

Abstract
AIMS
To investigate the effects of two single nucleotide polymorphisms (SNPs) in the human P2X7 receptor gene (P2RX7) – 1068G>A (A348T) and 1513A>C (E496A) – on P2X7 receptor function, using a specific receptor antagonist (GSK1370319A) and prospective genetic stratification.
METHODS
Lipopolysaccharide- and ATP-stimulated interleukin-1β production was determined in the presence or absence of GSK1370319A in blood culture from 32 prospectively genotyped subjects.
RESULTS
There was approximately 6.7-fold difference (P < 0.0001) in IC50 for inhibition of ATP-stimulated interleukin-1β release by GSK1370319A between individuals with the homozygous gain- (1068A) and loss-of-function (1513C) genotypes (expressing the 348T, 496E and 348A, 496A alleles, respectively).
CONCLUSIONS
Leukocyte P2X7 receptors had significantly altered pharmacodynamic responses to a specific antagonist (GSK1370319A), directly related to SNP genotype.
Keywords: bioresource, GSK1370319A, P2RX7 gene, P2X7 antagonist, single nucleotide polymorphism
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Analysis of allelic variation in large populations has identified numerous single nucleotide polymorphisms (SNPs) in the P2RX7 gene. In vitro transfection has demonstrated SNP-dependent alterations (gain or loss) of P2X7 receptor function, as measured by ATP-induced ethidium uptake and interleukin-1β production.
WHAT THIS STUDY ADDS
We provide definitive evidence of SNP-dependent alteration in P2X7 receptor pharmacodynamics to a specific antagonist (GSK1370319A) in a small sample of prospectively genotyped subjects. These effects on drug response underline the importance of genetic stratification in drug development of P2X7 receptor antagonists.
Introduction
There is considerable interest in use of human bioresource databases, based on ethically approved collections of DNA, for prospective recruit-by-genotype approaches to experimental medicine and clinical trials designed to investigate the functional consequences of allelic variation in a gene of interest [1–4]. We have employed this approach to investigate the genetically based functional variation in the P2X7 receptor.
The P2X7 receptor is a purinergic nonselective cation channel receptor gated by extracellular ATP [5, 6]. The receptor-associated ion channel appears to change permeability to cations during prolonged agonist exposure either by channel dilatation or by coupling to the pannexin hemichannel [7]. P2X7 receptor signalling is important in inflammasome formation, leading to cleavage-induced maturation of caspase-1 and subsequent release of interleukin-1β (IL-1β) and interleukin-18 following stimulation with endogenous receptor agonist, ATP [8, 9]. The P2X7 receptor is expressed on many cell types, including monocytes/macrophages, erythrocytes, osteoblasts, lung mast cells, fibroblasts and certain neuronal tissues. Involvement of the P2X7 receptor in IL-1β processing and release has led to considerable interest in development of selective P2X7 receptor antagonists for the treatment of inflammatory disorders and pain [10] and has motivated therapeutic development of selective receptor antagonists, including GSK1370319A (N-[(2,4-dichlorophenyl)methyl]-1-methyl-5-oxo-l-prolinamide).
The P2RX7 gene located on chromosome 12q24 [symbol approved by the HUGO Gene Nomenclature Committee (HGNC)][5] encodes the P2X receptor subtype 7 (P2X7) and is highly polymorphic, with 29 nonsynonymous amino-acid-altering single nucleotide polymorphisms (SNPs) reported [11]. Some P2RX7 SNPs have been associated with mood disorders, susceptibility to infections and bone diseases in humans [12]. Previous studies have reported effects of multiple P2RX7 SNPs on receptor expression and functional response to ATP [13], but SNP-dependent pharmacogenetic effects remain relatively poorly characterized.
The aim of this study was to investigate the effects on P2X7 receptor function of two SNPs in the human P2X7 receptor gene (P2RX7); 1068G>A (A348T) and 1513A>C (E496A). These two SNPs have been described as having the greatest opposite functional effects in terms of induced IL-1β production [14, 15] and downstream inflammatory responses. The study was conducted using a specific receptor antagonist (GSK1370319A) and prospective genetic stratification from a human bioresource.
Methods
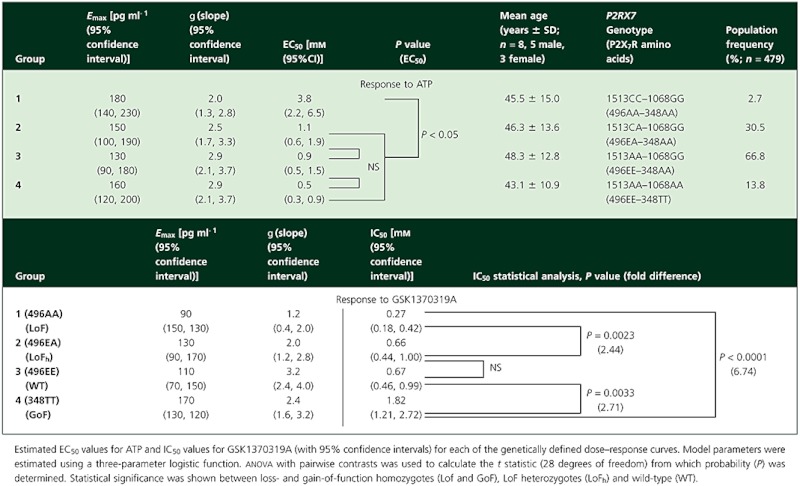
To investigate the effects of genetic variation in the P2RX7 gene on functional response to GSK1370319A, we focused on two SNPs resulting in the amino-acid changes Glu496Ala (E496A; 1513A>C, rs3751143) and Ala348Thr (A348T; 1068G>A, rs1718119). The SNP 1513A>C is believed to confer loss of function and 1068G>A gain of function of the P2X7 receptor, and they were selected for study on this basis. For 1513A>C, a full three-way cross was performed, comparing the homozygous loss of function (CC) with both heterozygous (AC) and dominant homozygous wild-type (AA), against a constant background of the homozygous wild-type (GG) of the 1068 variant. For 1068G>A, a two-way cross was performed, comparing homozygous gain of function (AA) with homozygous wild-type (GG), against a constant background of the homozygous (AA) 1513 variant (Table 1). Four other SNPs with putative functional effects at the P2X7 receptor (rs1653624, rs17525809, rs2230911 and rs28360447) were all wild-type matched.
Table 1.
Significance of functional single nucleotide polymorphisms (SNPs) in the P2RX7 gene on P2X7 receptor sensitivity to agonist (ATP) and to antagonism by GSK1370319A
Four groups of eight genetically stratified, age- and sex-matched healthy European Caucasian volunteers were recruited from a bioresource maintained at GSK's Clinical Unit in Cambridge, UK. The bioresource has DNA extracted from peripheral venous blood samples from volunteers who had given consent to be genotyped at any gene of interest, and recontacted if their genetic status makes them potentially eligible to participate in further studies. Four hundred and seventy-nine volunteers were genotyped for the listed SNPs of interest in the P2RX7 gene by a competitive allele specific PCR SNP genotyping system using Fluorescence Resonance Energy Transfer quencher cassette oligonucleotides (KBioscience, Hoddesdon, Hertfordshire, UK).
Ethical approval (LREC ref: 08/H0302/100, UK) and written informed consent were obtained prior to conducting this study. The P2X7 receptor function was assessed in the presence or absence of GSK1370319A in a lipopolysaccharide (LPS)–ATP stimulation, IL-1β release whole-blood assay. Blood was collected from P2X7-genotyped volunteers into citrate buffer (15% v/v) and incubated for 80 min with or without 1 µg ml−1 LPS (Escherichia coli serotype 7136; Sigma, St Louis, MO, USA) at 37°C. Thereafter, 50 µl aliquots of blood were added to 96-well plates together with 30 µl PBS or antagonist and the plates incubated for 40 min at 37°C, before adding 20 µl of ATP solution (final concentration 0.125–8.0 mm). The plates were mixed by shaking and incubated at 37°C for 30 min. Reactions were terminated by the addition of ice-cold HEPES-buffered RPMI-1640 (Invitrogen, Life Technologies Ltd., Paisley, UK). The 96-well plates were centrifuged at 300g for 10 min at 4°C, and the resulting supernatants were harvested, diluted appropriately, and their IL-1β content was determined using a human IL-1β ELISA (DLB50; R&D systems, Europe Ltd., Abingdon, UK) according to the manufacturer's instructions. In titration experiments, 4.0 mm ATP was found to provide optimal stimulation and was used in the reported experiments.
Results
Dose–response relationships were constructed for ATP-induced IL-1β release (absolute amount in picograms per millilitre) and percentage inhibition of IL-1β release as a function of the concentration of GSK1370319A for each of the genetically stratified groups (Figure 1), and the drug concentration causing 50% inhibition of IL-1β production at 4 mm ATP was estimated (IC50; Table 1). The maximal levels of IL-1β produced did not vary significantly between the groups, but the ATP concentration required was significantly higher for the loss-of-function homozygotes (1513CC and 1068GG). Conversely, for ATP, the amount of IL-1β produced was significantly higher for the gain-of-function homozygotes (1513AA and 1068AA). Loss-of-function genotype was associated with a leftward shift of the dose–response curve, whereas gain-of-function was associated with a rightward shift.
Figure 1.
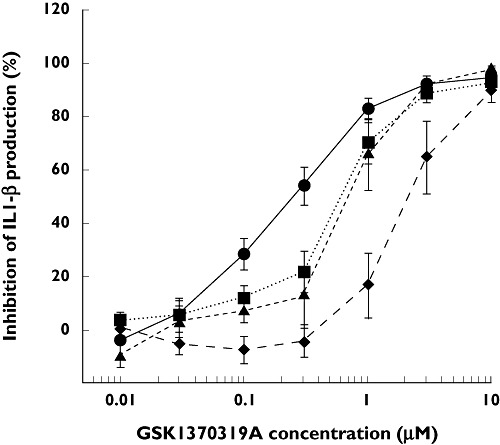
Influence of polymorphisms on pharmacodynamic effects of an antagonist at the P2X7 receptor. Dose–response curves for interleukin-1β (IL-1β) production following lipopolysaccharide/ATP stimulation of whole blood, drawn from four genetically stratified groups of healthy volunteers (n= 8 per group), incubated in the presence of variable concentrations of the P2X7 antagonist, GSK1370319A. Each data point represents the mean ± SEM of percentage IL-1β inhibition (derived from biological assay triplicates). 496AA-348AA (Loss of function hom) ( ); 496EA-348AA (Loss of function het) (
); 496EA-348AA (Loss of function het) ( ); 496EE-348AA (Wild type) (
); 496EE-348AA (Wild type) ( ); 496EE-348TT (Gain of function hom) (
); 496EE-348TT (Gain of function hom) ( )
)
Discussion
The P2X7 receptor is implicated in a number of disorders (reviewed in [16]), including inflammatory components of atherosclerosis, temporal lobe epilepsy, Parkinson's disease and Alzheimer's disease, diabetes and diabetic retinopathy, polycystic kidney disease, neurodegeneration and immunomodulation, and in neuropathic pain (where disruption of signalling via P2X7 abolishes pain). Formation of the innate inflammasome via P2X7 receptor engagement is a central process in initiating an inflammatory cascade, resulting in caspase-1-dependent release of IL-1β and interleukin-18 [9]. This is not only important in antibacterial immune function, but it also provides the basis of a physiologically and mechanistically relevant pharmacodynamic assay. We have demonstrated that the extent to which activation of such an immune response occurs is highly dependent on certain SNPs within the P2RX7 gene. Leukocyte P2X7 receptors had significantly altered functional capacity directly related to genotype. Thus, individuals homozygous for 1513C (expressing Ala496) had loss of function in terms of IL-1β production (i.e. increased sensitivity to GSK1370319A), whilst individuals homozygous for 1068A (expressing Thr348) showed gain of function, when tested against a controlled genetic background. In an HEK-293 transfection model, gain of function has been demonstrated using Thr348 [13], but to a lesser degree than we observe in ex vivo whole-blood assays.
The genetically determined molecular basis of the altered function of the P2X7 receptor is currently not well understood, but recent structure–function studies suggest that alterations in ATP and/or antagonist binding sites and allosteric modulation of ATP binding are all important determinants of receptor function [16]. Our results suggest that the SNP in P2RX7 resulting in E496A substitution caused significant alteration in sensitivity to ATP, whereas both E496A and A348T together resulted in an almost sevenfold difference in response to the specific antagonist GSK1370319A between the extremes of gain or loss of receptor function.
P2X7 receptor antagonists have been used in a number of clinical settings [16], and understanding their relative potency and pharmacogenetics is critical in establishing safe and effective criteria for dose setting. We provide evidence that the problem of lack of statistical power in addressing equivocal interpretation of genotype–phenotype association due to insufficiently stringent criteria [17], particularly in ex vivo and clinical pharmacogenetic studies, can be addressed by prospective genotyping. Use of DNA biobanks is increasing, e.g. http://www.cambridgebioresource.org.uk, and more widespread adoption of such platforms in clinical development may lead to greater personalization of medicines or genetic stratification of therapeutics in the longer term. As this study also demonstrates, if the drug target is accessible from a peripheral blood sample, then prospective stratification supports estimation of human pharmacogenetic effects on dose–response parameters of new compounds, before administration to man.
Acknowledgments
This work was funded by GlaxoSmithKline plc. The Juvenile Diabetes Research Foundation, the Wellcome Trust, and the European Union's 7th Framework Programme (FP7/2007–2013) under grant agreement n°241447 (NAIMIT) are acknowledged for support to J.A.T.
Competing Interests
S.M.McH., S.R., B.D., A.K., A.M.P., J.C.R., S.R.M., S.W., C.J.C. and E.T.B. are, or have been, employees of GSK and may hold shares in the company. The other authors have no competing interests to declare.
REFERENCES
- 1.Craigen JL, Mackus JM, Engleberts P, Miller SR, Speller S, Chamberlain L, Davis BG, McHugh SM, Bullmore ET, Cox CJ, Wetten S, Perdock G, Bakker JM, van de Winkel JGJ, Parren PWHI. Ofatumumab, a Human Mab Targeting a Membrane-Proximal Small-Loop Epitope On CD20, Induces Potent NK Cell-Mediated ADCC. Blood. 2009;114:1725. [Google Scholar]
- 2.Dendrou CA, Plagnol V, Fung E, Yang JHM, Downes K, Cooper JD, Nutland S, Coleman G, Himsworth M, Hardy M, Burren O, Healy B, Walker NM, Koch K, Ouwehand WH, Bradley JR, Wareham NJ, Todd JA, Wicker LS. Cell-specific protein phenotypes for the autoimmune locus IL2RA using a genotype-selectable human bioresource. Nat Genet. 2009;41:1011–5. doi: 10.1038/ng.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soranzo N, Spector TD, Mangino M, Kühnel B, Rendon A, Teumer A, Willenborg C, Wright B, Chen L, Li M, Salo P, Voight BF, Burns P, Laskowski RA, Xue Y, Menzel S, Altshuler D, Bradley JR, Bumpstead S, Burnett MS, Devaney J, Döring A, Elosua R, Epstein SE, Erber W, Falchi M, Garner SF, Ghori MJR, Goodall AH, Gwilliam R, Hakonarson HH, Hall AS, Hammond N, Hengstenberg C, Illig T, König IR, Knouff CW, McPherson R, Melander O, Mooser V, Nauck M, Nieminen MS, O'Donnell CJ, Peltonen L, Potter SC, Prokisch H, Rader DJ, Rice CM, Roberts R, Salomaa V, Sambrook J, Schreiber S, Schunkert H, Schwartz SM, Serbanovic-Canic J, Sinisalo J, Siscovick DS, Stark K, Surakka I, Stephens J, Thompson JR, Völker U, Völzke H, Watkins NA, Wells GA, Wichmann HE, Van Heel DA, Tyler-Smith C, Thein SL, Kathiresan S, Perola M, Reilly MP, Stewart AFR, Erdmann J, Samani NJ, Meisinger C, Greinacher A, Deloukas P, Ouwehand WH, Gieger C. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet. 2009;41:1182–90. doi: 10.1038/ng.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan GD, Neville MJ, Liverani E, Humphreys SM, Currie JM, Dennis L, Fielding BA, Karpe F. The in vivo effects of the Pro12Ala PPARγ2 polymorphism on adipose tissue NEFA metabolism: the first use of the Oxford Biobank. Diabetologia. 2006;49:158–68. doi: 10.1007/s00125-005-0044-z. [DOI] [PubMed] [Google Scholar]
- 5.Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–67. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 7.Pelegrin P, Surprenant A. The P2X7 receptor – Pannexin connection to dye uptake and IL-1b release. Purinergic Signal. 2009;5:129–37. doi: 10.1007/s11302-009-9141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: A key player in IL-1 processing and release. J Immunol. 2006;176:3877–83. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- 9.Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP-Y. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci. 2010;30:15811–20. doi: 10.1523/JNEUROSCI.4088-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll WA, Donnelly-Roberts D, Jarvis MF. Selective P2X7 receptor antagonists for chronic inflammation and pain. Purinergic Signal. 2009;5:63–73. doi: 10.1007/s11302-008-9110-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7 Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–6. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- 12.Fuller SJ, Stokes L, Skarratt KK, Gu BJ, Wiley JS. Genetics of the P2X7 receptor and human disease. Purinergic Signal. 2009;5:257–62. doi: 10.1007/s11302-009-9136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stokes L, Fuller SJ, Sluyter R, Skarratt KK, Gu BJ, Wiley JS. Two haplotypes of the P2X7 receptor containing the Ala-348 to Thr polymorphism exhibit a gain-of-function effect and enhanced interleukin-1β secretion. FASEB J. 2010;24:2916–27. doi: 10.1096/fj.09-150862. [DOI] [PubMed] [Google Scholar]
- 14.Gu BJ, Zhang W, Worthington RA, Gu BJ, Zhang W, Worthington RA, Sluyter R, Dao-Ung P, Petrou S, Barden JA, Wiley JS. A Glu-496 to Ala polymorphism leads to loss of function of the human P2X7 receptor. J Biol Chem. 2001;276:11135–42. doi: 10.1074/jbc.M010353200. [DOI] [PubMed] [Google Scholar]
- 15.Burnstock G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006;58:58–86. doi: 10.1124/pr.58.1.5. [DOI] [PubMed] [Google Scholar]
- 16.Evans RJ. Structural interpretation of P2X receptor mutagenesis studies on drug action. Br J Pharmacol. 2010;161:961–71. doi: 10.1111/j.1476-5381.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nebert DW, Zhang G, Vesell ES. From human genetics and genomics to pharmacogenetics and pharmacogenomics: past lessons, future directions. Drug Metab Rev. 2008;40:187–224. doi: 10.1080/03602530801952864. [DOI] [PMC free article] [PubMed] [Google Scholar]