteoblastic cells in response to BMP treatment (Katagiri et al., 1994). This process was shown to be mainly dependent on the Smad-signaling pathway because it was enhanced by transfection of Smad1 or Smad5 and inhibited by inhibitory Smads, i.e., Smad6 and Smad7 (Fujii et al., 1999). We examined the effects of c-Ski and c-Ski (ARPG) on BMP-induced osteoblastic differentiation. C2C12 cells were infected with adenoviruses carrying various cDNAs and treated with BMP-6 for 96 h. Osteoblast differentiation of infected cells was monitored by induction of alkaline phosphatase activity (Figure 2, top panel). LacZ-infected cells and uninfected cells exhibited BMP-dependent increase of alkaline phosphatase activity. c-Ski-infected cells as well as Smad6- or Smad7-infected cells exhibited decreased alkaline phosphatase activity, whereas c-Ski (ARPG)-infected cells efficiently differentiated into osteoblasts. We confirmed that the levels of expression of c-Ski and c-Ski (ARPG) were comparable by immunoblotting analysis (Figure 2, bottom panel) and concluded that c-Ski (ARPG) failed to inhibit BMP signaling.
Figure 2.

c-Ski (ARPG) failed to inhibit BMP-induced osteoblastic differentiation of C2C12 cells. C2C12 cells were infected with adenoviruses carrying various cDNAs, followed by BMP-6 treatment (200 ng/ml) for 96 h. Osteoblastic differentiation was monitored by induction of alkaline phosphatase activity (top). □, unstimulated samples; ▪, BMP-6–stimulated samples. Expression of recombinant proteins was determined by immunoblotting analysis of the cell lysates using anti-Flag M2 antibody (bottom).
We next examined the effects of c-Ski proteins on TGF-β–induced epithelial-to-mesenchymal transdifferentiation (EMT) of NMuMG mouse mammary epithelial cells. TGF-β treatment causes morphological transformation of cells from cuboidal epithelial morphology to elongated spindle-like fibroblastic morphology, accompanying reorganization of actin cytoskelton and downregulation of E-cadherin (Miettinen et al., 1994). Effects of adenovirally overexpressed proteins were examined by microscopic observation of cell morphology (Figure 3). Overexpression of LacZ did not affect this process, but that of Smad7 inhibited it (Figure 3, B and C). c-Ski as well as c-Ski (ARPG) inhibited TGF-β–induced morphological change of NMuMG cells (Figure 3, D and E). These results demonstrated the selectivity of inhibition by c-Ski (ARPG) in mammalian cells in culture.
Figure 3.
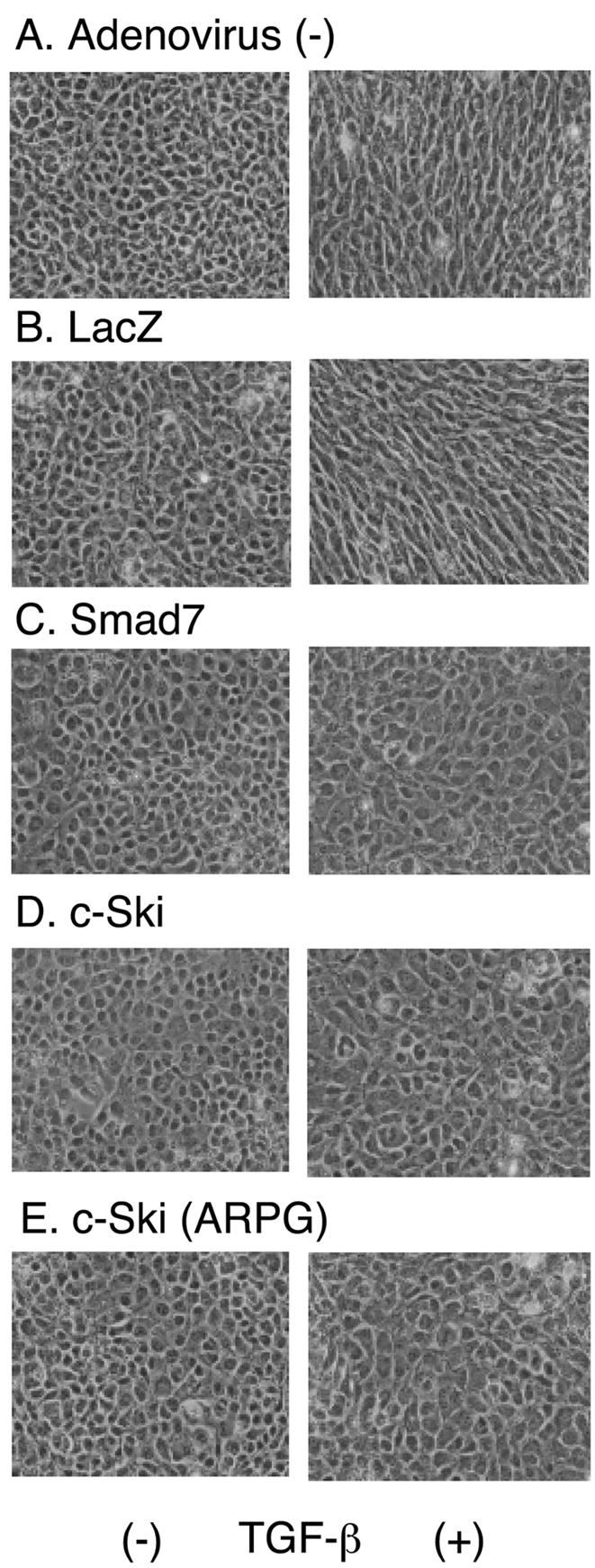
Inhibition of TGF-β–induced EMT in NMuMG cells by c-Ski and c-Ski (ARPG). NMuMG cells were infected with adenoviruses carrying various cDNAs, followed by TGF-β treatment (5 ng/ml). Twenty-four hours after ligand stimulation, cell morphology was observed under microscopy. Control cells (left panels) and TGF-β–treated cells (right panels) are shown for noninfected cells (A) and cells infected with adenoviruses carrying LacZ (B), Smad7 (C), c-Ski (D), and c-Ski (ARPG) (E).
c-Ski (ARPG) Inhibits Activin Signaling but Does Not Inhibit Endogenous BMP Signaling in Xenopus Embryos
To further explore the selectivity of c-Ski (ARPG) action on TGF-β superfamily signaling, effects of c-Ski proteins on endogenous BMP signaling were examined using Xenopus embryos. Overexpression of c-Ski in Xenopus embryos has been reported to induce secondary neural axis formation (Amaravadi et al., 1997) through suppression of endogenous BMP signaling (Wang et al., 2000). We thus used this system to determine effects of c-Ski (ARPG) on BMP signaling (Figure 4A and Table 1).
Figure 4.

Effects of c-Ski (ARPG) on signaling of TGF-β superfamily in Xenopus embryos. (A) Effect on endogenous BMP signaling using whole embryos. Equivalent amounts (500 pg) of RNAs encoding c-Ski, c-Ski (ARPG), or β-globin were injected near the ventral midline of four-cell Xenopus embryos. Resultant phenotypes in a representative experiment are shown (left, c-Ski; middle, c-Ski (ARPG); and right, β-globin). (B) Effect on endogenous BMP signaling in animal caps. RNA encoding c-Ski or c-Ski (ARPG) was injected into the animal pole of four-cell embryos. Animal caps were excised at brastulla stage 8 and cultured until stage 23. RNAs were then extracted from the animal caps and expression of marker genes (muscle actin and NCAM) was analyzed by RT-PCR. Histone H4 was used as a loading control. (C) Effect on activin signaling in animal caps. RNA encoding c-Ski or c-Ski (ARPG) was injected together with RNA encoding activin into the animal pole of four-cell embryos, and expression of marker genes was analyzed as described above.
Table 1.
Dorsalizing activity of c-Ski and c-Ski (ARPG) in whole Xenopus embryos
| Injected RNA (pg)
|
Dorsalized phenotypes (%)
|
|||||
|---|---|---|---|---|---|---|
| c-Ski | c-Ski (ARPG) | β-globin | Secondary axis | Hyperdorsalized | Total | N |
| 0 | 0 | 0 | 0 | 0 | 0 | 41 |
| 0 | 0 | 500 | 5 | 0 | 5 | 20 |
| 500 | 0 | 0 | 69 | 28 | 97 | 36 |
| 250 | 0 | 0 | 88 | 3 | 91 | 40 |
| 125 | 0 | 0 | 50 | 29 | 79 | 34 |
| 0 | 500 | 0 | 21 | 0 | 21 | 38 |
| 0 | 250 | 0 | 5 | 0 | 5 | 44 |
| 0 | 125 | 0 | 7 | 0 | 7 | 15 |
RNA encoding β-globin, c-Ski, or c-Ski (ARPG) was injected near the ventral midline of four-cell Xenopus embryos. The total number of uninjected or injected embryos surviving to tadpole stage (N) and the percentage of these embryos exhibiting either a secondary dorsal axis or hyperdorsalization with no apparent secondary axis are indicated. Data are pooled from multiple experiments.
Injection of 500 pg of RNA encoding c-Ski into ventral blastomeres of four-cell Xenopus embryos caused secondary dorsal axis formation and/or a hyperdorsalized phenotype in which the trunk and tail were severely reduced or lost (Figure 4A, left). In contrast, most of the embryos developed normally when 500 pg of RNA encoding c-Ski (ARPG) or β-globin was injected (Figure 4A, middle and right). These results suggested that c-Ski (ARPG) no longer inhibited BMP signaling.
We further examined the effect of c-Ski (ARPG) using an animal cap assay. RNA encoding c-Ski or c-Ski (ARPG) was injected into the animal pole of four-cell Xenopus embryos. Animal caps were excised at the blastula stage 8, cultured until stage 23, and then harvested. RNAs were extracted, and expression of mesodermal marker gene (muscle actin) and neural marker gene (NCAM) was analyzed by RT-PCR (Figure 4B). Injection of RNA encoding c-Ski induced NCAM in animal caps without inducing muscle actin, indicating that c-Ski caused neuralization through inhibition of BMP signaling but not through induction of mesoderm (Wang et al., 2000). c-Ski (ARPG) failed to induce NCAM (Figure 4B), indicating that c-Ski (ARPG) does not inhibit endogenous BMP signaling in animal caps.
Next, we examined effects of c-Ski and c-Ski (ARPG) on activin signaling. Injection of RNA encoding activin caused mesodermal induction in animal caps, resulting in expression of muscle actin (Figure 4C). Coinjection of c-Ski or c-Ski (ARPG) inhibited activin-induced expression of muscle actin, indicating that both c-Ski and c-Ski (ARPG) inhibited activin signaling. These results indicated that c-Ski (ARPG) inhibits activin signaling but does not inhibit BMP signaling in Xenopus embryos.
c-Ski (ARPG) Recruits HDAC1 to Smad3-Smad4 Complex, But Not to Smad5-Smad4 or to Smad1-Smad4 Complex
We next addressed the question how c-Ski (ARPG) selectively inhibits TGF-β/activin signaling. We first examined effects of the ARPG mutation on recruitment of HDAC1 to activated Smad complexes. It is established that Smad2/3-Smad4 complex mediates TGF-β/activin signaling, whereas Smad1/5-Smad4 complex mediates BMP signaling. Smad3, Smad5, or Smad1 was coexpressed in COS-7 cells with Smad4, c-Ski, mSin3A, HDAC1, and constitutively active form of TGF-β type I receptor (ALK-5) or BMP type IB receptor (ALK-6), followed by immunoprecipitation of Smad proteins. Coprecipitated proteins were visualized by immunoblotting (Figure 5A). HDAC1 was recruited to Smad3-Smad4 complex in the presence of c-Ski as well as c-Ski (ARPG), although c-Ski recruited more HDAC1 than did c-Ski (ARPG). However, HDAC1 was recruited to Smad5-Smad4 complex or Smad1-Smad4 complex only in the presence of wild-type c-Ski. This result explains selectivity for TGF-β/activin signaling in suppression by c-Ski (ARPG). Notably, c-Ski (ARPG) itself was not coprecipitated with Smad5-Smad4 complex nor Smad1-Smad4 complex. To explore this further, we examined the interaction of c-Ski (ARPG) with each of the Smad proteins.
Figure 5.

Interaction of c-Ski (ARPG) with Smad proteins. (A) Recruitment of HDAC1 to activated Smad complexes by c-Ski and c-Ski (ARPG) and (B) binding of c-Ski and c-Ski (ARPG) to each of the Smad proteins. COS-7 cells were transfected with indicated plasmids. Smad proteins (A) or c-Ski (B) was immunoprecipitated from cell lysates and coprecipitated proteins were visualized by immunoblotting. ALK-5TD and ALK-6QD are constitutively active forms of ALK-5 and ALK-6, respectively. (C) c-Ski (ARPG) suppression of TGF-β signaling was dependent on R-Smad binding. c-Ski (Δ40) lacks the N-terminal 40 amino acid residues and therefore cannot bind Smad2/3. c-Ski (ARPGΔ40) is a double mutant of (ARPG) and (Δ40). Luciferase reporter assay was conducted in HepG2 cells stimulated with TGF-β (2.5 ng/ml) using (CAGA)9-MLP-Luc. □, unstimulated samples; ▪, TGF-β–stimulated samples.
Lack of Smad4 Binding Confers TGF-β/Activin Selectivity to c-Ski (ARPG)
Smad3, Smad4, Smad5, or Smad1 was coexpressed in COS-7 cells with c-Ski and constitutively active type I receptors (ALKs), followed by immunoprecipitation of c-Ski and immunoblotting (Figure 5B). Smad3 was coprecipitated with c-Ski and with c-Ski (ARPG). In contrast, Smad4 was coprecipitated with c-Ski but only weakly with c-Ski (ARPG). Smad5 and Smad1 were coprecipitated with neither c-Ski nor c-Ski (ARPG) in the present experimental conditions. These results strongly suggested that lack of Smad4 binding confers the selectivity of inhibition to c-Ski (ARPG).
Because c-Ski (ARPG) lacks the ability to bind Smad4, it appeared likely that suppression of TGF-β signaling by c-Ski (ARPG) principally depends on its interaction with R-Smad. We therefore examined the effect of a mutation to abolish R-Smad binding on c-Ski (ARPG). The R-Smad binding region has recently been reported to be located between amino acid residues 16 and 40 (Qin et al., 2002) or the N-terminal 23 amino acid residues (Wu et al., 2002) of c-Ski. Although Smad2/3 also binds the middle region of c-Ski (amino acid residues 335–490 or 241–441), the affinity of the middle region appears to be lower than that of the N-terminal region (Akiyoshi et al., 1999; He et al., 2003; our unpublished observation). We thus constructed a truncated mutant lacking the N-terminal 40 amino acid residues (Δ40) as well as a double mutant with Δ40 and ARPG insertion and confirmed that the Δ40 mutation caused loss of R-Smad binding. As shown in Figure 5C, c-Ski (ARPG) as well as c-Ski (Δ40) inhibited TGF-β–induced transactivation of (CAGA)9-MLP-Luc, whereas the ARPG-Δ40 double mutant lost a significant amount of inhibitory activity. These results indicate that inhibition of TGF-β signaling by ARPG mutant is principally dependent on its interaction with Smad2/3.
To confirm that loss of inhibition of BMP signaling is due to lack of Smad4 binding, we used c-Ski W274E mutant, which was recently reported to lack Smad4 binding (Wu et al., 2002). We confirmed that the mutant binds to Smad3 but not to Smad4. c-Ski W274E inhibited TGF-β–induced transactivation of (CAGA)9-MLP-Luc reporter construct (Figure 6A, left), consistent with the finding obtained by Wu et al. (2002), who used AR3-Lux and constitutively active ALK-4. We also examined the effect of the W274E mutant on BMP-induced trans-activation of 3GC2-Lux and found that the W274E mutant did not inhibit trans-activation (Figure 6A, right). The W274E mutant recruited HDAC1 to Smad3-Smad4 complex but not to Smad5-Smad4 complex, entirely consistent with the result obtained using c-Ski (ARPG) (Figure 6B).
Figure 6.

Selective loss of suppression of BMP signaling by c-Ski W274E mutant. (A) Luciferase reporter assay in HepG2 cells stimulated with TGF-β or BMP-7 using (CAGA)9-MLP-Luc (left) and 3GC2-Lux (right), respectively. □, unstimulated samples; ▪, ligand-stimulated samples. (B) Recruitment of HDAC1 to activated Smad complexes. COS-7 cells were transfected with indicated plasmids. Smad proteins were immunoprecipitated from cell lysates and coprecipitated proteins (c-Ski and HDAC1) were visualized by immunoblotting. ALK-5TD and ALK-6QD are constitutively active forms of ALK-5 and ALK-6, respectively.
On the basis of results obtained with two different types of mutants lacking Smad4 binding, we concluded that lack of Smad4 binding in c-Ski results in selective loss of inhibition of BMP signaling.
Effect of c-Ski on Complex Formation between R-Smads and Co-Smads
Wu et al. (2002) recently proposed a novel model of c-Ski–mediated inhibition of Smad signaling: c-Ski binding causes disruption of oligomerization of R-Smads and Co-Smads, thus abrogating Smad function. We then examined how lack of Co-Smad or R-Smad binding in c-Ski affects the complex formation of R-Smads, Co-Smad, and c-Ski. We performed complex formation assays, in which differently tagged c-Ski, Smad3, and/or Smad4 were coexpressed and stimulated with constitutively active ALK-5, followed by immunoprecipitation of c-Ski or Smad proteins and visualization of coprecipitated proteins by immunoblotting.
First, we used c-Ski W274E, which was reported to lack Smad4 binding (Figure 7, A and B). In Figure 7A, c-Ski W274E was immunoprecipitated and coprecipitated proteins were analyzed. When c-Ski W274E was coexpressed with Smad3, Smad3 was coprecipitated with c-Ski W274E in a signal-dependent manner. When c-Ski W274E was coexpressed with Smad4, Smad4 was only very weakly coprecipitated with c-Ski W274E irrespective of the input of TGF-β signaling. When coexpressed both with Smad3 and Smad4, c-Ski W274E coprecipitated not only Smad3 but also Smad4 even in the absence of TGF-β signaling. Because c-Ski W274E does not efficiently interact with Smad4, coprecipitation of Smad4 is most likely mediated through Smad3, suggesting that c-Ski W274E interacted with Smad3-Smad4 heteromer. In Figure 7B, Smad4 was immunoprecipitated to examine c-Ski-Smad complexes. Smad4 only weakly interacted with c-Ski W274E, but interacted with Smad3 in a signal-dependent manner. Notably, c-Ski W274E was coprecipitated with Smad4 in the presence of Smad3. Again, c-Ski W274E was shown to interact with Smad3-Smad4 heteromer. We obtained essentially the same results when we used c-Ski (ARPG) instead of c-Ski W274E (Figure 7C and unpublished data). We thus concluded that disruption of R-Smad-Co-Smad heteromer is not the sole mechanism of inhibition of TGF-β/activin signaling by c-Ski (ARPG) or c-Ski W274E.
Figure 7.

Effects of c-Ski proteins on complex formation of Smad proteins. COS-7 cells were transfected with various combination of cDNAs (Smad3, Smad4, c-Ski, and ALK-5TD). c-Ski (wild-type or mutant) or Smad4 was immunoprecipitated and coprecipitated proteins were visualized by immunoblotting. Effect of c-Ski W274E on complex formation was examined by immunoprecipitating c-Ski W274E (A) or Smad4 (B). Effect of c-Ski (ARPG) (C), c-Ski ΔS2/3 (D), or wild-type c-Ski (E) was examined by immunoprecipitating c-Ski proteins. Effects of increasing amounts of c-Ski proteins on heteromer formation were also examined by immunoprecipitating Smad4 (F).
Next, we used c-Ski ΔS2/3, which does not interact with R-Smads (Wu et al., 2002). Smad3 and c-Ski ΔS2/3 were coexpressed and c-Ski ΔS2/3 was immunoprecipitated (Figure 7D). Smad3 was not coprecipitated in the absence of Smad4, whereas it was coprecipitated in a signal-dependent manner in the presence of Smad4. These results indicate that c-Ski ΔS2/3 interacts with Smad3-Smad4 heteromer.
We also noticed that c-Ski W274E interacted with Smad3-Smad4 heteromer in a signal-independent manner, whereas c-Ski ΔS2/3 interacted with Smad3-Smad4 heteromer in a signal-dependent manner. To elucidate whether this signal-independent interaction with Smad3 is a unique feature of W274E mutant, we examined complex formation using wild-type c-Ski (Figure 7E). Wild-type c-Ski interacted with Smad3 in a signal-dependent manner, but with Smad4 in a signal-independent manner. When coexpressed with both Smad3 and Smad4, c-Ski interacted with Smad3 even in the absence of signaling. We confirmed that Smad3 bound to Smad4 and c-Ski in the absence of TGF-β signaling is unphosphorylated. Taken together with the results obtained using c-Ski mutants, these findings suggest that c-Ski and Smad4 cooperate in stabilizing complexes composed of c-Ski and Smad3-Smad4 heteromer (see DISCUSSION).
The results presented here have raised the possibility that c-Ski mutants lacking Smad4 binding does not interfere with R-Smad-Co-Smad interaction. To address this issue directly, we examined the effects of c-Ski and c-Ski mutants on Smad3-Smad4 heteromer formation (Figure 7F). Flag-tagged Smad3 and 6Myc-tagged Smad4 were expressed in COS cells, followed by immunoprecipitation of Smad4. In the absence of c-Ski, Smad3 was coimmunoprecipitated in a signaling-dependent manner. We then examined how increasing levels of c-Ski affect the coprecipitation. When wild-type c-Ski was coexpressed, c-Ski as well as Smad3 was coprecipitated with Smad4; the amount of coprecipitated Smad3 was not decreased. To exclude the possibility of bridging effect of wild-type c-Ski that interacts both with Smad3 and Smad4, we next used c-Ski (ARPG) and c-Ski W274E that do not interact with Smad4. Coexpression of c-Ski mutants did not cause reduction of coprecipitated Smad3, indicating that heteromer formation between Smad3 and Smad4 was not abrogated by c-Ski proteins. c-Ski mutants were also coprecipitated upon immunoprecipitation of Smad4, confirming the interaction of c-Ski mutants with Smad3. We thus concluded that c-Ski mutants, c-Ski (ARPG) and c-Ski W274E, suppress TGF-β signaling without disrupting Smad heteromer formation.
DISCUSSION
Interaction with Smad4 Is Essential for Suppression of BMP Signaling by c-Ski
The ARPG mutant of Ski protein, in which four amino acids (Ala-Arg-Pro-Gly) are inserted in a potential amphipathic region, has been reported to cause altered cellular responses in mammalian cells (Colmenares and Stavnezer, 1989; Colmenares et al., 1991; Nomura et al., 1999). In the present study, we established that c-Ski (ARPG) selectively inhibits TGF-β/activin signaling using various assay systems.
We further found that c-Ski (ARPG) recruits HDAC1 to the Smad3-Smad4 complex but not the Smad5-Smad4 complex, a finding attributed to lack of Smad4 binding in this mutant. Recently, the crystal structure of c-Ski (219–312)-Smad4 MH2 domain was determined and detailed information on the c-Ski-Smad4 binding surface was reported (Wu et al., 2002). The L3 loop of Smad4 interacts with a loop structure in c-Ski termed the I-loop that is well conserved among c-Ski and SnoN of various species. Because the I-loop spans amino acid residues 259–281, it is distantly located from the ARPG mutation site, which is located around residue 181. Wang et al. (2000) previously reported that amino acid residues 222 and 223 are important in binding to Smad4. These residues have subsequently been shown to support the I-loop structure, although they are not directly involved in the interaction with Smad4 (Wu et al., 2002). Thus, ARPG insertion may also affect Smad4 binding in a similar manner.
Using a point mutant, c-Ski W274E (Wu et al., 2002) as well as c-Ski (ARPG), we found that lack of Smad4 binding is a key event in exhibiting selectivity of inhibition of TGF-β/activin signaling. Although Smad1 and Smad5 have been reported to interact with c-Ski (Wang et al., 2000), we did not detect the interaction in our hands (Figure 5B). Smad4 binding appears to be essential for c-Ski–mediated suppression of BMP signaling. This property is strikingly different from that of TGF-β signaling; loss of interaction with Smad4 does not lead to loss of inhibition of TGF-β signaling. The inhibition of TGF-β/activin signaling by c-Ski (ARPG) was dependent on its interaction with Smad2/3. The mode of inhibition of TGF-β/activin signaling and BMP signaling by c-Ski and c-Ski (ARPG) is summarized in Figure 8, A and B.
Figure 8.

Schematic model of inhibition of Smad signaling by c-Ski (A), c-Ski (ARPG) (B), v-Ski (C), and v-Ski (ARPG) (D). c-Ski possesses a Smad2/3 binding region as well as Smad4 binding region, whereas c-Ski (ARPG) lacks a Smad4 binding region, causing failure of c-Ski (ARPG) to inhibit BMP signaling. v-Ski, which has truncated structure in the N- and C- termini of c-Ski, lacks a Smad2/3 binding region and one of the mSin3A binding regions. v-Ski inhibits TGF-β/activin signaling as well as BMP signaling through interaction with Smad4. v-Ski (ARPG) does not inhibit Smad signaling because it no longer interacts with Smad proteins.
Mode of Binding of c-Ski to Smad2/3
Wu et al. (2002) suggested that binding of c-Ski to R-Smad or Co-Smad causes disruption of active Smad complexes. Because wild-type c-Ski can interact with Smad2/3 and Smad4 simultaneously, it would be difficult to directly demonstrate that Smad2/3-Smad4 heteromer is actually disrupted; only an approach using mutant proteins would be available for this purpose.
In the present study, we demonstrated that c-Ski mutants lacking Smad4 binding, c-Ski (ARPG) and c-Ski W274E, interact with Smad3 but do not disrupt Smad3-Smad4 heteromers. This result is consistent with our previous observation that c-Ski interacts with Smad2/3 through the upper surface of the MH2 toroid structure of Smad2/3 trimer (Mizuide et al., 2003). We further examined how c-Ski affects Smad heteromeric complex using c-Ski ΔS2/3, which interacts with Smad4 but not with R-Smads. We detected coprecipitation of c-Ski ΔS2/3 with Smad3-Smad4 heteromer. These results raise the possibility that wild-type c-Ski can also interact with Smad proteins without abrogating formation of active Smad complexes.
We also observed that c-Ski W274E induced formation of Smad3-Smad4 complexes even in the absence of signaling. Correia et al. (2001) reported that Smad3 tends to trimerize and that this tendency is dramatically enhanced by C-terminal phosphorylation of Smad3. Qin et al. (2002) found that the N-terminal region of c-Ski (residues 16–40) preferentially interacts with a Smad3 conformer that favors oligomerization. Interaction of c-Ski with Smad3 may stabilize the Smad3 conformer for oligomerization even in unphosphorylated form, which may be further stabilized by the presence of Smad4 through formation of Smad3-Smad4 heteromer. The stabilizing effect of Smad4 on unphosphorylated Smad3-c-Ski complex was also observed when we used wild-type c-Ski.
Recently, Prunier et al. (2003) reported that c-Ski induces Smad2/3-Smad4 heteromeric complex formation independent of TGF-β signaling. They concluded that c-Ski-Smad4 interaction is required for induction of Smad2/3-Smad4 complex formation based on results obtained using c-Ski del A, an N-terminally truncated mutant of c-Ski, which spans amino acid residues 335–728 (Akiyoshi et al., 1999). c-Ski del A lacks a Smad4 binding region as well as an N-terminal R-Smad binding region that has been reported to preferentially interact with the trimeric conformer of Smad3. However, c-Ski del A can weakly interact with Smad2/3 through its middle region (Akiyoshi et al., 1999). In the present study, we observed that Smad3-Smad4 heteromer formation was promoted by c-Ski W274E, which lacks interaction with Smad4, but was not promoted by c-Ski ΔS2/3, which lacks interaction with Smad2/3. The failure of c-Ski del A to induce signal-independent Smad3-Smad4 heteromer formation can be attributed to the lack of N-terminal R-Smad binding region but not to the lack of Smad4 binding.
Molecular Basis for the Loss of Transforming Activity of v-Ski (ARPG) Mutant
The ARPG mutation of Ski protein was first described as a transformation-defective mutation in v-Ski (Colmenares et al., 1991). Although the oncogenic mechanism of v-Ski is not fully understood, suppression of TGF-β–induced growth inhibition is thought to be a part of the oncogenic properties of Ski proteins (He et al., 2003). v-Ski has a truncated structure in both the N- and C- termini of c-Ski; v-Ski lacks a 23-amino acid region from its N-terminus, and a 292-amino acid region from its C-terminus (Stavnezer et al., 1989). Because the truncated region in the N-terminus constitutes the R-Smad binding site, v-Ski is unable to interact with R-Smads (He et al., 2003). The C-terminal truncated region includes one of the mSin3A binding regions (Nomura et al., 1999). Because the ARPG mutation was reported to cause loss of NCoR binding, v-Ski (ARPG) is thought to lack these two repressor regions, although presence of another repressor domain has been suggested (Nomura et al., 1999). We recently identified a novel mSin3A-dependent repressor region spanning amino acid residues 150–240 and found that this region is not affected by ARPG mutation (our unpublished observation). Thus, loss of transforming activity of v-Ski (ARPG) may not be due to the loss of HDAC recruitment. Remarkably, we found that ARPG insertion caused loss of Smad4 binding ability, suggesting that v-Ski (ARPG) would be unable to interact with both R-Smads and Co-Smad. This property can thus explain in part the loss of transforming activity in v-Ski (ARPG) mutant (Figure 8, C and D).
EMT Is a Smad-dependent Process
EMT by TGF-β is now regarded as one of the key events during tumor progression (Piek and Roberts, 2001), but the mechanism of this process is not fully understood. Piek et al. (1999) reconstituted components of TGF-β signaling in NMuMG cells by adenovirus infection and found that constitutively-active ALK-5 triggered EMT only when it was coexpressed with Smad2 and Smad4, or with Smad3 and Smad4. Oft et al. (2002) reported that the constitutively active form of Smad2 cooperates with activated H-Ras to induce EMT. These reports suggest that EMT is a Smad-dependent process. In contrast, Bhowmick et al. (2001) reported that expression of Smad7 or dominant-negative Smad3 did not inhibit EMT and proposed that the RhoA-dependent pathway plays an indispensable role in EMT. A role of phosphatidylinositol 3-kinase in EMT has also been reported (Bakin et al., 2000). In the present study, we observed that overexpression of Smad7 as well as that c-Ski inhibited EMT. Because c-Ski specifically targets Smad signaling at the level of transcription, our results strongly suggest that TGF-β–induced EMT is dependent on Smad-mediated transcription. However, they do not exclude contribution by other pathways. Further studies are thus required to elucidate the mechanism of EMT in detail.
Use of c-Ski (ARPG) as an Antagonist of TGF-β/Activin Signaling Targeting Smad-dependent Transcriptional Activation
Because overactivity of TGF-β signaling is implicated in many pathological processes, development of useful antagonists of TGF-β signaling is highly desired. Successful use of various types of TGF-β antagonists has been reported in mouse pathological models. Recombinant soluble Fc-TGF-β type II receptor fusion protein was used to suppress breast cancer metastasis (Muraoka et al., 2002; Yang et al., 2002). Adenoviruses carrying cDNA encoding Smad7 were used to prevent pulmonary fibrosis induced by bleomycin administration (Nakao et al., 1999), although Smad7 also blocks BMP signaling. Recently, a low-molecular-weight synthetic compound, SB-431542, which selectively inhibits ALK-4/5/7 kinase but not ALK-1/2/3/6 kinase, was developed (Inman et al., 2002), although its effects in vivo have not yet been examined. Considering the multifunctional properties of TGF-β, however, pathway-specific antagonists would be more favorable for clinical applications (Akhurst, 2002). One unique feature of c-Ski (ARPG) is that it selectively suppresses TGF-β/activin signaling at the level of transcription and thus is not supposed to inhibit Smad-independent signaling. In the present study, we demonstrated the selective action of c-Ski (ARPG) in vivo using Xenopus embryo system; c-Ski (ARPG) did not inhibit endogenous BMP signaling in whole embryos as well as in animal caps, whereas it inhibited activin signaling in animal caps. It would be interesting to explore effects of this unique mutant on other cellular responses to TGF-β and activin.
Acknowledgments
We thank N. Nomura and S. Pearson-White for c-Ski and c-Ski (ARPG), R. Eisenman for mSin3A, E. Seto for HDAC1, and J.-M. Gauthier for (CAGA)9-MLP-Luc. We are also grateful to H. Hirano for secretarial assistance and M. Saitoh and M. Kondo for useful discussions and suggestions. This study was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and by a grant from Boehringer Ingerheim.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03–07–0478. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-07-0478.
References
- Akhurst, R.J. (2002). TGF-β antagonists: why suppress a tumor suppressor? J. Clin. Invest. 109, 1533-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi, S., Inoue, H., Hanai, J., Kusanagi, K., Nemoto, N., Miyazono, K., and Kawabata, M. (1999). c-Ski acts as a transcriptional co-repressor in transforming growth factor-β signaling through interaction with Smads. J. Biol. Chem. 274, 35269-35277. [DOI] [PubMed] [Google Scholar]
- Amaravadi, L.S., Neff, A.W., Sleeman, J.P., and Smith, R.C. (1997). Autonomous neural axis formation by ectopic expression of the protooncogene c-ski. Dev. Biol. 192, 392-404. [DOI] [PubMed] [Google Scholar]
- Asahina, I., Sampath, T.K., Nishimura, I., and Hauschka, P.V. (1993). Human osteogenic protein-1 induces both chondroblastic and osteoblastic differentiation of osteoprogenitor cells derived from newborn rat calvaria. J. Cell Biol. 123, 921-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attisano, L., and Wrana, J.L. (2000). Smads as transcriptional co-modulators. Curr. Opin. Cell Biol. 12, 235-243. [DOI] [PubMed] [Google Scholar]
- Bakin, A., Tomlinson, A.K., Bhowmick, N.A., Moses, H.L., and Artega, C.L. (2000). Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275, 36803-36810. [DOI] [PubMed] [Google Scholar]
- Bhowmick, N.A., Ghiassi, M., Bakin, A., Aakre, M., Lundquist, C.A., Engel, M.E., Arteaga, C.L., and Moses, H.L. (2001). Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 12, 27-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, K.W., Blumberg, B., Steinbeisser, H., and De Robertis, E.M. (1991). Molecular nature of Spemann's organizer: the role of the Xenopus homeobox gene gooscoid. Cell 67, 1111-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenares, C., and Stavnezer, E. (1989). The ski oncogene induces muscle differentiation in quail embryo cells. Cell 59, 293-303. [DOI] [PubMed] [Google Scholar]
- Colmenares, C., Teumer, J.K., and Stavnezer, E. (1991). Translation-defective v-ski induces MyoD and myogenin expression but not myotube formation. Mol. Cell. Biol. 11, 1167-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia, J., Chacko, B.B., Lam, S.S., and Lin, K. (2001). Sedimentation studies reveal a direct role of phosphorylation in Smad3:Smad4 homo- and hetero-trimerization. Biochemistry 40, 1473-1482. [DOI] [PubMed] [Google Scholar]
- Dennler, S., Itoh, S., Vivien, D., ten Dijke, P., Huet, S., and Gauthier, J.-M. (1998). Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 17, 3091-3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck, R., Zhang, Y., and Feng, X.H. (1998). Smads: transcriptional activators of TGF-β responses. Cell 95, 737-740. [DOI] [PubMed] [Google Scholar]
- Fujii, M. et al. (1999). Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell 10, 3801-3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli, S., Doneda, L., Nomura, N., and Larizza, L. (1993). Expression of the c-ski proto-oncogene in human melanoma cell lines. Melanoma Res. 3, 23-37. [DOI] [PubMed] [Google Scholar]
- Goldman, L.A., Cutrone, E.C., Kotenko, S.V., Krause, C.D., and Langer, J.A. (1996). Modifications of vectors pEF-BOS, pcDNA1 and pcDNA3 result in improved convenience and expression. Biotechniques 21, 1013-1015. [DOI] [PubMed] [Google Scholar]
- He, J., Tegen, A.B., Krawitz, A.R., Martin, G.S., and Luo, K. (2003). The transforming activity of Ski and SnoN is dependent on their ability to repress the activity of Smad proteins. J. Biol. Chem. 278, 30540-30547. [DOI] [PubMed] [Google Scholar]
- Inman, G.J., Nicolas, F.J., Callahan, J.F., Harling, J.D., Gaster, L.M., Reith, A.D., Laping, N.J., and Hill, C.S. (2002). SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 63, 65-74. [DOI] [PubMed] [Google Scholar]
- Ishida, W., Hamamoto, T., Kusanagi, K., Yagi, K., Kawabata, M., Takehara. K., Sampath, T.K., Kato, M., and Miyazono, K. (2000). Smad6 is a Smad1/5-induced Smad inhibitor: characterization of bone morphogeneic protein-responsive element in the mouse Smad6 promoter. J. Biol. Chem. 275, 6075-6079. [DOI] [PubMed] [Google Scholar]
- Katagiri, T. et al. (1994). Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J. Cell Biol. 127, 1755-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata, M., Inoue, H., Hanyu, A., Imamura, T., and Miyazono, K. (1998). Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. EMBO J. 17, 4056-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y., Turck, C.M., Teumer, J.K., and Stavnezer, E. (1986). Unique sequence, ski, in Sloan-Kettering avian retroviruses with properties of a new cell-derived oncogene. J. Virol. 57, 1065-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., Sun, Y., Weinberg, R.A., and Lodish, H.F. (2001). Ski/Sno and TGF-β signaling. Cytokine Growth Factor Rev. 12, 1-8. [DOI] [PubMed] [Google Scholar]
- Luo, K., Stroschein, S.L., Wang, W., Chen, D., Martens, E., Zhou, S., and Zhou, Q. (1999). The Ski oncoprotein interacts with the Smad proteins to repress TGFβ signaling. Genes Dev. 13, 2196-2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J., and Chen, Y.-G. (2000). Controlling TGF-β signaling. Genes Dev. 14, 637-644. [PubMed] [Google Scholar]
- Miettinen, P.J., Ebner, R., Lopez, A.R., and Derynck, R. (1994). TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J. Cell Biol. 127, 2021-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T., and Miyazono, K. (2002). Two major Smad pathways in TGF-β superfamily signalling. Genes Cells 7, 1191-1204. [DOI] [PubMed] [Google Scholar]
- Miyazono, K., Kusanagi, K., and Inoue, H. (2001). Divergence and convergence of TGF-β/BMP signaling. J. Cell. Physiol. 187, 265-276. [DOI] [PubMed] [Google Scholar]
- Mizuide, M. et al. (2003). Two short segments of Smad3 are important for specific interaction of Smad3 with c-Ski and SnoN. J. Biol. Chem. 278, 531-536. [DOI] [PubMed] [Google Scholar]
- Muraoka, R.S. et al. (2002). Blockade of TGF-β inhibits mammary tumor cell viability, migration, and metastases. J. Clin. Invest. 109, 1551-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao, A., Fujii, M., Matsumura, R., Kumano, K., Saito, Y., Miyazono, K., and Iwamoto, I. (1999). Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Invest. 104, 5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, T., Snyder, M.A., Grewal, S.S., Tsuneizumi, K., Tabata, T., and Christian, J.L. (1998). Xenopus Smad8 acts downstream of BMP-4 to modulate its activity during vertebrate embryonic patterning. Development 125, 857-867. [DOI] [PubMed] [Google Scholar]
- Nomura, N., Sasamoto, S., Ishii, S., Date, T., Matsui, M., and Ishizaki, R. (1989). Isolation of human cDNA clones of ski and the ski-related gene, sno. Nucleic Acid Res. 17, 5489-5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, T., Khan, M.M., Kaul, S.C., Dong, H.D., Wadhwa, R., Colmenares, C., Kohno, I., and Ishii, S. (1999). Ski is a component of the histone deacetylase complex required for transcriptional repression by Mad and thyroid hormone receptor. Genes Dev. 13, 412-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oft, M., Akhurst, R.J., and Balmain, A. (2002). Metastasis is driven by sequential elevation of H-ras and Smad2 level. Nat. Cell Biol. 4, 487-494. [DOI] [PubMed] [Google Scholar]
- Ota, T., Fujii, M., Sugizaki, T., Ishii, M., Miyazawa, K., Aburatani, H., and Miyazono, K. (2002). Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-β in human umbilical vein endothelial cells. J. Cell. Physiol. 193, 299-318. [DOI] [PubMed] [Google Scholar]
- Piek, E., and Roberts, A.B. (2001). Suppressor and oncogenic roles of transforming growth factor-β and its signaling pathways in tumorigenesis. Adv. Cancer Res. 83, 1-54. [DOI] [PubMed] [Google Scholar]
- Piek, E., Moustakas, A., Kurisaki, A., Heldin, C.-H., and ten Dijke, P. (1999). TGF-β type-I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J. Cell Sci. 112, 4557-4568. [DOI] [PubMed] [Google Scholar]
- Prunier, C., Pessah, M., Ferrand, N., Seo. S. R., Howe, P., and Atfi, A. (2003). The oncoprotein Ski acts as an antagonist of TGF-β signaling by suppressing Smad2 phosphorylation. J. Biol. Chem. 278, 26249-26257. [DOI] [PubMed] [Google Scholar]
- Qin, B.Y., Lam, S.S., Correia, J.J., and Lin, K. (2002). Smad3 allostery links TGF-β receptor kinase activation to transcriptional control. Genes Dev. 16, 1950-1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, J.A., Bales, E., Xu, W., Okan, N.A., Bandyopadhyay, D., and Medrano, E.E. (2001). Cytoplasmic localization of the oncogenic protein Ski in human cutaneous melanomas in vivo: functional implications for transforming growth factor β signaling. Cancer Res. 61, 8074-8078. [PubMed] [Google Scholar]
- Shi, Y., and Massagué, J. (2003). Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685-700. [DOI] [PubMed] [Google Scholar]
- Stavnezer, E., Brodeur, D., and Brennan, L. (1989). The v-ski oncogene encodes a truncated set of c-ski coding exons with limited sequence and structural relatedness to v-myc. Mol. Cell. Biol. 9, 4038-4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y., Liu, X., Eaton, E.N., Lane, W.S., Lodish, H.F., and Weinberg, R.A. (1999). Interaction of the Ski oncoprotein with Smad3 regulates TGF-β signaling. Mol. Cell 4, 499-509. [DOI] [PubMed] [Google Scholar]
- Wang, W., Mariani, F.V., Harland, R.M., and Luo, K. (2000). Ski represses bone morphogenic protein signaling in Xenopus and mammalian cells. Proc. Natl. Acad. Sci. USA 97, 14394-14399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J.W., Krawitz, A.R., Chai, J., Li, W., Zhang, F., Luo, K., and Shi, Y. (2002). Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski. Insights on Ski-mediated repression of TGF-β signaling. Cell 111, 357-367. [DOI] [PubMed] [Google Scholar]
- Xu, W., Angelis, K., Danielpour, D., Haddad, M.M., Bischof, O., Campisi, J., Stavnezer, E., and Medrano, E.E. (2000). Ski acts as a co-repressor with Smad2 and Smad3 to regulate the response to type β transforming growth factor. Proc. Natl. Acad. Sci. USA 97, 5924-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. et al. (2002). Lifetime exposure to soluble TGF-β antagonist protects mice against metastasis without adverse side effects. J. Clin. Invest. 109, 1607-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]