

Abstract
AIM
To evaluate dosing and intervention strategies for the phase II programme of a VEGF receptor inhibitor using PK–PD modelling and simulation, with the aim of maximizing (i) the number of patients on treatment and (ii) the average dose level during treatment.
METHODS
A previously developed PK–PD model for lenvatinib (E7080) was updated and parameters were re-estimated (141 patients, once daily and twice daily regimens). Treatment of lenvatinib was simulated for 16 weeks, initiated at 25 mg once daily. Outcome measures included the number of patients on treatment and overall drug exposure. A hypertension intervention design proposed for phase II studies was evaluated, including antihypertensive treatment and dose de-escalation. Additionally, a within-patient dose escalation was investigated, titrating up to 50 mg once daily unless unacceptable toxicity occurred.
RESULTS
Using the proposed antihypertension intervention design, 82% of patients could remain on treatment, and the mean dose administered was 21.5 mg day−1. The adverse event (AE) guided dose titration increased the average dose by 4.6 mg day−1, while only marginally increasing the percentage of patients dropping out due to toxicity (from 18% to 20.8%).
CONCLUSIONS
The proposed hypertension intervention design is expected to be effective in maintaining patients on treatment with lenvatinib. The AE-guided dose titration with blood pressure as a biomarker yielded a higher overall dose level, without relevant increases in toxicity. Since increased exposure to lenvatinib seems correlated with increased treatment efficacy, the adaptive treatment design may thus be a valid approach to improve treatment outcome.
Keywords: hypertension, lenvatinib, modelling and simulation, oncology, pharmacodynamics, proteinuria
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Drugs targeting the VEGF-pathway often show increase in blood pressure (BP) and proteinuria as dose limiting toxicity. However, high dose levels are also considered crucial for anti-tumour activity. In preclinical models, lenvatinib affects tumour cell proliferation and tumour vascularization, while therapeutic effects were observed in phase I and II trials.
WHAT THIS STUDY ADDS
Adverse event guided dose titration, including treatment with anti-hypertensive treatment and dose de-escalations can be expected to keep about 80% of patients on treatment with lenvatinib for 16 weeks. Additionally, within subject dose escalation in subjects not experiencing toxicity can considerably increase dose intensity.
Introduction
Many of the recently approved anti-cancer drugs or drugs that are currently in clinical development target tumour angiogenesis. They aim to limit blood flow to the tumour, and thereby induce tumour growth inhibition [1]. These agents generally show mild toxicity compared with classical cytotoxic drugs. For an important subset of these drugs, i.e. the inhibitors of vascular endothelial growth factor (VEGF) or its receptor, increases in blood pressure (BP) in patients are observed during treatment [2–4]. Additionally, in a significant fraction of patients, proteinuria is observed as a side effect [5–7]. While generally, hypertension can be controlled with antihypertensive (AH) medication, the occurrence of (higher grades of) proteinuria is usually treated with dose delays or dose reductions and is therefore considered dose-limiting. However, a large fraction of patients does not experience dose-limiting hypertension and/or proteinuria at the standard dosage and may therefore tolerate higher doses. As blood pressure has also been suggested to be a biomarker of efficacy [8–10], an adaptive treatment design with within patient dose escalation guided by (the absence of) adverse events may be able to achieve higher drug exposure.
Currently, dosing strategies for tyrosine kinase inhibitors (TKIs) are defined as the highest tolerated dose, assuming that higher exposure is correlated with higher efficacy. The relationship between higher drug exposure and improved clinical response has been established convincingly for imatinib [11–15] and sunitinib [16]. For other TKIs such relationships have not yet been shown in clinical trials, although positive correlations between plasma exposure and tumour growth inhibition have been established in xenograft studies for, for example, sorafenib [17, 18] and erlotinib [19, 20]. It may however be that in clinical trials with some TKIs, maximal activity was already achieved at all tested dose levels. At the present time, it seems rational to aim for the highest possible drug exposure in patients when designing dosing regimens for TKIs [21].
In this study we used a model-based approach to optimize dosing strategies for the novel investigational anti-cancer agent lenvatinib, which is a tyrosine kinase inhibitor of VEGFR and several other tyrosine kinase receptors. Lenvatinib has shown anti-tumour activity in preclinical tests [22, 23] and phase I and II clinical trials [7, 24–27], and is currently under evaluation in phase II/III trials. Hypertension and proteinuria were the main and dose limiting toxicities in these trials. Previously, a population pharmacokinetic–pharmacodynamic (PK–PD) model was constructed that linked lenvatinib plasma exposure to increases in blood pressure, as well as the occurrence of different grades of proteinuria [28]. In the analysis presented here, this model was updated and refined using data obtained from patients in an additional phase I trial. Simulations from this model were then performed to evaluate a proposed protocol for hypertension management and evaluate a dose titration design, with the aim of maximizing drug exposure. The primary goal of the simulation study was to treat patients on treatment as long as possible. Maintaining continuous dosing at effective dosages is important, since this is considered to be required to achieve optimal inhibition of tumour angiogenesis [29, 30]. Additionally, we evaluated whether an adverse event (AE) guided dose titration design was able to increase average dose levels in patients, while maintaining tolerability.
Methods
Construction of PK–PD model and update
The PK–PD model used in this analysis was described earlier [31] and is based on data from one phase I trial of lenvatinib [7]. Briefly, this trial was a two-site phase I dose finding study of lenvatinib in patients with advanced malignancies. Patients were ≥18 years of age with solid tumours or lymphoma and a Karnofsky score ≥70%. Patients were treated with lenvatinib once daily, using fixed doses. No other anti-cancer drugs were administered during or less than 4 weeks before the start of treatment. Doses were escalated from 0.2 mg day−1 up to 32 mg day−1 in cohorts of three–six patients. Intra-patient dose escalation was not allowed. Blood pressure (BP) measurement and urinalysis were performed weekly. Relevant exclusion criteria were brain tumours, uncontrolled infections, reduced bone marrow reserve, clinically significant cardiac impairment and bleeding or thrombotic disorders. Also excluded were patients using therapeutic dosages of anticoagulants, patients treated with other investigational drugs within 30 days prior to start of study, patients with proteinuria ≥2+ (CTC grade ≥2), and patients with poorly controlled hypertension (repeated measurements higher than 160/90 mmHg) at screening. The study was approved by a local medical ethics committee.
The previously constructed PK–PD model is shown in Figure 1. It consists of a two compartment PK model, with combined first and zero order absorption and linear distribution and elimination. It includes two separate indirect effect models for the systolic blood pressure (BPsys) and diastolic blood pressure (BPdia). Correlations between BPsys and BPdia for baseline and residual errors are incorporated in the model. AH medication was accounted for by a negative effect on input rate of the indirect effect models. To account for differences in dosing and efficacy of AH medication, and combinations of AH drugs, a cumulative measure of daily doses relative to the WHO defined daily dose was introduced (defined daily dose equivalents, DDDE):
| 1 |
with n being the number of AH medications, DDi the prescribed dose for the specific AH drug, and DDDi the WHO defined daily dose for that drug (http://www.whocc.no/atcddd/indexdatabase/, accessed November 11, 2008). A Markov model, driven by plasma concentrations of lenvatinib, described the probabilities of the occurrence of the different grades of proteinuria over time.
Figure 1.
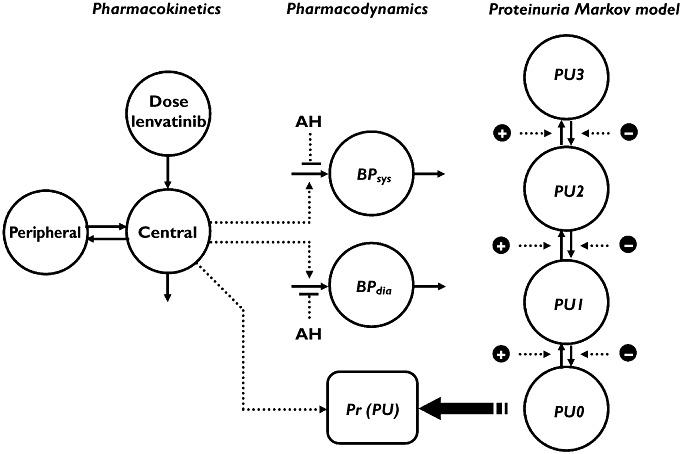
PK–PD model for lenvatinib used in the simulations. PU proteinuria
Parameter estimates of the model were initially based only on data from 67 patients. Data from an additional 74 patients from a subsequent phase I trial of lenvatinib [32] studying twice daily administration in patients with advanced solid tumours were used to update parameter estimates. Briefly, this second trial studied escalating doses of lenvatinib ranging from 0.1 mg to 12 mg twice daily in patients with advanced solid tumours. Inclusion and exclusion criteria were the same as described for the former study. Similar to the earlier trial, BP measurements and urinalysis were performed weekly. The two-site study was approved by a local ethics committee. Model fit of the blood pressure model was re-evaluated using goodness-of-fit plots, visual predictive checks (VPCs), mirror plots of the VPC [33], and plots of normalized prediction distribution errors (NPDE) [34]. The likelihood ratio test based on the objective function value (OFV) was used to evaluate significance of improvement in model fit, in which a significance level of P < 0.01 was used.To evaluate the ability of the model to describe the observed proteinuria data, numerical predictive checks were performed on the observed numbers of the different grades of proteinuria, as well as the observed number of transitions from one grade to another, as described in our previous paper [28].
Simulations: AH intervention designs
The main goal of this analysis was to evaluate the performance of an AE-intervention scheme that was designed earlier, which was intended to be applied in a phase II trial. This scheme (design C, Figure 2) included the administration of AH therapy and the implementation of dose reductions in several steps at the occurrence of AEs. Additionally, two more conservative schemes (A and B) were evaluated. Finally, a dose-titration scheme (D) was evaluated, which implemented within-patient dose escalation in the absence of adverse events. The designs were implemented as follows: (also see Table 1 for an overview of features):
Figure 2.
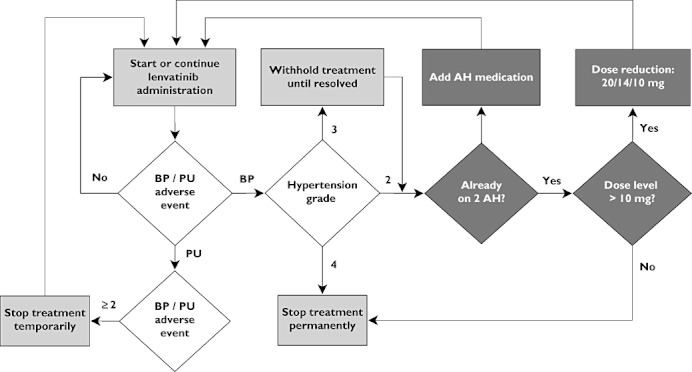
Schematic representation of AE-intervention scheme intended for phase II (design C). The same design was also used in design D for the AE-interventions, although that design also incorporated within patient dose escalations. In design B, no dose reductions were applied, while in design A, no dose reductions or AH medication was applied, and treatment was stopped permanently at the occurrence of adverse events
Table 1.
Evaluated treatment designs
| A. Conventional design | B. AH therapy | C. AH therapy and dose de-escalation | D. Adaptive treatment | |
|---|---|---|---|---|
| Dose reductions | • | • | ||
| AH interventions, at AH = 1 or 2 DDDE | • | • | • | |
| Dose delays due to proteinuria | • | • | • | • |
| Treatment stop due to hypertension ≥ grade 2 | • | |||
| Treatment stop due to uncontrollable hypertension | • | • | • | |
| Within-patient dose escalation | • |
AH, antihypertensive.
Conservative design: lenvatinib treatment discontinued at the occurrence of unacceptable hypertension toxicity (grade ≥2).
AH treatment, followed by discontinuation lenvatinib treatment if hypertension toxicity persisted.
AH treatment, followed by lenvatinib dose reduction if hypertension toxicity persisted = proposed phase II hypertension management design)
Adverse event guided dose titration intra-patient lenvatinib dose escalation and de-escalation, combined with AH therapy (as in design C).
In all designs, if proteinuria grade ≥3 was detected, lenvatinib administration was stopped temporarily, until proteinuria was normalized again to grade 0 (no proteinuria).
Sixteen weeks of lenvatinib therapy were simulated, with treatment initiated at 25 mg once daily (the maximum tolerated dose (MTD) based on phase I rising dose studies) for each patient. Measurements of BP and proteinuria were simulated at the start of treatment, and weekly thereafter, as was done in the phase I clinical trials. Primary measures of outcome for the simulations were a) the fraction of patients remaining on treatment, and b) the average lenvatinib daily dose over the full treatment period.
Grade 2 hypertension toxicity was defined as increased diastolic BP of ≥20 mmHg or BP ≥150/100 mmHg, equal to Common Terminology Criteria for Adverse Events v3.0 (CTCAE) defined by the NCI [35]. In designs B, C and D, when grade ≥2 hypertension occurred, AH treatment was started at 1 DDDE, and if this did not result in sufficient decrease of BP on subsequent occasions, AH medication was intensified to 2 DDDE. Dose de-escalation (designs C/D) was performed in the following order: 25 mg → 20 mg → 14 mg → 10 mg. If a patient's BP could not be controlled even a dose of 10 mg once daily, treatment was discontinued permanently. In the AE-guided dose titration design (D), at the end of each 4 week treatment cycle, the dose level was increased if no toxicity or a toxicity lower than grade 2 occurred, and if a patient had not received a prior dose reduction or was not on maximal AH therapy (2 DDDE). Dose escalation (design D) was performed in the following order: 32 mg → 40 mg → 50 mg. The simulated profile of one typical patient is shown in Figure 3, showing dose levels, AH administration, blood pressure and proteinuria toxicity levels.
Figure 3.
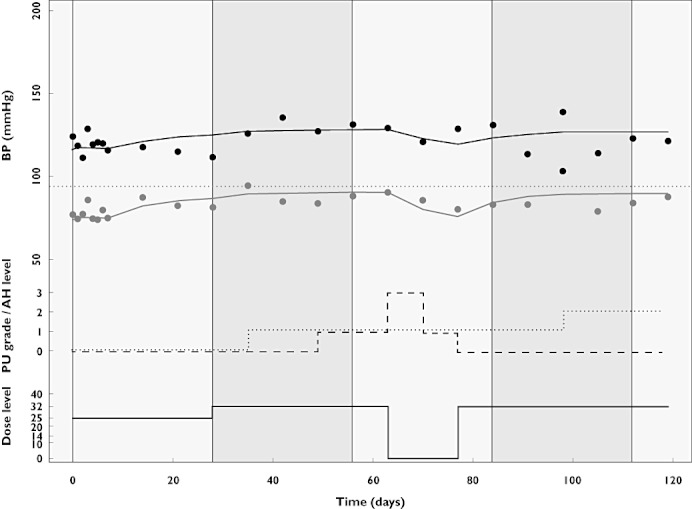
Simulated profile of BP (upper part), proteinuria (PU) toxicity levels (dashed line), dose level (solid line) and AH levels (dotted line) for a typical patient
Simulations were performed first for the point estimates of the established model. The same simulations were also performed repeatedly (n= 100) with parameter estimates sampled from the variance-covariance matrix of the PK, BP and proteinuria (PU) model. Formal statistical testing of superiority between designs was not performed since statistical tests generally depend on the number of observations (n), which in simulations can be increased arbitrarily. However, in Table 1, the CV(%) due to uncertainty in model parameters is presented, which is not dependent on n. CV(%) was calculated as the SD of the mean outcome variable (obtained in simulations performed with different parameter samples), divided by the mean outcome variable.
Software
Model estimation was performed with NONMEM (version VI, level 2.0, ICON Development Solutions, Ellicott City, MD, USA) [36], using Pirana as modelling environment (http://www.pirana-software.com) [37]. The first-order conditional estimation method with interaction (FOCE-I) was used for estimation of the PK model and the PK–PD model for blood pressure. For estimation of a Markov model fitted to the categorical observations of proteinuria, the Laplacian estimation method was used. All stochastic simulations were performed using R (version 2.10.1, http://www.r-project.cran.org), supplied with the deSolve and MASS packages for numerical solving of differential equations and sampling from multivariate normal distributions. Simulations were performed for cohorts of 500 patients per treatment design, which allowed the study of the mean fraction of patients on treatment and the mean average dose with sufficient precision.
Results
PK–PD model update
Data were obtained from 141 subjects, combining 2893 plasma concentrations of lenvatinib, 2580 BP measurements and proteinuria data from 2174 urine analyses. Demographics of the included subjects are shown in Table 2. Fifty-eight subjects received treatment with AH medication somewhere during the study period. The previously developed PK model structure provided adequate fit to the updated dataset and PK parameters were found to be similar to those obtained earlier. The PK–PD model could predict adequately the BP data, as judged by a VPC generated using parameter estimates from the previous analysis which shows that predicted and observed median, 5th and 95th percentiles agree very closely (supporting information Figure S1). Despite the adequate prediction, we did attempt to improve this model, by re-evaluating model structure and re-estimating the parameters. When the same model structure was used, parameter estimates were similar for most parameters (Table 3). The effect of AH medication was however estimated to be considerably lower than previously estimated, and both the effect of AH medication and the kin of the indirect response model could only be estimated with large uncertainty (>100%). Result from a limited bootstrap (limited to a 50 replicates due to computational constraints) were comparable with the uncertainty estimates obtained from the covariance step. From the bootstrap, fairly high uncertainty (∼100%) was estimated for the effect of AH and input rate of the BP model, while uncertainty estimates were intermediate (∼30%) for the effect of lenvatinib on BPsys/dia, and low for the other fixed effect estimates (<5%). The combined direct and indirect drug effect on proteinuria found in the previous analysis could not be reproduced, as only a direct effect could be identified from the data. A VPC of the final BP model showed good correspondence between observed and model predicted data. Mirror plots of the VPC also showed no indication of model mis-specification, although a slight overprediction of the 95th percentile was present for systolic blood pressure (Figure 4). The distribution of NPDEs very closely followed the normal distribution, while a plot of NPDE vs. time showed no bias (supportiang information Figure S2A–C),
Table 2.
Demographics of subjects enrolled in studies 1 and 2
| Study 1 | Study 2 | |||
|---|---|---|---|---|
| Median/n | Range | Median/n | Range | |
| Men | 43 | – | 39 | – |
| Women | 38 | – | 36 | – |
| Weight (kg) | 74 | (48.3–121) | 78.6 | (45.6–137.4) |
| Height (cm) | 170 | (142–200) | 168.9 | (143.5–193.0) |
| BSA (m2) | 1.73 | (1.11–3.33) | 1.87 | (1.05–3.15) |
| Age (years) | 54 | (25–84) | 61 | (28–85) |
| Baseline BPsys (mmHg) | 130 | (99–170) | 130 | (100–180) |
| Baseline BPdia (mmHg) | 78 | (51–105) | 78 | (55–100) |
| Dose range | 0.2–32 mg once daily | 0.1–12 mg twice daily | ||
BSA, body surface area; CV, coefficient of variation.
Table 3.
Model parameters estimated for the PK-PD model for BP on data from both phase I studies and from only Study 1
| Parameter | Description | Study 1 | Studies 1 and 2 | Units | |||
|---|---|---|---|---|---|---|---|
| Estimate | RSE | Estimate | RSE | ||||
| BP model | BPsys,base | Baseline systolic BP | 126 | (2%) | 127 | (5%) | mmHg |
| BPdia,base | Baseline diastolic BP | 76.8 | (1%) | 77.4 | (10%) | mmHg | |
| k in | Input rate indirect effect model | 0.304 | (19%) | 0.395 | (141%) | mmHg h−1 | |
| EE7080,S | Drug effect systolic input rate | 0.543 | (19%) | 0.520 | (38%) | ng−1 ml | |
| EE7080,D | Drug effect diastolic input rate | 0.904 | (13%) | 0.695 | (26%) | ng−1 ml | |
| EAH | AH effect | 36.4 | (53%) | 12.3 | (137%) | mmHg h−1 DDDE−1 | |
| BSV | BPsys | BSV in baseline systolic blood pressure | 10 | (12%) | 10 | (6%) | % |
| BPdia | BSV in baseline diastolic blood pressure | 9 | (13%) | 9 | (8%) | % | |
| BSVcorr,BP | Correlation in baseline between BPsys∼ BPdia | 55% | (16%) | 61 | (%) | % | |
| EE7080,sys | BSV in drug effect on systolic input rate | 67 | (41%) | 66 | (17%) | % | |
| EE7080,dia | BSV in drug effect on diatolic input rate | 39 | (47%) | 66 | (17%) | % | |
| BSVcorr,E | Correlation in drug effect sizes BPsys∼ BPdia | 32 | (71%) | 100 | (%) | % | |
| BSVRE | BSV in residual error magnitude | 12.4 | (23%) | – | (%) | % | |
| RE | RE | Exponential residual error | 9.0 | (3%) | 9.7 | (%) | % |
| RES∼ RED | Correlation residual errors for BPsys∼ BPdia | 52.4 | (3%) | 50.0 | (%) | % | |
| Markov proteinuria model | Eindir | Indirect effect size | 1.09·10−3 | (29%) | – | ng−1 h−1 ml | |
| t 1/2,indir | Half-life effect compartment | 98 | (47%) | – | days | ||
| Edir | Direct effect size, slope | 2.2 | (21%) | – | ng−1 ml | ||
| Emax,dir | Maximal relative effect size | – | 0.675 | (39%) | – | ||
| EC50,dir | Concentration at half maximal effect | – | 114 | (103%) | ng ml−1 | ||
| k0→1 | Transition rate from state 0 to 1 | 0.141 | (23%) | 0.134 | (19%) | week−1 | |
| k1→0 | Transition rate from state 1 to 0 | 1.19 | (20%) | 0.912 | (16%) | week−1 | |
| k1→2 | Transition rate from state 1 to 2 | 0.361 | (41%) | 0.313 | (25%) | week−1 | |
| k2→1 | Transition rate from state 2 to 1 | 1.90 | (44%) | 1.09 | (33%) | week−1 | |
| k2→3 | Transition rate from state 2 to 3 | 0.20 | (fixed) | 0.538 | (127%) | week−1 | |
| k3→2 | Transition rate from state 3 to 2 | 4.97 | (78%) | 2.87 | (132%) | week−1 | |
BSV, between subject variability; RE, residual error; RSE, relative standard error.
Figure 4.
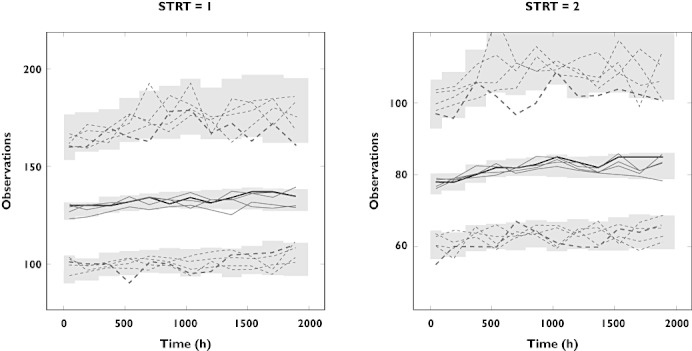
VPC, including mirror plots, of model for blood pressure based on full dataset (two studies combined). Observed data are shown by the thick black line (median) and dashed lines (5th and 95th percentile). Dashed lines represent simulated (mirror plots), while grey areas show the 95% confidence interval of the 5th, 50th and 95th percentile of the simulated data
Figure 5 shows the results of numerical predictive checks for the number of proteinuria toxicities (A) and the number of transitions between proteinuria toxicity grades (B). Previously, the Markov model provided a better description of the longitudinal profile of proteinuria occurences than a proportional odds model. For the updated model, the observed number of proteinuria toxicity grades was well within the 95% prediction intervals for all grades, although median predicted occurrences of grade 2 and ≥3 toxicities were slightly higher than the observed number. The observed transitions between toxicity grades were within the 95% prediction interval, except the transitions from grade 2 to 1, and from 3 to 2. The number of observations for these transitions were however low (26 and 7, respectively).
Figure 5.
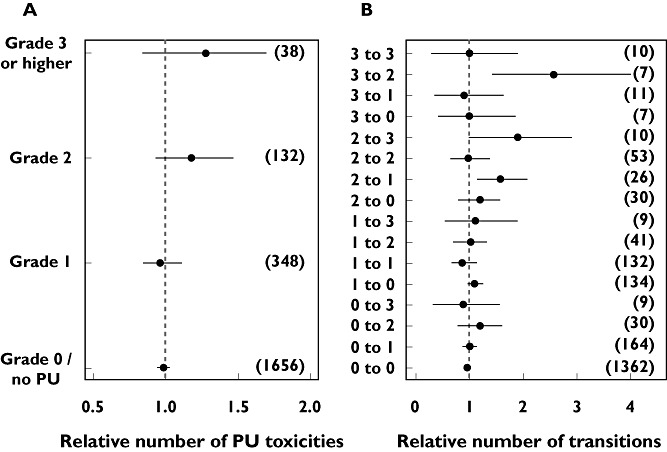
Numerical predictive checks of the number of proteinuria (PU) toxicities and the number of transitions from one grate to another, both relative to the respective observed numbers. Dots present median number of 200 simulations and the lines indicate the 95% confidence interval. The observed number of toxicities/transitions is shown between brackets
Simulations
In Table 4, the simulation results for the four designs are summarized. Designs A, B and C were effective in keeping 37.8%, 53.2% and 82.0% of patients on treatment after 16 weeks. A plot of the fraction of subjects on treatment over time during treatment is shown in Figure 6. At the end of the treatment period, in design C, 65.4%, 9.3%, 16.6%, and 9.8% of subjects on treatment continued at dose levels of 25, 20, 14 and 10 mg, respectively. As the effect of AH medication could only be estimated with large uncertainty (>100%), the simulations were repeated for a ‘worst case’ scenario in which the AH effect was set at only 50% of the estimated value, and a ‘best case’ in which the AH effect was set at 200% of the estimated value. These scenarios resulted in 80.2% and 84.2% of subjects being able to remain on treatment, vs. the 82.0% predicted using the actual parameter estimates. Design C resulted in 23% of subjects experiencing proteinuria of grade 2 or higher during the four cycle treatment period. This was higher than encountered in designs A and B, and was caused by an increase of the number of subjects on treatment, as well as an increase of the average dose level.
Table 4.
Simulation results of evaluated treatment designs
| A. Conventional design | B. AH therapy | C. AH therapy and dose de-escalation | D. Adaptive treatment | |
|---|---|---|---|---|
| Mean dose over treatment period (mg day−1)* | 13.6 (7%) | 18.2 (7%) | 21.5 (4%) | 26.1 (5%) |
| Mean dose level† | 9.5 (10%) | 13.3 (9%) | 17.5 (5%) | 24.4 (6%) |
| % subjects on treatment† | 37.8 (11%) | 53.2 (9%) | 82 (5%) | 79.2 (5%) |
| % subjects at dose ≥25 mg† | 37.8 (11%) | 53.2 (9%) | 53.6 (9%) | 49.0(7%) |
| % subjects receiving AH medication† | 0 | 60.4 (7%) | 60.4 (7%) | 80.2 (4%) |
| Increase in mean BPsys(mmHg)† | 2.23 (13%) | 5.1 (8%) | 7.0 (6%) | 6.7 (6%) |
| Increase in mean BPdia (mmHg)† | 2.96 (10%) | 7.9 (5%) | 10.1 (4%) | 9.7 (5%) |
| % subjects proteinuria grade ≥2 event during treatment | 16.0 (15%) | 19.8 (17%) | 23.2(17%) | 26.2 (15%) |
Including patient at dose level 0 mg;
at t= 16 weeks. Between brackets () is the CV% due to uncertainty in parameter estimates. AH, antihypertensive.
Figure 6.
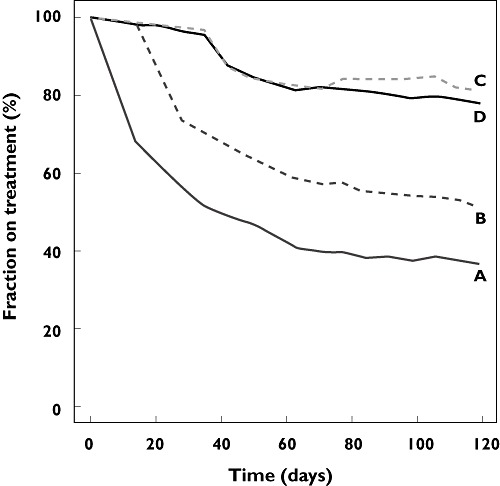
Fraction of subjects on treatment in the evaluated designs. Designs: (A) treatment discontinued at the occurrence of unacceptable toxicity (grade 2); (B) AH treatment, followed by treatment discontinuation at the persistence of hypertension toxicity; (C) AH treatment, followed by dose reductions at the persistence of hypertension toxicity; (D) Toxicity-guided intra-patient dose escalation and de-escalation, combined with AH therapy
Examination of the longitudinal plot of the mean dose level (Figure 7) shows that the dose titration design was able to increase the overall dose level considerably compared to the other designs. Over 16 weeks of treatment, this design resulted in an increase in mean dose level of 4.6 mg day−1 compared with design C. Due to higher drug exposure, a median increase in BPdia of 1.6 mmHg was observed, and the fraction of subjects who experienced proteinuria of grade >2 was increased from 23.2% to 26.2%. Consequently, the fraction of subjects able to continue treatment for 16 weeks was decreased slightly compared with the control group, from 82.0% to 79.2%. Also, the fraction of patients who required AH treatment was higher for the adaptive treatment design. Due to a slight increase in the occurrence of proteinuria, dose interruptions were made in a larger fraction of patients. This explains why the fraction of subjects treated at a dose level ≥25 mg was slightly lower (49.0% vs. 53.6%) for the AE-guided dose titration design than for design C.
Figure 7.
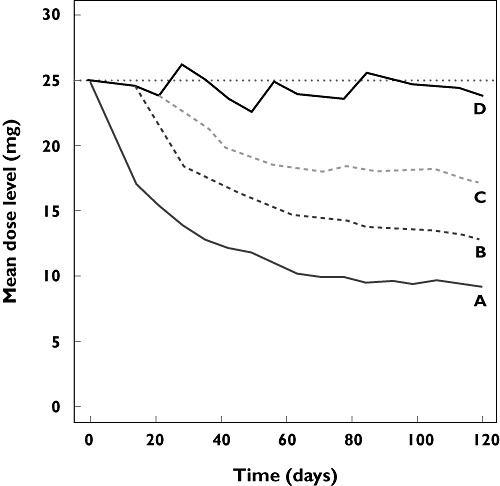
Mean dose levels in the evaluated designs A, B, C and D
Discussion
The analysis described in this article presents how modelling and simulation and the use of a biomarker can be used in the development of new drugs, specifically for the evaluation of AE-intervention designs for phase II. Our simulation results (based on early clinical data) showed that a hypertension intervention design, consisting of AH medication and dose de-escalations (design C), was able to keep a large fraction of subjects on treatment with an experimental oncology drug. Additionally, the simulations indicate that that an AE-guided dose titration (design D), may be able to increase drug exposure without considerable additional toxicity. For the drug studied here (lenvatinib/E7080, a multi-receptor TKI), maintaining continuous dosing at effective dosages is important, since this is considered to be required to achieve optimal inhibition of tumour angiogenesis [29, 30]. Additionally, BP has been reported to correlate with efficacy in treatment with several VEGFR inhibitors [8, 9, 38–41], although reports are ambiguous [42]. These results indicate that the implementation of dose titration, aimed at maximizing sustained dose levels and guided by hypertension adverse events, may be effective in optimizing treatment efficacy of these drugs. It must be noted that in the simulations it was assumed that hypertension and proteinuria were the only dose limiting toxicities, which from current evidence indeed seems to be the case. These findings need to be confirmed in a clinicial setting.
Furthermore, to be able to perform modelling and simulation with the aim of optimizing trial design, a relationship must exist between drug exposure and some pharmacodynamic measurement or outcome variable. Data from phase I trials has shown that higher lenvatinib exposure is correlated with better efficacy outcome. This relationship needs to be studied in more detail in future studies, but in the absence of a clear description of an exposure–response relationship or a distinct biomarker, it seems reasonable to aim for the highest tolerable dose [43]. By definition, for a subset of subjects, the MTD is lower than the individual maximal tolerated dose, and BP-guided within patient dose titration may therefore be considered in patients who are not experiencing toxicity.
A previously developed PK–PD model for BP and proteinuria was used in our current simulation analysis. The previously developed model adequately predicted the new BP data on a population level, However, in an attempt to reduce bias in parameter estimates, data from an additional phase I trial were used to update the PK–PD model. The same model structure was used, except for a slight modification in the relationship between drug exposure and proteinuria toxicity. From the current dataset, an indirect effect, incorporated through an effect compartment, could not be shown. Such an indirect effect component would model a delay in drug effect on the occurrence of proteinuria toxicity. It is likely that direct and indirect effects can only be distinguished adequately using intermittent dosing of lenvatinib, and/or if proteinuria is monitored more often than once a week, because more information on the dynamics of the PK–PD system is required. Although in the analysis of the combined dataset more data were available to estimate the model parameters, it was noticed that uncertainty estimates for the fixed effects parameters of the BP model were inflated. The uncertainty estimates for the random effects were however smaller in the analysis using the combined dataset, while those for the proteinuria model were broadly similar between analyses. Generally, one would expect parameters to be estimated more precisely when more data is included. Decrease of precision could indicate, for example model misspecification, differences in patient characteristics between the old data and the included dataset, or parameter estimation in a non-global optimum. However, parameter estimation was stable between sets of different initial parameter estimates, and final parameter estimates were also broadly similar between both analyses. A limited bootstrap analysis showed similar uncertainty estimates as obtained with the covariance step. These observations do however call for attention when adding further study data in the future.
We would like to stress that the focus of this analysis was on the population level, i.e. to predict the percentage of patients experiencing elevated blood pressure and requiring dose adaptations, and not on the specific prediction of individual profiles. We demonstrated that the model is suited for this task, by using the VPC and NPDE (BP) and the numerical predictive check (proteinuria) in model evaluation. Predictive performance of the model could not be evaluated on an independent dataset. However, we did show that the model that was previously developed was able to predict data from a subsequent trial. The final validation of the outcome of this evaluation study would obviously be to compare the predictions from our simulations to the outcome measures of the actual trial. In the actual lenvatinib phase I trials, hypertension and proteinuria were established as the main, dose limiting toxicities. Therefore, the dose de-escalation and interruption protocols simulated in this study provide a realistic representation of the clinical setting with respect to drop-out and treatment interruption due to toxicity.
The AH intervention design intended for phase II trials (design C) showed that combined treatment with AH medication, followed by dose reductions, allowed a large fraction (82.0%) of subjects to continue treatment with lenvatinib during the 16 weeks treatment period. The period of 16 weeks was chosen as it was considered a reasonable median treatment period in subjects with metastatic melanoma, which is now being studied as a potential indication for lenvatinib. The sensitivity analysis showed that the effect of best and worst case scenarios for the AH effect had little impact on the outcome variables. Additionally, the simulations, in which uncertainty in parameter estimates was included, also showed only moderate variability in outcome variables. One reason for this finding is that the CV% was calculated as the variability of the mean of multiple simulation studies, and thus CV% will not be inflated relevantly by including more uncertainty.
The simulation results indicated that dose reductions as implemented in the proposed hypertension intervention design (design D) could be an effective strategy to allow subjects to continue treatment with this drug. The mean daily dose level using this dose-titration design was expected to be increased by 21.4% to 26.1 mg day−1, although a slightly larger fraction of subjects received a temporary dose delay or dropped out completely due to hypertension or proteinuria toxicity. Phase I data from mixed tumour types indicated a statistically significant correlation between lenvatinib exposure and efficacy [43]. Hence continuous treatment without dose reductions could be beneficial in terms of tumour response. The concentration–effect relationship of lenvatinib, however, needs to be established in specific tumour types in further studies. Clinical efficacy of lenvatinib has recently been established in differentiated thyroid cancer [26, 27] and increase of the dose might lead to higher response rates in patients.
In the treatment simulations described in this study, bias may have been introduced by patient drop out due to toxicity. Subjects experiencing toxicity were de-escalated to lower doses, possibly leading to decreased drug efficacy. Reduced drug exposure may lead to tumour progression in those subjects, which, in a clinical trial setting, will lead to study drop-out. Therefore, the fraction of subjects on treatment may have been overestimated in our simulations. Since the relationship between drug exposure and efficacy for lenvatinib has only partially been established, we were not able to account for this bias in the simulations. However, if the occurrence of hypertension and/or proteinuria correlate with drug efficacy, as has been suggested for other TKIs [8, 9], this bias may be low. Subjects who are experiencing these toxicities will also be the ones responding to the treatment, and will not be at increased risk of drop-out due to treatment failure.
In conclusion, this article presents a model-based analysis that supported the design of a phase II trial, based on data from earlier clinical trials. For lenvatinib, the hypertension intervention design proposed for phase II is expected to allow a large fraction of subjects to continue treatment. The implementation of an AE-guided dose titration design is expected to allow the administration of higher dosages to subjects, while only marginally increasing toxicity.
Competing Interests
RK was supported by a grant from the Eisai network of companies. AG and RCS are employed by Eisai, Ltd, the company developing E7080.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Visual predictive check for PK–BP model. Model based on data from first clinical trial, predicting into new design/data. Observed data are shown by the thick black line (median) and dashed lines (5th and 95th percentile). Grey areas showthe 95% confidence interval of the 5th, 50th and 95th percentile of the simulated data
NPDE, plotted as distribution (A), vs. time (h) (B) and vs. predicted BP (C)
REFERENCES
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–34. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O'Farrell A-M, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–93. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 4.Thomas AL, Morgan B, Drevs J, Unger C, Wiedenmann B, Vanhoefer U, Laurent D, Dugan M, Steward WP. Vascular endothelial growth factor receptor tyrosine kinase inhibitors: PTK787/ZK 222584. Semin Oncol. 2003;30(3):32–8. doi: 10.1016/s0093-7754(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 5.Zhu X, Wu S, Dahut WL, Parikh CR. Risks of proteinuria and hypertension with bevacizumab, an antibody against vascular endothelial growth factor: systematic review and meta-analysis. Am J Kidney Dis. 2007;49:186–93. doi: 10.1053/j.ajkd.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 6.Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol. 2005;23:5474–83. doi: 10.1200/JCO.2005.04.192. [DOI] [PubMed] [Google Scholar]
- 7.Glen H, Boss D, Evans TR, Roelvink M, Saro JM, Bezodis P, Copalu W, Das A, Crosswell G, Schellens JH. A phase I dose finding study of E7080 in patients (pts) with advanced malignancies. J Clin Oncol. 2007;25(18) [Google Scholar]
- 8.Maitland ML, Moshier K, Imperial J, Kasza KE, Karrison T, Elliott W, Undevia SD, Stadler W, Desai AA, Ratain MJ. Blood pressure (BP) as a biomarker for sorafenib (S), an inhibitor of the vascular endothelial growth factor (VEGF) signaling pathway. J Clin Oncol. 2006;24(18) [Google Scholar]
- 9.Levy BI. Blood pressure as a potential biomarker of the efficacy of angiogenesis inhibitors. Ann Oncol. 2009;20:200–3. doi: 10.1093/annonc/mdp018. [DOI] [PubMed] [Google Scholar]
- 10.Lindauer A, Di Gion P, Kanefendt F, Tomalik-Scharte D, Kinzig M, Rodamer M, Dodos F, Sörgel F, Fuhr U, Jaehde U. Pharmacokinetic/Pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin Pharmacol Ther. 2010;87:601–8. doi: 10.1038/clpt.2010.20. [DOI] [PubMed] [Google Scholar]
- 11.Widmer N, Decosterd LA, Leyvraz S, Duchosal MA, Rosselet A, Debiec-Rychter M, Csajka C, Biollaz J, Buclin T. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br J Cancer. 2008;98:1633–40. doi: 10.1038/sj.bjc.6604355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Bernard M-A, Lassalle R, Marit G, Reiffers J, Begaud B, Moore N, Molimard M, Mahon F-X. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109:3496–9. doi: 10.1182/blood-2006-07-036012. [DOI] [PubMed] [Google Scholar]
- 13.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 14.Peng B, Hayes M, Resta D, Racine-Poon A, Druker BJ, Talpaz M, Sawyers CL, Rosamilia M, Ford J, Lloyd P, Capdeville R. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22:935–42. doi: 10.1200/JCO.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 15.Larson RA, Druker BJ, Guilhot F, O'Brien SG, Riviere GJ, Krahnke T, Gathmann I, Wang Y. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111:4022–8. doi: 10.1182/blood-2007-10-116475. [DOI] [PubMed] [Google Scholar]
- 16.Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2009;66:357–71. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- 17.Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, Bortolon E, Ichetovkin M, Chen C, McNabola A, Wilkie D, Carter CA, Taylor ICA, Lynch M, Wilhelm S. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol. 2007;59:561–74. doi: 10.1007/s00280-006-0393-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–8. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 19.Hidalgo M. Erlotinib: preclinical investigations. Oncology (Huntingt) 2003;11(12):11–6. [PubMed] [Google Scholar]
- 20.Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, Iwata KK, Morin MJ. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999;291:739–48. [PubMed] [Google Scholar]
- 21.Sleijfer S, Wiemer E. Dose selection in phase I studies: why we should always go for the top. J Clin Oncol. 2008;26:1576–8. doi: 10.1200/JCO.2007.15.5192. [DOI] [PubMed] [Google Scholar]
- 22.Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, Uenaka T, Asada M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122:664–71. doi: 10.1002/ijc.23131. [DOI] [PubMed] [Google Scholar]
- 23.Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14:5459–65. doi: 10.1158/1078-0432.CCR-07-5270. [DOI] [PubMed] [Google Scholar]
- 24.Yamada K, Hirata T, Fujiwara Y, Nokihara H, Yamamoto N, Yamada Y, Koizumi F, Nishio K, Koyama N, Tamura T. Phase I dose escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. J Clin Oncol. 2008;26(15) doi: 10.1158/1078-0432.CCR-10-2638. [DOI] [PubMed] [Google Scholar]
- 25.Yamada K, Yamamoto N, Yamada Y, Nokihara H, Fujiwara Y, Hirata T, Koizumi F, Nishio K, Koyama N, Tamura T. Phase I dose-escalation study and biomarker analysis of E7080 in patients with advanced solid tumors. Clin Cancer Res. 2011;17:2528–37. doi: 10.1158/1078-0432.CCR-10-2638. [DOI] [PubMed] [Google Scholar]
- 26.Gild ML, Bullock M, Robinson BG, Clifton-Bligh R. Multikinase inhibitors: a new option for the treatment of thyroid cancer. Nat Rev Endocrinol. 2011;7:617–24. doi: 10.1038/nrendo.2011.141. [DOI] [PubMed] [Google Scholar]
- 27.Sherman SI, Jarzab B, Cabanillas ME, Licitra LF, Pacini F, Martins R, Robinson B, Ball D, McCaffrey J, Shah MH, Bodenner D, Allison R, Newbold K, Elisei R, O'Brien J, Schlumberger M. A phase II trial of the multitargeted kinase inhibitor E7080 in advanced radioiodine (RAI)-refractory differentiated thyroid cancer (DTC) J Clin Oncol. 2011;29 [Google Scholar]
- 28.Keizer RJ, Gupta A, Mac Gillavry MR, Jansen M, Wanders J, Beijnen JH, Schellens JHM, Karlsson MO, Huitema ADR. A model of hypertension and proteinuria in cancer patients treated with the anti-angiogenic drug E7080. J Pharmacokinet Pharmacodyn. 2010;37:347–63. doi: 10.1007/s10928-010-9164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kisker O, Becker CM, Prox D, Fannon M, D'Amato R, Flynn E, Fogler WE, Sim BK, Allred EN, Pirie-Shepherd SR, Folkman J. Continuous administration of endostatin by intraperitoneally implanted osmotic pump improves the efficacy and potency of therapy in a mouse xenograft tumor model. Cancer Res. 2001;61:7669–74. [PubMed] [Google Scholar]
- 30.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–39. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 31.Keizer R, Gupta A, Jansen M, Wanders J, Beijnen J, Schellens J, Karlsson M, Huitema A. 2009. Modeling of hypertension in response to anti-angiogenic therapy; PAGE 18 (2009) Abstr 1467. Available at http://www.page-meeting.org/?abstract=1467 (last accessed 1 May 2010).
- 32.Hong D, Koetz B, Kurzrock R, Senzer N, Hanekom W, Naing A, Wheler J, Mink J, Ren M, Nemunaitis J. Phase I dose-escalation study of E7080, a selective tyrosine kinase inhibitor, administered orally to patients with solid tumors. J Clin Oncol. 2010;28 [Google Scholar]
- 33.Karlsson M, Holford N. 2008. A tutorial on visual predictive checks. Abstracts of the Annual Meeting of the Population Approach Group in Europe (Internet). Marseille; . Available at http://www.page-meeting.org/default.asp?abstract=1434 (last accessed 1 May 2010).
- 34.Brendel K, Comets E, Mentre F. Abstracts of the Annual Meeting of the Population Approach Group in Europe; 2007. Normalised prediction distribution errors for the evaluation of nonlinearmixed-effects models. Copenhagen: . Available at http://www.page-meeting.org/default.asp?abstract=1085 (last accessed 1 May 2010). [Google Scholar]
- 35.NCI. 2006. Common Terminology Criteria for Adverse Events. 3.0 ed.
- 36.Beal SL, Sheiner LB. In: Beal SL, Sheiner LB, editors. Icon Development Solutions; 1989. NONMEM Users Guides, eds . Ellicott City, MD: [Google Scholar]
- 37.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101:72–9. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 38.De Stefano A, Cannella L, Carlomagno C, Crispo A, Bianco R, Marciano R, Pepe S, De Placido S. Correlation between bevacizumab-related hypertension and response in mCRC patients. J Clin Oncol. 2010;28 [Google Scholar]
- 39.Radaideh S, Gerber D, Dunphy F, Yan J, Xie Y, Fenske E, Shouldis J, Kindler H, Dowell J. Association of hypertension (HTN) and clinical outcome in a phase II trial of cisplatin (C), pemetrexed (P), and bevacizumab (B) in patients (pts) with untreated malignant mesothelioma. J Clin Oncol. 2010;28 [Google Scholar]
- 40.Wilhelm K, Atkinson B, Khakoo A, Tannir N, Jonasch E. A retrospective evaluation of antiangiogenic therapy induced hypertension in metastatic renal cell carcinoma. J Clin Oncol. 2010;28 [Google Scholar]
- 41.Kim R, Byrne M, Hammel J, El-Gazzaz G, Aucejo F. Association of hypertension with overall outcome in patients taking sorafenib in advanced hepatocellular carcinoma (HCC) J Clin Oncol. 2010;28 [Google Scholar]
- 42.Hurwitz H, Douglas P, Middleton J, Sledge G, Johnson D, Reardon D, Chen D, Rosen O. Analysis of early hypertension (HTN) and clinical outcome with bevacizumab (BV) J Clin Oncol. 2010;28 doi: 10.1634/theoncologist.2012-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta A, Koetz B, Hanekom W, O'Brien JP, Wanders J, Jansen M. 453 Population pharmacokinetics (PK) and exposure/response relationships of the receptor tyrosine kinase inhibitor E7080 in phase I studies. Eur J Cancer Suppl. 2010;8:143. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Visual predictive check for PK–BP model. Model based on data from first clinical trial, predicting into new design/data. Observed data are shown by the thick black line (median) and dashed lines (5th and 95th percentile). Grey areas showthe 95% confidence interval of the 5th, 50th and 95th percentile of the simulated data
NPDE, plotted as distribution (A), vs. time (h) (B) and vs. predicted BP (C)