

Abstract
AIMS
To determine the effect of febuxostat on cytochrome P450 2C8 (CYP2C8) activity using rosiglitazone as a CYP2C8 substrate.
METHODS
Healthy subjects received febuxostat 120 mg daily (regimen A) or matching placebo (regimen B) for 9 days along with a single oral dose of rosiglitazone 4 mg on day 5 in a double-blind, randomized, cross-over fashion (≥7 day washout between periods). Plasma samples for analysis of the impact of febuxostat on the pharmacokinetics (PK) of rosiglitazone and its metabolite, N-desmethylrosiglitazone, were collected for 120 h after co-administration.
RESULTS
Of the 39 subjects enrolled, 36 completed the study and were included in the PK analyses. Rosiglitazone PK parameters were comparable between regimens A and B. Median time to maximal plasma concentration, mean maximal plasma concentration (Cmax), area under the concentration-time curve (AUC) from time zero to the last quantifiable concentration (AUC0–tlqc), AUC from time zero to infinity (AUC0–∞), and terminal elimination half-life for regimen A were 0.50 h, 308.6 ng ml−1, 1594.9 ng h ml−1, 1616.0 ng h ml−1 and 4.1 h, respectively, and for regimen B they were 0.50 h, 327.6 ng ml−1, 1564.5 ng h ml−1, 1584.2 ng h ml−1 and 4.0 h, respectively. Point estimates for the ratio of regimen A to regimen B (90% confidence intervals) for rosiglitazone Cmax, AUC0–tlqc and AUC0–∞ central values were 0.94 (0.89–1.00), 1.02 (1.00–1.04) and 1.02 (1.00–1.04), respectively.
CONCLUSIONS
Co-administration of febuxostat had no effect on rosiglitazone or N-desmethylrosiglitazone PK parameters, suggesting that febuxostat can be given safely with drugs metabolized through CYP2C8.
Keywords: CYP2C8, drug–drug interaction, febuxostat, pharmacokinetics, rosiglitazone
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Febuxostat is a xanthine oxidase inhibitor approved for lowering serum urate levels in hyperuricaemic gout patients. It is extensively metabolized both by conjugation via uridine diphosphate glucuronosyltransferase enzymes and by oxidation via cytochrome P450 enzymes.
WHAT THIS STUDY ADDS
Using rosiglitazone as a CYP2C8 substrate, we have demonstrated that febuxostat does not impede CYP2C8 metabolic activity, because febuxostat had no impact on rosiglitazone pharmacokinetic parameters. We therefore suggest that febuxostat can be safely co-administered with CYP2C8-metabolized drugs.
Introduction
Circulating and tissue levels of urate can be elevated by overproduction and/or underexcretion of urate [1]. Sustained elevation of the tissue level of urate above the saturation limit (approximately 6.8 mg dl−1, 400 µm) may lead to the deposition of crystals of monosodium urate. The acute inflammatory response to these crystals within joints manifests as acute attacks of gout, a painful and debilitating arthritis [2, 3]. Reduction of urate levels is essential for good long-term management of gout [4, 5].
The most common mode for urate-lowering therapy in gout is use of the xanthine oxidase inhibitors [6] febuxostat and allopurinol. Febuxostat is a potent, nonpurine xanthine oxidase inhibitor [7, 8], approved in the USA [9], Canada [10], Europe [11] and Japan [12] for the management of hyperuricaemia in patients with gout. Clinical studies in both healthy volunteers and hyperuricaemic subjects with gout have demonstrated the ability of febuxostat to reduce serum urate in a dose-dependent manner [13–17].
The pharmacokinetic (PK) profile of febuxostat has been previously described in healthy subjects following multiple oral daily dose administration of febuxostat in the range of 10–240 mg [18]. Following administration of a 120 mg once-daily dose of febuxostat for 14 days, the mean maximal plasma concentration (Cmax) was 5.31 µg ml−1, and mean time to maximal plasma concentration (Tmax) was 1.0 h, with a harmonic mean half-life (T1/2) of 11.9 h [18]. Febuxostat is extensively metabolized both by conjugation via uridine diphosphate glucuronosyltransferase (UGT) enzymes, including UGT1A1, UGT1A3, UGT1A9 and UGT2B7, and by oxidation via cytochrome P450 (CYP) enzymes, including CYP1A2, CYP2C8 and CYP2C9, and non-P450 enzymes [9, 18].
Febuxostat undergoes metabolism by multiple CYP pathways. As a result, clinically relevant interactions between febuxostat and drugs metabolized by these CYP enzymes are unlikely. However, the possibility of CYP2C8 inhibition by febuxostat was demonstrated in an in vitro human liver microsomal study [inhibition constant (Ki) = 20 µm Teijin Pharma Ltd, Y. Adachi, T. Ambo and Y. Kametani; unpublished data]. From clinical studies, the mean Cmax of febuxostat following multiple 80 mg oral doses is 9.4 µm (Takeda Global Research & Development Center, Inc. unpublished data), which is approximately twofold lower than the observed Ki value for CYP2C8 inhibition. Based on the criteria given in the US Food and Drug Administration (FDA) guidance for predicting a potential in vivo drug interaction using in vitro metabolism data [19] (0.1 < [I]/Ki ratio<1, where [I] represents the plasma concentration of the inhibitor), febuxostat may possibly affect the PK of drugs predominantly metabolized by CYP2C8, because the calculated [I]/Ki ratio was 0.47. Gout patients have high rates of comorbidities and therefore high concomitant medication use [17, 20, 21]. Knowledge of potential drug–drug interactions due to CYP2C8 inhibition is needed for safe co-administration of febuxostat with CYP2C8-metabolized drugs in order to avoid increased plasma levels and possible drug toxicity.
Rosiglitazone is an insulin-sensitizing oral antidiabetic agent of the thiazolidinedione class used in the treatment of patients with type 2 diabetes mellitus [22]. Rosiglitazone is rapidly and completely absorbed, with an absolute bioavailability estimated to be more than 99% after oral administration [23]. The major route of rosiglitazone metabolism is N-demethylation and hydroxylation, mainly by CYP2C8 and to a lesser extent by CYP2C9, followed by conjugation with sulfate and glucuronic acid [24]. Rosiglitazone has been established and recommended in FDA guidance as a suitable agent to assess the impact of a drug on CYP2C8 activity in human subjects [19].
The primary objective of this study was to evaluate the effect of multiple doses of febuxostat on CYP2C8 metabolic activity via the effects on the PK of a single oral dose of rosiglitazone and its metabolite, N-desmethylrosiglitazone. The secondary objective was to assess the safety of a single dose of rosiglitazone with or without multiple doses of febuxostat in healthy subjects.
Methods
Subjects
Qualifying healthy male and female subjects, aged 18–55 years inclusive, were enrolled after their health status had been determined by assessments performed during the screening period. Subjects were to have a body mass index between 18 and 30 kg m−2 inclusive, and normal renal function (estimated creatinine clearance > 90 ml min−1, calculated by the Cockcroft–Gault formula). Nonsterilized, sexually active male subjects had to use an acceptable method of barrier contraception. In addition, female subjects had to be either postmenopausal for at least 2 years, surgically sterilized or, if of childbearing age and sexually active, using an acceptable method of barrier contraception and could not be pregnant or lactating. Subjects were excluded for any one of the following reasons: prior history of gout, xanthinuria or secondary hyperuricaemia; evidence of cardiovascular, central nervous system, hepatic, haematopoietic, renal or endocrine dysfunction; serious allergy, asthma, hypoxaemia, hypertension, seizures or allergic skin rash; any finding from medical history, physical examination or laboratory tests that would contraindicate taking febuxostat or rosiglitazone or that would interfere with the conduct of the study; current or recent (within 6 months) gastrointestinal disease; history of cancer not in remission for at least 5 years; positive test for hepatitis B or C or human immunodeficiency viral infection; consumption of any food or beverage containing caffeine or xanthine, grapefruit or grapefruit juice, or charbroiled foods within 14 days prior to check-in; use of nicotine, alcohol or any prescription or enzyme-altering medication that could interfere with the study data; or a recent blood donation or loss of blood volume of 450 ml or more.
Study design
This was a phase 1, double-blind, randomized, two-period cross-over study conducted at a single clinical site in the USA. The study was approved by the site's Institutional Review Board and was conducted in accordance with requirements of the Declaration of Helsinki and the International Conference on Harmonization. Potential subjects reviewed and signed informed consent forms prior to undergoing any study procedures. Subjects were screened for eligibility from days −28 to −1, and were eligible to enrol if they met all of the inclusion and none of the exclusion criteria. Eligibility was assessed with data collected from complete physical examinations, electrocardiograms, clinical laboratory evaluations and recording of demographics, social behavioural characteristics, medical histories and medication use (past and present).
Enrolled subjects were randomized 1:1 to one of two dosing sequence groups: regimen A (period 1) followed by regimen B (period 2) or regimen B (period 1) followed by regimen A (period 2). Regimen A comprised febuxostat 120 mg (Uloric®;Takeda Pharmaceuticals North America, Inc., Deerfield, IL, USA), which consisted of one overencapsulated 80 mg tablet and one overencapsulated 40 mg tablet, once daily for nine consecutive days, and a single oral 4 mg tablet of rosiglitazone (Avandia®; GlaxoSmithKline, Research Triangle Park, NC, USA) on day 5. Regimen B comprised two matching placebo capsules once daily for nine consecutive days and a single oral 4 mg tablet of rosiglitazone on day 5. For both periods, subjects were confined until all procedures were completed, beginning on day −1 until day 10. There was a washout interval of at least 7 days between the last dose of study drug in period 1 and the first dose of study drug in period 2.
The safety and efficacy of febuxostat 120 mg was extensively characterized during the febuxostat clinical development programme, and the 120 mg dose is the highest marketed dose worldwide (marketed in the European Union). Therefore, 120 mg of febuxostat was selected for this interaction study to maximize the potential to observe an effect of febuxostat on the pharmacokinetics of rosiglitazone [19]. The recommended starting dose for rosiglitazone is 4 mg daily. As rosiglitazone is a low therapeutic index drug, a single 4 mg dose was selected for this study.
On days 1–9 of each period, subjects received either febuxostat or placebo at approximately 08.00 h after a 10 h fast, and on day 5 subjects received a single dose of rosiglitazone along with febuxostat or placebo. During each period, blood samples were collected to determine febuxostat trough plasma concentrations predose on days 5, 6 and 7. Blood samples for the determination of rosiglitazone and its metabolite N-desmethylrosiglitazone were collected at scheduled time points for 120 h following administration of rosiglitazone on day 5. During confinement, all subjects received the same standardized breakfast, lunch, dinner and snack, except on day 5 when they did not receive breakfast prior to or following administration of rosiglitazone. Subjects refrained from strenuous activities or exercise during the course of study.
Pharmacokinetic sample collection and evaluations
For the PK analyses of rosiglitazone and N-desmethylrosiglitazone, 4 ml blood samples were obtained on day 5 of each period within 30 min before dosing and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, 16, 24, 36, 48, 72, 96 and 120 h post-rosiglitazone dose. The PK analyses were performed using the WinNonlin Enterprise Version 5.2.1 (Pharsight Co, Inc., Mountain View, CA, USA). Plasma concentrations of rosiglitazone and its metabolite N-desmethylrosiglitazone were determined using high-performance liquid chromatography/tandem mass spectrometry assays performed by Pharmaceutical Product Development, Inc. (Richmond, VA, USA). Briefly, in 96-well plates, 25 µl of a 500 ng ml−1 internal standard working solution was added to 50.0 µl aliquot of each collected plasma sample. Analytes were then isolated through supported liquid/liquid extraction (SLE) using 60:40 hexane/ethyl acetate, v/v. The organic solvent was then passed through an Isolute SLE+ plate to another 96-well plate and evaporated under a nitrogen stream at room temperature. The resulting residue was reconstituted with 500 µl of 50:50 acetonitrile/10 mm ammonium acetate with 0.02% trifluoroacetic acid, v/v. The final extractions were then analysed via high-performance liquid chromatography/tandem mass spectrometry detection. The detection assays were validated against concentration ranges from 1.0 ng ml−1 to 500 ng ml−1 for the quantification of both rosiglitazone and N-desmethylrosiglitazone. Precision and accuracy were evaluated by replicate analyses of human plasma quality control pools prepared at five concentrations spanning the calibration range. Precision was measured as the percentage coefficient of variation (%CV) of the set of values for each pool. Accuracy was expressed as the percentage difference of the mean value for each pool from the theoretical concentration. The intra-assay %CV ranged from 2.10 to 7.59 for rosiglitazone and from 1.04 to 9.37 for N-desmethylrosiglitazone. The intra-assay accuracy ranged from −6.33 to 6.84 for rosiglitazone and from −5.90 to 12.7 for N-desmethylrosiglitazone. The lower limit of quantification for both rosiglitazone and N-desmethylrosiglitazone was 1.00 ng ml−1.
The observed Cmax and Tmax of rosiglitazone and N-desmethylrosiglitazone were determined from the concentration–time profiles for all evaluable subjects for each treatment period. The terminal elimination rate constant (λz) was calculated as the negative slope of the log-linear regression of the natural concentration–time curve profile during the terminal phase. The area under the plasma concentration–time curve (AUC) from time zero to the time of the last quantifiable concentration (tlqc; AUC0–tlqc) was calculated using the linear trapezoidal rule. The AUC from time zero to infinity (AUC0–∞) was calculated as AUC0–∞= AUC0–tlqc+ lqc/λz. The terminal elimination half-life (T1/2) was calculated as T1/2= ln(2)/λz. The apparent oral clearance (CL/F; for rosiglitazone only) was calculated as CL/F= dose/AUC0–∞. The apparent volume of distribution during the terminal phase (Vz/F; for rosiglitazone only) was calculated as Vz/F= (CL/F)/λz.
Statistical analyses
All statistical analyses were conducted using SAS version 9.1 or higher (SAS Institute, Cary, NC, USA). For each regimen, descriptive statistics were used to summarize the plasma PK parameters of rosiglitazone and its metabolite. Only PK data from subjects who completed the both periods were included in the summaries. Analysis of variance (ANOVA) models were performed on rosiglitazone and N-desmethylrosiglitazone Tmax, λz, and the natural logarithms of Cmax, AUC0–tlqc and AUC0–∞, with factors for sequence, subject nested within sequence, period and regimen. The effect of febuxostat on the PK of rosiglitazone and N-desmethylrosiglitazone was assessed via point estimates and 90% confidence intervals (CIs) for the ratio of the central values of Cmax and AUCs for regimen A to regimen B. A conclusion of ‘no effect’ of febuxostat on the PK of rosiglitazone was reached if the 90% CIs for the ratio of rosiglitazone Cmax and AUCs central values were within the 0.80–1.25 interval [19]. ANOVA models were also performed on the natural logarithms of metabolite to parent ratios for Cmax, AUC0–tlqc and AUC0–∞. The models included factors of sequence, subject within sequence, period and regimen. The factor of subject within sequence was random, and other factors were fixed.
Safety analyses
All subjects who received at least one dose of study drug were included in the safety analyses. All treatment-emergent adverse events and clinical laboratory evaluations were summarized with descriptive statistics by regimen.
Sample size
A sample size of 38 subjects (19 in each sequence group) was planned. This allowed for up to two dropouts (approximately 5% dropout rate), while providing at least 90% probability of concluding ‘no effect’ of febuxostat on rosiglitazone Cmax provided the true difference between rosiglitazone Cmax central values was no more than 5%. The sample size was calculated using an intrasubject variance of 0.058, which was obtained from previously published studies [25–29]. The power for AUC was also at least 95%.
Results
Baseline demographics and disposition of subjects
Thirty-eight subjects were randomized in 1:1 ratio into sequence groups AB and BA. One subject withdrew from the study after one dose due to difficult blood draw, and was replaced by a new subject. Therefore, 39 subjects were enrolled and dosed in this study (27 men and 12 women); 20 were in sequence AB (regimen A then B) and 19 in sequence BA (regimen B then A). All subjects in sequence BA completed both periods. However, three subjects in sequence AB discontinued prematurely. One subject discontinued after one dose of febuxostat in period 1 owing to poor venous access. One subject discontinued before starting period 2 owing to an estimated creatinine clearance of <90 ml min−1, and one subject discontinued owing to elevated liver function tests. These three subjects were excluded from the PK analysis because they did not complete both periods. The sequence factor in the ANOVA models was used to assess the possible unequal first-order carry-over between the two periods. As the washout period was sufficiently long, there should not be any carry-over effect from period 1 to period 2. Hence, the imbalance in the sequence groups should not affect assessment of the effect of febuxostat on rosiglitazone PK. The mean (SD) age of all subjects was 34.4 (8.88) years, and 84.6% (n= 33) were Caucasian. Mean (SD) body mass index was 25.5 (3.07) kg m−2.
Pharmacokinetics
Mean plasma concentration–time profiles for rosiglitazone and N-desmethylrosiglitazone are illustrated in Figures 1 and 2. The plasma concentration–time profiles for both rosiglitazone and N-desmethylrosiglitazone when rosiglitazone was co-administered with febuxostat or placebo were comparable between regimens.
Figure 1.
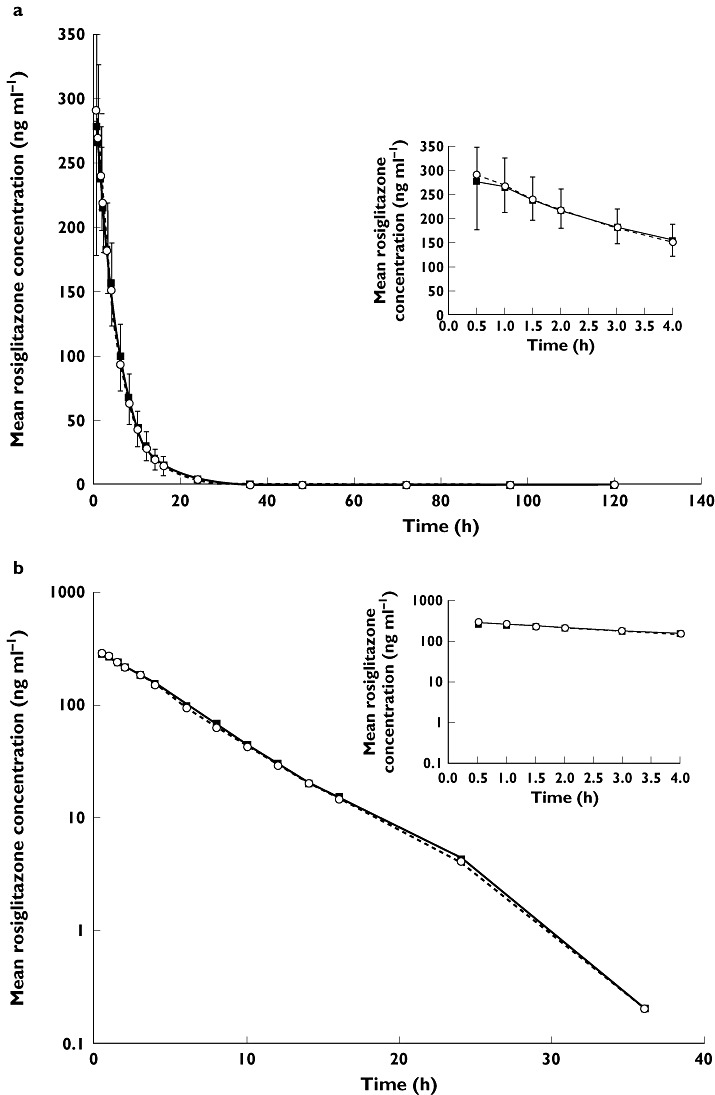
Mean rosiglitazone plasma concentration–time profiles following a single 4 mg dose of rosiglitazone co-administered with 120 mg febuxostat or matching placebo; linear (a) and logarithmic plots (b). Error bars represent SD; upper bars, febuxostat phase, and lower bars, placebo phase. Rosiglitazone + Febuxostat ( ); Rosiglitazone + Placebo (
); Rosiglitazone + Placebo ( )
)
Figure 2.
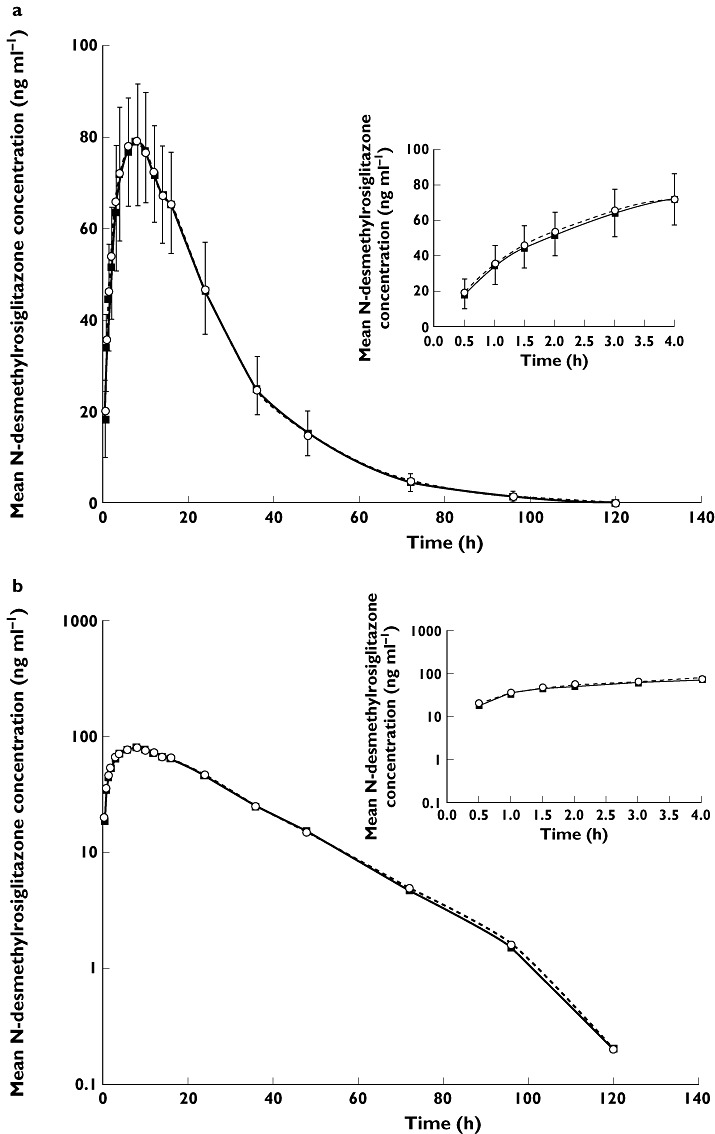
Mean N-desmethylrosiglitazone plasma concentration–time profiles following a single 4 mg dose of rosiglitazone co-administered with 120 mg febuxostat or matching placebo; linear (a) and logarithmic plots (b). Error bars represent SD; upper bars, febuxostat phase and lower bars, placebo phase. Rosiglitazone + Febuxostat ( ); Rosiglitazone + Placebo (
); Rosiglitazone + Placebo ( )
)
Following oral co-administration of a 4 mg dose of rosiglitazone with either febuxostat or placebo, rosiglitazone was rapidly absorbed with a median Tmax value of 0.5 h and eliminated with a mean T1/2 of approximately 4 h for both regimens. Table 1 summarizes the PK parameters of both rosiglitazone and its metabolite, N-desmethylrosiglitazone. All PK parameters for both rosiglitazone and N-desmethylrosiglitazone were comparable between regimen A and regimen B. N-desmethylrosiglitazone exhibited a relatively slower elimination from the systemic circulation, with a greater mean T1/2 compared with that of rosiglitazone.
Table 1.
Summary of rosiglitazone and N-desmethylrosiglitazone pharmacokinetic parameter estimates following administration of a single 4 mg dose of rosiglitazone co-administered with febuxostat 120 mg or matching placebo
| Regimen A (febuxostat) | Regimen B (placebo) | |
|---|---|---|
| Rosiglitazone (n= 36) | ||
| Tmax (h)* | 0.50 (0.50–6.00) | 0.50 (0.50–2.00) |
| Cmax (ng ml−1) | 308.6 ± 57.37 | 327.6 ± 67.04 |
| AUC0–tlqc (ng h ml−1) | 1594.9 ± 310.36 | 1564.5 ± 296.97 |
| AUC0–∞ (ng h ml−1) | 1616.0 ± 313.83 | 1584.2 ± 303.82 |
| T1/2 (h) | 4.1 ± 0.66 | 4.0 ± 0.60 |
| CL/F (l h−1) | 2.6 ± 0.49 | 2.6 ± 0.51 |
| Vz/F (l) | 15.0 ± 2.57 | 15.1 ± 2.56 |
| N-Desmethylrosiglitazone (n= 36) | ||
| Tmax (h)* | 8.0 (3.0–16.0) | 8.0 (4.0–14.0) |
| Cmax (ng ml−1) | 81.9 ± 11.87 | 83.0 ± 13.39 |
| AUC0–tlqc (ng h ml−1) | 2499.1 ± 450.32 | 2495.1 ± 434.23 |
| AUC0–∞ (ng h ml−1) | 2541.2 ± 450.24 | 2538.4 ± 436.66 |
| T1/2 (h) | 14.9 ± 2.09 | 15.4 ± 2.45 |
Unless indicated otherwise, all parameters are reported as means ± SD. Abbreviations: Tmax, time to maximal plasma concentration; Cmax, maximal plasma concentration; AUC0–tlqc, area under the curve from time 0 to the time of last quantifiable concentration; AUC0–∞, area under the curve from time 0 to infinity; T1/2, terminal elimination half-life; CL/F, oral clearance; and Vz/F, volume of distribution.
Tmax is reported as the median (minimal minus maximal values).
The bioavailability of rosiglitazone and N-desmethylrosiglitazone in regimen A (febuxostat) vs. regimen B (placebo) is represented in Table 2 as point estimates and 90% CIs for the ratios of the central values for Cmax, AUC0–tlqc and AUC0–∞. The point estimates were close to 1, and the 90% CIs for the ratios were within the ‘no effect’ criteria of 0.80–1.25. Box plots comparing rosiglitazone Cmax and AUC0–∞ values from the two treatment regimens are shown in Figure 3.
Table 2.
Bioavailability of rosiglitazone and N-desmethylrosiglitazone co-administered with febuxostat relative to rosiglitazone co-administered with placebo
| Parameter | Point estimate | 90% Confidence interval |
|---|---|---|
| Rosiglitazone; regimen A vs. regimen B | ||
| C max | 0.9447 | 0.8924–0.9999 |
| AUC0–tlqc | 1.0209 | 1.0024–1.0398 |
| AUC0–∞ | 1.0219 | 1.0032–1.0410 |
| N-Desmethylrosiglitazone; regimen A vs. regimen B | ||
| C max | 0.9882 | 0.9687–1.0081 |
| AUC0–tlqc | 1.0010 | 0.9829–1.0194 |
| AUC0–∞ | 1.0005 | 0.9836–1.0177 |
| Metabolite-to-parent ratios | ||
| C max ratio | 1.0461 | 0.9888–1.1067 |
| AUC0–tlqc ratio | 0.9805 | 0.9635–0.9977 |
| AUC0–∞ ratio | 0.9790 | 0.9627–0.9957 |
Abbreviations: Cmax, maximal plasma concentration; AUC0–tlqc, area under the curve from time 0 to the time of last quantifiable concentration; AUC0–∞, area under the curve from time 0 to infinity.
Figure 3.
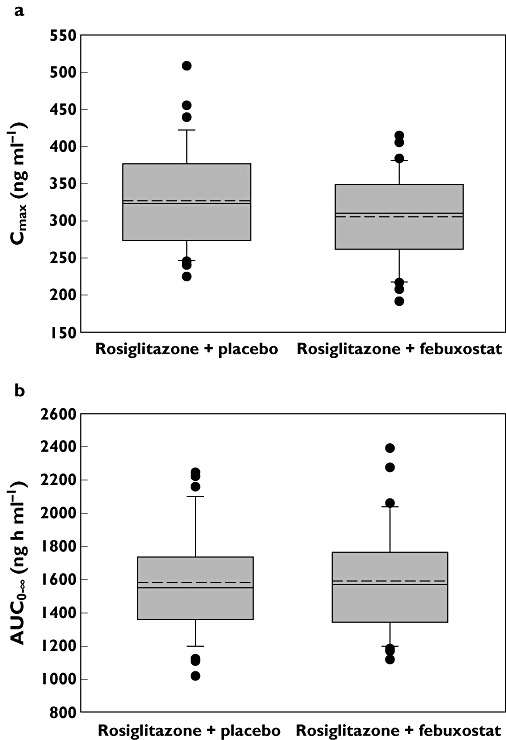
Box plots of rosiglitazone Cmax(a) and AUC0–∞ (b) following administration of a single 4 mg dose of rosiglitazone co-administered with febuxostat 120 mg or matching placebo. The upper and lower boundaries of the box represent the 75th and 25th percentiles, respectively. This continuous line within the box marks the mean and the dashed line marks the median. Error bars indicated the 90th and 10th percentiles. Filled circles represent all data points outside the 90th to 10th percentiles
The factor of period was statistically significant in the ANOVA models for rosiglitazone AUCs and metabolite-to-parent AUC ratios. Period in the ANOVA accounted for the differences between periods. As only the subjects who had complete data for both periods were included in the ANOVA model, the period effect on each of the regimens was considered to be balanced; hence, the existence of the period effect did not affect the comparison of regimen A and regimen B, but rather confirmed the appropriateness of the cross-over design. The sequence factor was statistically significant in the ANOVA models for N-desmethylrosiglitazone AUCs. The significant test for sequence may raise the concern of unequal carry-over effect. However, because of the sufficient washout interval and imbalance in the sequence group, it is very likely that the statistically significant sequence effect was due to the difference of the subjects between sequence groups and not the unequal carry-over effect. Period and sequence factors are not statistically significant in other ANOVA models.
Safety
Safety analyses were carried out on the 39 subjects who received at least one dose of study drugs. The percentage of subjects with one or more treatment-emergent adverse events were 30.8% (12 of 39) and 23.7% (nine of 38) for regimens A and B, respectively. The most frequently reported treatment-emergent adverse events, reported in ≥5% of subjects for either regimen, were upper abdominal pain (5.1 and 2.6%), flatulence (5.1 and 2.6%), nausea (5.1% and 0), nasopharyngitis (5.1% and 0), headache (10.3 and 5.3%), dizziness (0 and 5.3%) and oropharyngeal pain (5.1% and 0) in regimens A and B, respectively. There were no discontinuations due to adverse events. Overall, there were no clinically relevant changes for any laboratory value. There were no deaths or serious adverse events in the study.
Discussion
In vitro analysis of the effect of febuxostat on CYP2C8 activity indicated an [I]/Ki ratio of 0.47. As an estimated [I]/Ki ratio of >0.1 is considered to be indicative of possible CYP inhibition [19], this suggested that febuxostat could possibly affect the PK of drugs predominantly metabolized by CYP2C8 and warranted in vivo follow-up. Rosiglitazone is an approved in vivo probe for assessing the effect of a new drug on CYP2C8 activity [19] because its bioavailability is significantly increased when administered in the presence of a known CYP2C8 inhibitor, such as gemfibrozil, and significantly reduced when co-administered with rifampin, a known CYP2C8 inducer [22]. In the present study, the PK parameters of rosiglitazone and its metabolite, N-desmethylrosiglitazone, were not affected when rosiglitazone was co-administered with febuxostat. As febuxostat can be metabolized by various CYP isozymes, these results are not unexpected and suggest that febuxostat can be co-administered with other drugs that are metabolized by CYP2C8 without affecting their PK parameters. Other drugs for which CYP2C8 has a major role in their biotransformation are as follows: amiodarone (antiarrhythmic agent); amodiaquine and choloroquine (antimalarial drugs); cervistatin (lipid-lowering drug); the chemotherapeutic drug, paclitaxel; retinoic acid; repaglinide and troglitazone (antidiabetics); and tazarotenic acid [30].
Maximal plasma concentration and AUC for both rosiglitazone and N-desmethylrosiglitazone were similar between the febuxostat and placebo regimens. Likewise, the T1/2 and clearance (CL/F) for both analytes were unaffected by co-administration of rosiglitazone with febuxostat. All PK parameters observed for rosiglitazone during this study were within the range of those previously reported for a 4 mg dose of rosiglitazone in healthy adults [31]. In addition, co-administration of rosiglitazone with febuxostat was well tolerated. Therefore, febuxostat does not alter in vivo CYP2C8 metabolic activity, suggesting that it can be co-administered safely with drugs metabolized by CYP2C8 without dose adjustments.
Acknowledgments
Assistance with manuscript preparation was provided by Meryl Gersh, PhD, of AlphaBioCom, LLC, in King of Prussia, PA, USA and funded by Takeda Pharmaceuticals North America, Inc.
Competing Interests
This study was fully funded by Takeda Global Research & Development Center, Inc. All authors are employees of the sponsor.
REFERENCES
- 1.Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143:499–516. doi: 10.7326/0003-4819-143-7-200510040-00009. [DOI] [PubMed] [Google Scholar]
- 2.Becker MA, Chohan S. We can make gout management more successful now. Curr Opin Rheumatol. 2008;20:167–72. doi: 10.1097/BOR.0b013e3282f54d03. [DOI] [PubMed] [Google Scholar]
- 3.Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51:321–5. doi: 10.1002/art.20405. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Doherty M, Bardin T, Pascual E, Barskova V, Conaghan P, Gerster J, Jacobs J, Leeb B, Liote F, McCarthy G, Netter P, Nuki G, Perez-Ruiz F, Pignone A, Pimentao J, Punzi L, Roddy E, Uhlig T, Zimmermann-Gorska I. EULAR evidence based recommendations for gout. Part II: management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT) Ann Rheum Dis. 2006;65:1312–24. doi: 10.1136/ard.2006.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Ruiz F, Liote F. Lowering serum uric acid levels: what is the optimal target for improving clinical outcomes in gout? Arthritis Rheum. 2007;57:1324–8. doi: 10.1002/art.23007. [DOI] [PubMed] [Google Scholar]
- 6.Sarawate CA, Brewer KK, Yang W, Patel PA, Schumacher HR, Saag KG, Bakst AW. Gout medication treatment patterns and adherence to standards of care from a managed care perspective. Mayo Clin Proc. 2006;81:925–34. doi: 10.4065/81.7.925. [DOI] [PubMed] [Google Scholar]
- 7.Takano Y, Hase-Aoki K, Horiuchi H, Zhao L, Kasahara Y, Kondo S, Becker MA. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005;76:1835–47. doi: 10.1016/j.lfs.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 8.Okamoto K, Eger BT, Nishino T, Kondo S, Pai EF, Nishino T. An extremely potent inhibitor of xanthine oxidoreductase. Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. J Biol Chem. 2003;278:1848–55. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- 9.Takeda Pharmaceuticals North America, Inc. 2011.
- 10.Takeda Canada, Inc. 2010.
- 11.Menarini International Operations. 2008. . Adenuric® Product Monograph. Luxembourg Menarini International Operations Luxembourg S.A.
- 12.Teijin Pharma Limited. 2011.
- 13.Becker MA, Kisicki J, Khosravan R, Wu J, Mulford D, Hunt B, MacDonald P, Joseph-Ridge N. Febuxostat (TMX-67), a novel, non-purine, selective inhibitor of xanthine oxidase, is safe and decreases serum urate in healthy volunteers. Nucleosides Nucleotides Nucleic Acids. 2004;23:1111–6. doi: 10.1081/NCN-200027372. [DOI] [PubMed] [Google Scholar]
- 14.Becker MA, Schumacher HR, Jr, Wortmann RL, MacDonald PA, Palo WA, Eustace D, Vernillet L, Joseph-Ridge N. Febuxostat, a novel nonpurine selective inhibitor of xanthine oxidase: a twenty-eight-day, multicenter, phase II, randomized, double-blind, placebo-controlled, dose-response clinical trial examining safety and efficacy in patients with gout. Arthritis Rheum. 2005;52:916–23. doi: 10.1002/art.20935. [DOI] [PubMed] [Google Scholar]
- 15.Becker MA, Schumacher HR, Jr, Wortmann RL, MacDonald PA, Eustace D, Palo WA, Streit J, Joseph-Ridge N. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–61. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 16.Schumacher HR, Jr, Becker MA, Wortmann RL, Macdonald PA, Hunt B, Streit J, Lademacher C, Joseph-Ridge N. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540–8. doi: 10.1002/art.24209. [DOI] [PubMed] [Google Scholar]
- 17.Becker MA, Schumacher HR, Espinoza LR, Wells AF, Macdonald P, Lloyd E, Lademacher C. The urate-lowering efficacy and safety of febuxostat in the treatment of the hyperuricemia of gout: the CONFIRMS trial. Arthritis Res Ther. 2010;12:R63. doi: 10.1186/ar2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khosravan R, Grabowski BA, Wu JT, Joseph-Ridge N, Vernillet L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin Pharmacokinet. 2006;45:821–41. doi: 10.2165/00003088-200645080-00005. [DOI] [PubMed] [Google Scholar]
- 19.US Food and Drug Admisitration. 2006. Guidance for industry: drug interaction studies study design, data analysis, and implications for dosing and labeling. US Dept of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Publication No. 6695. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072101.pdf (last accessed 16 June 2011) Rockville, MD,
- 20.Annemans L, Spaepen E, Gaskin M, Bonnemaire M, Malier V, Gilbert T, Nuki G. Gout in the UK and Germany: prevalence, comorbidities and management in general practice 2000–2005. Ann Rheum Dis. 2008;67:960–6. doi: 10.1136/ard.2007.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keenan RT, O'Brien WR, Lee KH, Crittenden DB, Fisher MC, Goldfarb DS, Krasnokutsky S, Oh C, Pillinger MH. Prevalence of contraindications and prescription of pharmacologic therapies for gout. Am J Med. 2011;124:155–63. doi: 10.1016/j.amjmed.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 22.GlaxoSmithKline. 2011.
- 23.Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, Cowley H. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–80. [PubMed] [Google Scholar]
- 24.Baldwin SJ, Clarke SE, Chenery RJ. Characterization of the cytochrome P450 enzymes involved in the in vitro metabolism of rosiglitazone. Br J Clin Pharmacol. 1999;48:424–32. doi: 10.1046/j.1365-2125.1999.00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller AK, DiCicco RA, Freed MI. The effect of ranitidine on the pharmacokinetics of rosiglitazone in healthy adult male volunteers. Clin Ther. 2002;24:1062–71. doi: 10.1016/s0149-2918(02)80019-4. [DOI] [PubMed] [Google Scholar]
- 26.Miller AK, Inglis AM, Culkin KT, Jorkasky DK, Freed MI. The effect of acarbose on the pharmacokinetics of rosiglitazone. Eur J Clin Pharmacol. 2001;57:105–9. doi: 10.1007/s002280100275. [DOI] [PubMed] [Google Scholar]
- 27.Kim KA, Park PW, Kim HK, Ha JM, Park JY. Effect of quercetin on the pharmacokinetics of rosiglitazone, a CYP2C8 substrate, in healthy subjects. J Clin Pharmacol. 2005;45:941–6. doi: 10.1177/0091270005278407. [DOI] [PubMed] [Google Scholar]
- 28.Kim KA, Park PW, Kim KR, Park JY. Effect of multiple doses of montelukast on the pharmacokinetics of rosiglitazone, a CYP2C8 substrate, in humans. Br J Clin Pharmacol. 2007;63:339–45. doi: 10.1111/j.1365-2125.2006.02764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niemi M, Backman JT, Neuvonen PJ. Effects of trimethoprim and rifampin on the pharmacokinetics of the cytochrome P450 2C8 substrate rosiglitazone. Clin Pharmacol Ther. 2004;76:239–49. doi: 10.1016/j.clpt.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Totah RA, Rettie AE. Cytochrome P450 2C8: substrates, inhibitors, pharmacogenetics, and clinical relevance. Clin Pharmacol Ther. 2005;77:341–52. doi: 10.1016/j.clpt.2004.12.267. [DOI] [PubMed] [Google Scholar]
- 31.Chu KM, Hu OY, Pao LH, Hsiong CH. Pharmacokinetics of oral rosiglitazone in Taiwanese and post hoc comparisons with Caucasian, Japanese, Korean, and mainland Chinese subjects. J Pharm Pharm Sci. 2007;10:411–9. doi: 10.18433/j3159d. [DOI] [PubMed] [Google Scholar]