

Abstract
Mice deficient for CELF4, a neuronal RNA-binding protein, have a complex seizure disorder that includes both convulsive and non-convulsive seizures, and is dependent upon Celf4 gene dosage and mouse strain background. It was previously shown that Celf4 is expressed predominantly in excitatory neurons, and that deficiency results in abnormal excitatory synaptic neurotransmission. To examine the physiological and molecular basis of this, we studied Celf4-deficient neurons in brain slices. Assessment of intrinsic properties of layer V cortical pyramidal neurons showed that neurons from mutant heterozygotes and homozygotes have a lower action potential (AP) initiation threshold and a larger AP gain when compared with wild-type neurons. Celf4 mutant neurons also demonstrate an increase in persistent sodium current (INaP) and a hyperpolarizing shift in the voltage dependence of activation. As part of a related study, we find that CELF4 directly binds Scn8a mRNA, encoding sodium channel Nav1.6, the primary instigator of AP at the axon initial segment (AIS) and the main carrier of INaP. In the present study we find that CELF4 deficiency results in a dramatic elevation in the expression of Nav1.6 protein at the AIS in both null and heterozygous neurons. Together these results suggest that activation of Nav1.6 plays a crucial role in seizure generation in this complex model of neurological disease.
Key points
Inappropriate electrochemical signalling between neurons is a central problem in epilepsy. Only a fraction of all heritable epilepsies are solved, and many are due to ion channel mutations.
Ion channels are cell surface molecules that initiate and coordinate electrochemical signalling, by allowing the right combination of ions to flow in and out of a neuron.
Ion channels may be regulated by other genes. We recently determined that the RNA-binding protein, ‘CELF4’, affects expression of many molecules, including ion channels, to coordinate electrochemical signalling.
Here we conclude that a sodium channel called Nav1.6 is the primary instigator of abnormal excitation when CELF4 is impaired.
Nav1.6 expression is increased in the axon initial segment – a part of the neuron that initiates electrical activity – altering intrinsic excitability. We surmise that in a brain that already has difficulty regulating other molecules due to CELF4 deficiency, this leads to uncontrolled network excitation and seizures.
Introduction
Idiopathic epilepsy (IE) is a common neurological disease characterized by recurrent spontaneous seizures without an obvious cause, other than a significant genetic component. Although approximately 20 IE genes have been identified to date (Nicita et al. 2012), these are mostly from Mendelian families and represent a small fraction of the heritability of IE – which is generally accepted to be genetically complex (Ottman, 2005). Recurrent seizures like those that occur in IE also feature significantly in other common idiopathic diseases of impaired neurological function, such as autism spectrum disorder (Spence & Schneider, 2009; Kohane et al. 2012), attention-deficit hyperactivity disorder (Hesdorffer et al. 2004), schizophrenia (Chang et al. 2011), mental retardation (Morgan et al. 2003), and in a variety of chromosomal structural abnormalities that are neurological in nature (Kumada et al. 2005). Although there has been much progress in the past few years towards identifying causal variants for these diseases, like epilepsy, the candidates that have been validated so far comprise only a fraction of total heritability (Congdon et al. 2010).
Mice deficient for CELF4 (CUGBP, ELAV-like family member 4), a brain-specific neuronal RNA-binding protein (RBP), exhibit both convulsive and non-convulsive (absence-like) seizures, and also have a low seizure threshold. The expression and penetrance of these phenotypes vary, depending both on Celf4 gene dosage and mouse strain background (Yang et al. 2007; Wagnon et al. 2011). Homozygous mutants are more severe than heterozygotes, with poor survival (especially on the C57BL/6J strain background), and the survivors are smaller and exhibit spontaneous convulsions at an earlier age (Wagnon et al. 2011). Thus, while the Celf4 mutant phenotypes are ultimately initiated by single gene mutation, because of the complexity of seizure phenotypes, their dependence on genetic context and the lack of obvious cellular pathology, Celf4 mutant mice provide an interesting and tractable model for common IEs. In addition to their seizure disorder, Celf4 mutants are hyperactive, gain weight and may have other behavioral abnormalities (Yang et al. 2007), making them potentially relevant to other seizure-associated diseases of synaptic dysfunction. Interestingly, CELF4 resides in the middle of the genomic interval involved in del(18q) syndrome, which has symptoms including seizures, mental retardation and other pathological features, and there is a very strong correlation between haploinsufficiency and seizures in these patients as we will discuss later. In support of this correlation a very recent case report described a male patient with similar clinical features and harboring a de novo translocation within the CELF4 gene itself (Halgren et al. 2012). However, it is not yet known whether more subtle genetic variants in CELF4 are associated with more common neurological disease.
RBPs selectively identify and bind to subsets of RNAs via motif-specific RNA-binding domains, and perform essential roles in each step of RNA metabolism, from transcription, pre-mRNA splicing and polyadenylation to RNA modification, transport, localization, translation and turnover (Glisovic et al. 2008). Dysregulation of RBPs can therefore affect the expression of many genes, often leading to disease (Lukong et al. 2008). Notably, dysregulation of RBPs is implicated in numerous complex human neurological diseases, including epilepsy, Alzheimer's disease, spinal muscular atrophy, amyotrophic lateral sclerosis and frontotemporal dementia (Wisniewski et al. 1985; Moore et al. 2001; Ule, 2008; Liu-Yesucevitz et al. 2011; King et al. 2012). CELF4 itself is one of six mammalian members of the CELF/BRUNO/CUGBP gene family. While CELF1 and CELF2 are broadly expressed, expression of CELF3–CELF6 is highly restricted, with CELF4 confined to the CNS in adults. CELF proteins are involved in multiple aspects of mRNA metabolism, from pre-mRNA splicing and RNA editing to deadenylation and regulation of translation (Dasgupta & Ladd, 2012). Mutations in UNC-75, the C. elegans CELF3–CELF6 orthologue, cause defects in neurotransmission and these are rescued by expression of human CELF4, suggesting that UNC-75 and CELF4 modulate synaptic transmission through regulation of RNA in the brain (Loria et al. 2003). This observation also suggests that the function of CELF4 is highly conserved.
We previously determined that murine Celf4 is expressed mainly in excitatory neurons in the cerebral cortex and hippocampus, and that conditional deletion of Celf4 from excitatory but not inhibitory neurons is associated with low seizure threshold and handling-associated seizures. Conditional deletion of Celf4 from the adult brain was sufficient to induce these convulsive seizure phenotypes, but interestingly did not cause absence seizures, which required deletion in the first postnatal week. We also noted that Celf4 null mutants have enhanced excitatory neurotransmission, but that inhibition is unaffected, suggesting that excessive excitation underlies their seizure disorder (Wagnon et al. 2011).
Here we pursue the mechanism by which Celf4 loss leads to excessive neuronal excitation and seizures, via a detailed examination of neuronal physiology in acute brain slices from Celf4 mutants. Patch-clamp recordings of layer V cortical pyramidal neurons reveal abnormal intrinsic properties that are consistent with hyperexcitability. Celf4 mutant neurons also have an increase in persistent sodium current (INaP) and a hyperpolarizing shift in the voltage dependence of steady-state activation. We pursued the voltage-gated sodium channel Nav1.6 as the instigator of these phenomena, as it is the primary determinant of action potential (AP) initiation at the axon initial segment (AIS) and the main carrier of INaP in cortical pyramidal neurons. We note that mRNA encoding Nav1.6, Scn8a is a direct target of CELF4 and that expression of Nav1.6 is dramatically increased at the AIS. We also show that the reduced seizure threshold of Celf4 mutants is completely dependent on normal Scn8a gene dosage. Together these findings strongly suggest that Celf4-dependent expression of Nav1.6 plays a crucial role in this genetically complex seizure model. These results may also shed light on the understanding of seizure phenotypes in del(18q) syndrome, and suggest modulation of Nav1.6 function as a potential therapeutic target.
Methods
Ethical approval
All animal procedures followed the Association for Assessment and Accreditation of Laboratory Animal Care guidelines, and were approved by the Institutional Animal Care and Use Committee.
Animals
All animals were fed standard National Institutes of Health diet containing 6% fat and acidified water ad libitum. Celf4 germline and conditional mutants were obtained and genotyped as described previously (Wagnon et al. 2011). All mice were analysed on an isogenic 129S1/SvImJ background, except for mice tested for electroconvulsive threshold (ECT), which were on a congenic C57BL/6J strain background. Temporal conditional deletion of Celf4 was done by crossing B6J mice with the Celf4 null conditional allele with Ubc-Cre mice obtained from JAX (B6.Cg-Tg(UBCcre/ERT2)1Ejb/J); Lacz staining following tamoxifen treatment deletion of a reporter shows generally broad and widespread staining in mouse brain (http://cre.jax.org/UBC-creERT/UBC-creERT.html); we have confirmed this web publication ourselves, observing very strong staining throughout the brain using a Lacz reporter strain (data not shown). Unless otherwise stated, following use in experiments mice were killed as adults by the CO2 asphyxiation method (AAALAC/IACUC approved).
Administration of tamoxifen
Tamoxifen (Sigma-Aldrich T5648, St Louis, MO, USA) was dissolved at 20 mg ml−1 in corn oil by mixing overnight at room temperature and stored at 4°C for up to 1 week. The tamoxifen was administered to adult mice by oral gavage using a curved 22G animal feeding needle once a day for five consecutive days at the following doses, 6 mg day−1 (0.30 ml) for 20–24 g body weight, and 7 mg day−1 (0.35 ml) for 25–30 g body weight. Mice were tested 12 days after the last treatment to ensure total loss of the gene product and avoid acute effects of tamoxifen treatment. Corn oil alone was administered as above as a sham treatment control. In parallel to treatment of Celf4 conditional mice, using the oral gavage protocol and a ROSA26-Lacz reporter we also examined adult brain expression and confirmed that it is widespread, almost ubiquitous (data not shown).
Whole-cell patch-clamp recording and data analysis
Acute brain slices were prepared from Celf4−/−, Celf4+/− and wild-type littermates between P14 and P21. Briefly, mice were anaesthetized with tribromoethanol (250 mg kg−1, i.p.) and decapitated. Brains were quickly removed and transferred into ice-cold solution containing (in mm): sucrose, 210; KCl, 3.0; CaCl2, 1.0; MgSO4, 3.0; NaH2PO4, 1.0; NaHCO3, 26; glucose, 10; saturated with 95% O2 and 5% CO2. Coronal slices were cut at 300 μm on a vibratome (VT 1200; Leica Microsystems, GmbH, Wetzlat, Germany) and kept in artificial cerebral spinal fluid (ACSF) containing (in mm): NaCl, 124; KCl, 3.0; CaCl2, 1.5; MgSO4, 1.3; NaH2PO4, 1.0; NaHCO3, 26; glucose, 20; saturated with 95% O2 and 5% CO2 at room temperature (21–23°C). Slices were allowed to recover for at least 1 h before any recording.
Whole-cell patch-clamp recordings were made at the soma of layer V pyramidal neurons of the visual cortex using a 40× water immersion objective (40×/0.80W; Carl Zeiss Microscopy, Thornwood, USA). All recordings were made at 31–33°C. Patch pipettes were pulled from thick wall borosilicate glass (1.5/0.86 mm; Sutter Instruments, Novato, USA) on a horizontal puller (P-97; Sutter Instruments). Resistance of electrodes was between 2 and 4 MΩ. The series resistance (Rs), usually between 10 and 20 MΩ, was monitored throughout the recording, and data were not included in further analysis when Rs varied by 20% or more during recording. For persistent sodium current recording, the pipette solution contains (in mm): CsCH3SO3, 110; ATP-Mg, 4; Hepes, 10; CsCl, 15; EGTA, 0.5; tetraethylammonium (TEA)-Cl, 20. The bath solution contains (in mm): NaCl, 139; KCl, 3.0; CaCl2, 1.5; MgSO4, 1.3; NaH2PO4, 1.0; Hepes, 10; CdCl2, 0.2; glucose, 20; saturated with 100% O2. Liquid junction potential (9.6 mV) and voltage drop across series resistance for each cell were corrected in the computation of sodium channel conductance. All other recordings used ACSF in the bath, and the pipette solution contains (in mm): potassium gluconate, 125; KCl, 5; Hepes, 10; ATP, 4; GTP, 0.4; EGTA, 0.2; MgSO4, 1. Liquid junction potential was not corrected. TTX (1 μm) was added to the bath solution. The numbers of mice used in patch-clamp experiments are as follows: Fig. 1, eight wild-type (22 cells), six heterozygous (27 cells), nine homozygous (22 cells); Fig. 2, six wild-type (12 cells), four heterozygous (10 cells), four homozygous (13 cells). All recordings were made with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, USA) and Axograph X (Axograph Scientific, Berkleley, USA). Data were filtered at 2 kHz, sampled at 10 kHz using an ITC-18 interface. All analysis was performed using custom routings in Matlab. Values are given as mean ± SEM.
Figure 1. Decreased AP initiation threshold and enhanced neuronal excitability.
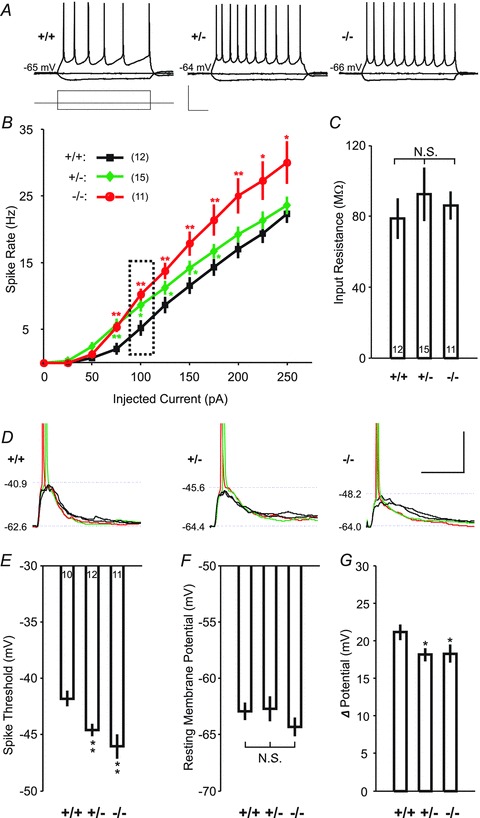
A, sample EPSP traces elicited by depolarizing current pulses (amplitude: −50 pA and 100 pA; duration: 800 ms) in a pyramidal cell for a wild-type (left), heterozygous (middle) or homozygous (right) mice. Scale bars: 40 mV or 200 pA, 200 ms. B, changes of the neuronal excitability, as measured by the number of APs triggered by injection of various depolarizing currents in wild-type (black), heterozygous (green) and homozygous (red) mice. Dashed box indicates the sample traces for spikes in A. C, input resistances of neurons for the same data sets as shown in A have no significant difference between wild-type, heterozygous and homozygous mice. D, sample traces of evoked EPSPs (with spike in red and green, without spike in black) by electrical stimulation with spiking probability less than 1 in wild-type, heterozygous or homozygous mice. Dashed lines indicate resting membrane potentials (lower) and threshold of APs (upper) for each cell. Overshoots of spikes are truncated. Scale bars: 20 mV, 20 ms. E–G, comparison of the membrane potential thresholds for AP initiation, resting membrane potentials and their difference for the same data sets as shown in D. *P < 0.05, **P < 0.01, t test.
Figure 2. Increase in INaP elicited by a slow ramp protocol in cortical layer V pyramidal neurons.
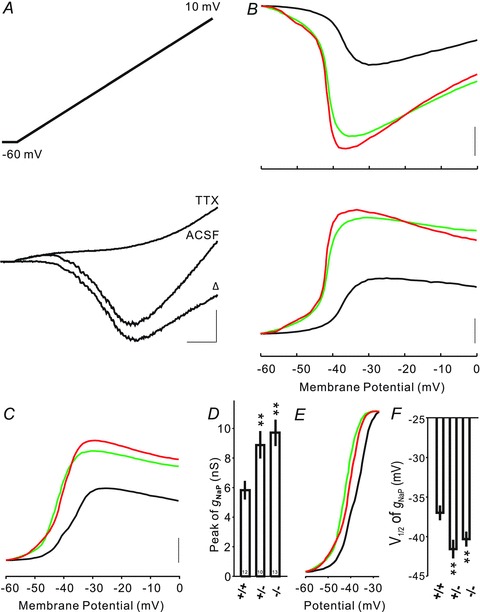
A, slow voltage ramps (35 mV s−1) were used to elicit INaP (top panel). Current traces elicited in a neuron in a wild-type mouse in the absence (artificial cerebrospinal fluid (ACSF) only) and presence (TTX) of TTX (bottom panel). The TTX-sensitive INaP was isolated by subtraction (Δ). Scale bar: 200 pA/200 ms. B, example of INaP–voltage curves (UP) for CELF4 +/+ (in black), +/− (in green) and −/− (in red) neurons. INaP–voltage curves were converted to gNaP–voltage curves (DOWN) for the same three cells. Scale bar: 200 pA and 2 nS. C, averaged gNaP–voltage curves for CELF4 +/+ (black), +/− (green) and −/− (red) mice. Scale bar: 2 nS. D, bar plots comparing peak of voltage-dependent conductance elicited by voltage ramps in CELF4 +/+, +/− and −/− neurons. gNaP is significantly increased in CELF4 +/− and −/− neurons. E, normalized gNaP–voltage relationships for CELF4 +/+ (black), +/− (green) and −/− (red) neurons. F, bar plots comparing half-maximal activation (V1/2) of gNaP in CELF4 +/+, +/− and −/− neurons. Shifts to hyperpolarizing were significant in CELF4 +/− and −/− mice. *P < 0.05, **P < 0.01, Mann–Whitney–Wilcoxon test.
Seizure threshold test
Susceptibility to generalized seizures was measured by using the ECT test essentially as previously described (Frankel et al. 2001). Briefly, mice were restrained, a drop of anaesthetic containing 0.5% tetracaine and 0.9% NaCl was placed onto each eye, and a preset current was applied via silver transcorneal electrodes using an electroconvulsive stimulator (Ugo Basile model 7801). As previously described, the stimulator was modified to produce rectangular wave pulses with fixed duration (0.2 s) and width (1.6 ms), and the range of current delivered was controlled by varying the pulse frequency (281–299 Hz) and amplitude (7.0–10.5 mA). The lowest stimulus that produced generalized seizure endpoint (minimal clonic forebrain seizure) was recorded for each subject and averaged across genotypes. The use of variable pulse frequency is a minor modification of methods used in prior studies (e.g. Wagnon et al. 2011), so that stimulus may be delivered in more refined increments. Whereas previously stimulus was expressed in mA (DC), we now express it as the integrated root mean square mA (AC), defined as the product of pulse duration (s), pulse width (ms), the square root of pulse frequency (Hz1/2) and current (mA). Twenty-seven male mice in total were tested beginning at 7 weeks old: 11 wild-type, three Celf4 single heterozygotes, 10 Scn8a8J single heterozygotes and three double heterozygotes from five different matings.
Immunohistochemistry
Mice were anaesthetized with a lethal dose of tribromoethanol and perfused with PBS (pH 7.4). Brains were removed from the skull, cut coronally and frozen in O.C.T. (Tissue-Tek, Torrance, CA, USA). Then, 15 μm thick sections from the cortex were cut with a freezing microtome and mounted onto lysine-coated Colorfrost/Plus microscope slides (Fisher Scientific, Pittsburgh, PA, USA). Slides were dried for 30 min at room temperature. Tissue sections were re-hydrated in PBS, fixed 10 min in 1% paraformaldehyde in PBS, rinsed three times in PBS, blocked in 0.3% Triton X-100, 1% bovine serum albumin and 10% normal goat serum in PBS for 2 h at room temperature, and incubated in primary antibodies diluted in 0.3% Triton-X-100, 1% bovine serum albumin and 3% normal goat serum in PBS for 2 days at 4°C. Sections were washed three times for 5 min each in PBST (PBS, 0.05% Tween-20) and incubated for 2 h at room temperature in secondary antibody. The sections were washed as before, followed by a wash in PBS and incubated in DAPI diluted in PBS for 5 min. The sections were mounted in Prolong Gold antifade reagent (P36930; Invitrogen, Carlsbad, CA, USA). The following primary antibodies were used: rabbit anti-CELF4 (G-19; 1:400; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); mouse anti-Nav1.6 (K87A/10; 1:250; UC Davis/NIH NeuroMab Facility, Davis, CA, USA); rabbit anti-AnkyrinG (AnkG; H-215; 1:150; Santa Cruz Biotechnology). The following secondary antibodies were used to visualize the immunoreactions: Alexa488-conjugated goat anti-mouse (A11017; 1:1000; Invitrogen) and Alexa555-conjugated goat anti-rabbit (A21430; 1:1000; Invitrogen). Images were collected using a Leica SP5 confocal microscope equipped with a 63× PlanApo objective (N.A. 1.4). The same laser and detection parameters were used for wild-type and mutant sections.
Nine mice were used in three replicates of staining. For each replicate, slices from wild-type, heterozygous and homozygous mice were mounted on the same slide and stained at the same time. To measure the immunofluorescence intensity of Nav1.6 and AnkG quantitatively, each well-defined AIS was traced, using AnkG as a mask, starting from the axon hillock and extending to the tapered AIS end (note that any AIS that was clearly truncated with a blunt end was excluded from further analysis), and the fluorescence intensity of each traced pixel was determined. Two image channels were simultaneously measured – green for Nav1.6 and red for AnkG. To examine the expression level of the proteins, the average fluorescence intensity of each of the three replicates was normalized to wild-type. To examine the distribution along the AIS, fluorescence intensity as a percentage of the total for each genotype was plotted along the length of the AIS.
iCLIP
Two each of Celf4 null and wild-type mouse whole brain tissue was split into hemispheres, dissociated in PBS, UV-crosslinked and collected by centrifugation. The iCLIP method was done as previously described (Konig et al. 2011). Briefly, crosslinked brain tissue dissociated in lysis buffer was sonicated and subjected to partial RNase I digestion (final dilution 1:100,000). CELF4 complexes were immunopurified with 0.9 μg anti-CELF4 polyclonal antibody (HPA037986, Sigma) conjugated to 100 μl protein A Dynabeads (Invitrogen). Immobilized on the beads, RNAs bound to CELF4 were dephosphorylated at their 3′-end for ligation of the DNA linker 5′-rAppAGATCGGAAGAGCGGTTCAG/ddC/-3′. Following 5′-end radiolabelling, CELF4-RNA complexes were size-separated by SDS–PAGE and transferred onto nitrocellulose membrane. The regions corresponding to 60–200 kDa on the autoradiogram were excised from the nitrocellulose, and bound RNAs were released by proteinase K treatment. RNAs were reverse-transcribed and cDNAs were size-selected from a 6% TBE-urea gel (Invitrogen). Purified cDNAs were circularized, linearized by restriction digestion and polymerase chain reaction amplified for high-throughput sequencing. To determine mRNA abundance in the brain of the same mouse strain, we used TopHat and Cufflinks to analyse public RNAseq data (ENA:ERP000614) and calculate FPKM value for each gene. We then normalized the iCLIP data relative to the total number of sequenced iCLIP cDNAs, and the mRNA abundance, by normalizing cDNA counts at each position in the following manner: Normalized cDNA count = (10,000,000 × cDNA count)/(total iCLIP cDNA count × FPKM). We generated wiggle files (window = 100) by calculating the total normalized cDNA count in each window.
Results
Elevated excitability in layer V pyramidal neurons in Celf4 mutant mice
CELF4 is expressed predominantly in excitatory neurons in the cerebral cortex and hippocampus, and miniature excitatory, but not inhibitory, postsynaptic currents are affected (Wagnon et al. 2011). In addition to synaptic function, CELF4 deficiency may alter the intrinsic excitability of pyramidal neurons. We examined this in layer V pyramidal neurons in the visual cortex by first testing whether APs are affected by Celf4 genotype, as elevated excitability may result in an increase in AP number. A series of 800 ms depolarizing current steps were applied to the neurons, and the number of APs elicited by each current step was examined in Celf4 null (−/−), heterozygous (+/−) and wild-type (+/+) neurons, respectively (Fig. 1A). The relationship between current amplitude and AP frequency was indeed significantly steeper in both null and heterozygous neurons compared with wild-type, which may indicate strong facilitation of evoked spiking in the mutants (Fig. 1B). We also examined the input resistance in each cell, and no significant difference was observed between genotypes under resting membrane potential (P > 0.2, t test; Fig. 1C).
We further examined mutant cortical pyramidal neurons for their AP threshold membrane potential. This was done by using whole-cell recording to record EPSPs in response to a range of presynaptic stimulation intensities (with spiking probability between 0 and 1) making use of twisted twin wires placed in cortical layer II/III (Fig. 1D). The AP thresholds were determined by measuring the voltage at which the first derivative (dV/dt) of the voltage trace is 50 mV s−1. We found that AP thresholds for Celf4 heterozygous and null neurons are −44.59 ± 0.75 mV and −46.06 ± 1.54 mV, respectively, significantly lower than −41.81 ± 1.09 mV in wild-type (P < 0.01 for each, t test; Fig. 1E). We also compared the resting membrane potentials, but there was no significant difference (−62.92 ± 1.00 mV for +/+, −62.72 ± 1.52 mV for +/− and −64.34 ± 1.13 mV for −/−, P > 0.1 for each, t test; Fig. 1F). This experiment indicated that the reduced AP initiation voltage is not a result of change in resting membrane potential, but that a smaller membrane potential rise (18.13 ± 1.25 mV for +/− and 18.28 ± 1.66 mV for −/−, respectively) is needed to trigger an AP in the mutants than in wild-type (21.11 ± 1.37 mV, P < 0.05 for each, t test; Fig. 1G). These AP results together showed that excitation of the cortical layer V pyramidal neurons by either presynaptic transmitter release or postsynaptic injection of depolarizing currents was promoted by complete loss (null) or reduction (heterozygosity) of Celf4.
Increased persistent Na+ current (INaP) in Celf4 mutant neurons
Voltage-gated sodium channels are well known to be associated with seizure disorders in human and in animal models (Meisler et al. 2010; Wimmer et al. 2010). We thought that the differences in AP threshold and spiking could be due to an increase in the persistent Na+ channel current (INaP). Therefore we measured INaP by using whole-cell recordings in layer V cortical pyramidal neurons from Celf4 null and heterozygous mice, comparing them with wild-type. For this, we used slow voltage ramps (from −60 mV to 10 mV at 35 mV s−1; Fig. 2A, top panel) to elicit slow membrane currents with voltage-sensitive Ca2+ currents blocked by 200 μm Cd2+ in the bath and K+ currents blocked by cesium and TEA in the intracellular solution. To obtain the INaP–voltage relationship, we plotted the TTX-sensitive currents by subtracting the current responses before and after quickly applying TTX into the bath for each measured neuron (Fig. 2A, bottom panel). We then converted the INaP–voltage curves (Fig. 2C, top panel) to conductance (gNaP) versus voltage relationships (Fig. 2B, bottom panel). Figure 2C shows the averaged gNaP–voltage curves for each genotype. The peak of gNaP elicited during voltage ramps was 5.80 ± 0.63 nS, 8.87 ± 0.93 nS and 9.69 ± 0.89 nS in wild-type, heterozygous and null neurons, respectively. INaP is significantly increased by 53% in Celf4 heterozygous and by 67% in null neurons compared with wild-type (Fig. 2D). Figure 2E depicts the normalized and averaged gNaP–voltage relationships for each genotype. By fitting gNaP–voltage curves with a Boltzmann function, the voltage of half-maximal activation (V1/2) was calculated for each cell. We observed a significant shift to hyperpolarizing voltage in V1/2 of gNaP in both the mutant genotypes (−37.0 ± 0.9 mV for wild-type compared with −41.6 ± 1.2 mV for heterozygous and −40.3 ± 0.9 mV for null; Fig. 2F). In summary, voltage-clamp recordings demonstrate that INaP is substantially increased by the absence or even the reduction of CELF4 in cortical pyramidal neurons. We also found a voltage shift to more negative V1/2 activation of INaP.
Given that: (a) Nav1.6 controls the AP initiation threshold and increased accumulation of Nav1.6 at the AIS lowers the AP initiation threshold (Hu et al. 2009); and (b) Nav1.6 is the main carrier of INaP in cortical pyramidal and other neurons (Raman et al. 1997; Maurice et al. 2001; Enomoto et al. 2007; Royeck et al. 2008; Osorio et al. 2010), these results – lowered AP threshold and increased INaP in mutant cortical pyramidal neurons – together suggest that Nav1.6 abundance is increased by loss or reduction of CELF4.
The gene encoding Nav1.6 is a CELF4 target and is overexpressed in Celf4 mutants
CELF4 is a brain-specific RBP whose mechanism of action has not yet been described in vivo. As part of a related study, we used crosslink immunoprecipitation and high-throughput sequencing of brain cDNA to identify mRNA targets of CELF4 (Wagnon et al. 2012). Briefly, by examining the CELF4 iCLIP cDNA density on transcripts in wild-type brain compared with null, we observed that CELF4 specifically binds to the 3′-untranslated region of many mRNAs to a U/UG consensus motif similar to that known for CELF family proteins, with a strong enrichment for targets involved in synaptic function. In that study, we used functional annotation clustering and the overall change in gene expression as independent measures to estimate a threshold for the number of significant targets compared with random expectation. Both approaches suggest that CELF4 nominally binds a vast number of mRNAs – up to 15–20% of the approximately 14,000 transcripts expressed. This includes Scn8a, which encodes Nav1.6 (Fig. 3A). Interestingly, the abundance of Scn8a RNA transcript was increased modestly in brain cDNA of Celf4 null compared with wild-type – by about 20% (data not shown). Given that Nav1.6 is the primary determinant of AP initiation and main contributor of INaP in excitatory neurons, we thought there might be a more significant accumulation of Nav1.6 at neuronal sites critical for AP initiation and sought to determine this by immunostaining.
Figure 3. CELF4 binding sites in 3′-UTR of the Scn8a and the Ank3 genes.
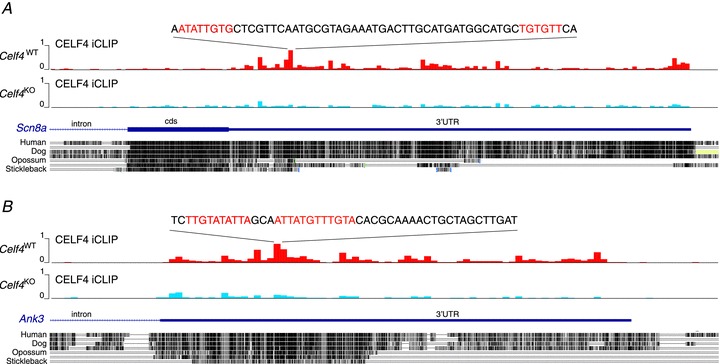
A, a graphic adapted from a custom track on the UCSC Genome Browser that displays the normalized iCLIP cDNA counts (y-axis, 0–1 scale where 0 is the minimum and 1 is the maximum) in a wiggle format (100 nt windows) on the 3′-end of the Scn8a gene (encoding Nav1.6) from wild-type (Celf4WT, red) and knockout (Celf4KO, blue) brain, respectively. The sequence at the strongest crosslink cluster is shown, with the CELF4-target motifs coloured in red, based on a pentamer [ACG]-[TA]-[Tc]-G-T, showing the highest significance as determined from a related study (Wagnon et al. in press). The high conservation of the CELF4 binding site is evident by the alignment of the sequence of vertebrate genomes. Intron (thin line), coding sequence (cds, thick line) and 3′-UTR (medium thick line) are shown. B, same as above, for Ank3 (encoding ANKG).
The AIS is the preferred site for the AP initiation in excitatory neurons and plays a critical role in excitability because this specialized axonal region contains a high density of Nav1.6 and other Na+ channel subtypes (Moore et al. 1983; Mainen et al. 1995; Rapp et al. 1996; Hu et al. 2009). We performed immunostaining of layer V pyramidal neurons of visual cortex to map the expression of Nav1.6 and Na+ channel-associated protein AnkG, which is also known to be enriched at the AIS (Fig. 4A). To measure the immunofluorescence intensity of Nav1.6 and AnkG quantitatively, each well-defined AIS was traced, using AnkG as a mask, starting from the axon hillock and extending to the tapered AIS end (note that any AIS that was clearly truncated with a blunt end was excluded from further analysis), and the fluorescence intensity of each traced pixel was determined. Two image channels were simultaneously measured – green for Nav1.6 and red for AnkG. We observed a very significant, Celf4 gene dosage-dependent increase in Nav1.6 expression at the AIS, using a relatively large sampling (n= 178 for +/+, 181 for +/− and 179 for −/−). Importantly, this increase was also observed in Celf4 heterozygotes (Fig. 4A and B). The total fluorescence intensity for Nav1.6 was 52.6% higher in heterozygotes and 73.8% higher in null (P < 0.0001 for each, t test). The relative distribution of Nav1.6 along the AIS was unaltered in mutant neurons (Fig. 4C).
Figure 4. Elevated expression of Nav1.6 at AIS in germline Celf4 mutant.
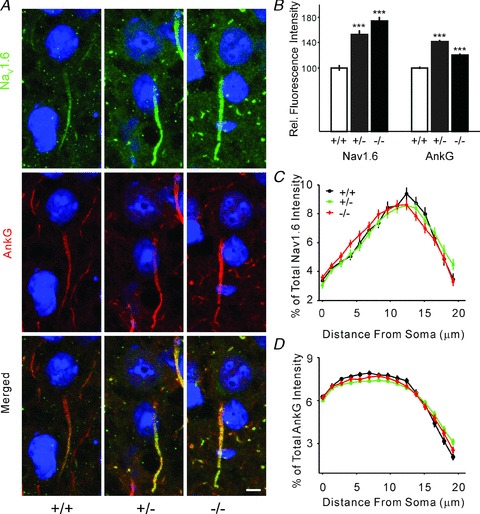
A, double immunostaining for Nav1.6 (green) and AnkG (red) in the mouse visual cortex. Images are summed projections of confocal z-stacks. Scale bar = 5 μm. B, plot of the averaged fluorescence intensity for Nav1.6 channels normalized to wild-type. Bars represent mean ± SEM. C, distribution of Nav1.6 along the AIS. Plot of the percentage of the averaged fluorescence intensity for Nav1.6 channels for each genotype as a function of distance from the soma at the AIS. D, distribution of AnkG along the AIS. Plot of the percentage of the averaged fluorescence intensity for AnkG for each genotype as a function of distance from the soma at the AIS.
Interestingly, AnkG protein was also more highly expressed in the AIS of Celf4 heterozygotes across the entire AIS, with a slightly larger increase in Celf4 heterozygotes (Fig. 4A, B and D). Ank3 (encoding AnkG), also a likely CELF4 target (Fig. 3B), is a scaffolding protein that recruits and concentrates sodium channels at the AIS. Thus, the apparently selective increase in AnkG at the AIS may contribute to or facilitate increased accumulation of Nav1.6 (Srinivasan et al. 1988; Zhou et al. 1998; Garrido et al. 2003; Lemaillet et al. 2003). AnkG also plays a role in suppressing INaP via its interactions with Nav1.6, which may explain why AnkG abundance is higher in heterozygotes compared with null, as it would be expected to respond to increased INaP (Shirahata et al. 2006). That is, as a CELF4 target, Ank3 may be further regulated in Celf4 heterozygotes that at least have some CELF4 protein.
To exclude the possibility that the increase in Nav1.6 or AnkG at the AIS was the result of cumulative compensatory effects during early or postnatal development, we examined somatic Celf4 mutants. Thus, mice homozygous for a Celf4 conditional allele and carrying the tamoxifen-inducible UbcERT-cre transgene (both used previously to establish the temporal etiology of seizures in Celf4 mutants; Wagnon et al. 2011), were allowed to proceed normally through development until 6 weeks old, treated with tamoxifen (Methods) and immunostained as above. A significant increase in Nav1.6 expression was also observed in the AIS of Celf4 somatic mutants (46.8%, P < 0.0001; Fig. 5A and B). This result suggests that the increase in Nav1.6 AIS expression is more likely the result of an acute effect of CELF4 loss than a long-term compensatory change.
Figure 5. Elevated expression of Nav1.6 at AIS in somatic Celf4 mutants.
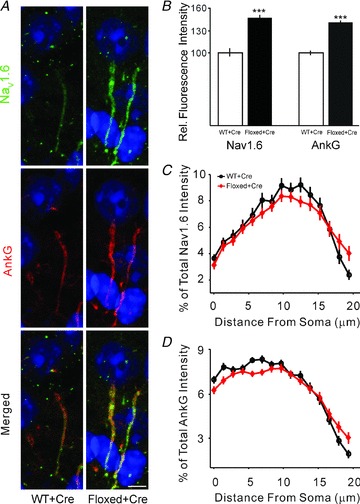
A, antibody double staining for Nav1.6 (green) and AnkG (red) in the mouse visual cortex. Experimental mice were UBC cre+ and Celf4flox/flox homozygotes. Control mice were Celf4+/+ with cre expression. Experimental mice and littermate controls were treated with tamoxifen (tam) by oral gavage at 6 weeks old for 5 sequential days, and brain tissue was harvested 14 days after completion of treatment. Images are projections of confocal z-stacks. Scale bar = 5 μm. B, plot of the averaged fluorescence intensity for Nav1.6 channels normalized to wild-type. Bars represent mean ± SEM. C, distribution of Nav1.6 along the AIS. Plot of the percentage of the averaged fluorescence intensity for Nav1.6 channels for each genotype as a function of distance from the soma at the AIS. D, distribution of AnkG along the AIS. Plot of the percentage of the averaged fluorescence intensity for AnkG for each genotype as a function of distance from the soma at the AIS.
Discussion
Celf4-deficient mice suffer a neurological disorder that is characterized primarily by complex seizure phenotypes – including grand-mal-like (convulsive) seizures, absence or petit-mal-like (non-convulsive) seizures and a low convulsive seizure threshold (Yang et al. 2007; Wagnon et al. 2011). Although more complex behaviors have not yet been assessed in detail, Celf4 mutants are also hyperactive, and males in particular gain weight excessively (Yang et al. 2007). Here we report that abnormal expression of sodium channel Nav1.6 in Celf4 mutants is associated with an increase of INaP and neuronal hyperexcitability in layer V cortical pyramidal neurons, accompanied by a lower AP threshold and a higher AP gain. Although Nav1.6 is only one of many molecules whose expression is altered as a result of Celf4 deficiency, our results suggest that it is an important determinant of the seizure disorder.
INaP and its role in epilepsy
In addition to the well-described fast-inactivating component of transient Na+ current (INaT), cortical neurons also exhibit a low-voltage-activated, slowly inactivating, TTX-sensitive INaP, which plays a role in determining neuronal excitability and synaptic integration (Chandler & Meves, 1966; Crill, 1996). INaP operates in the subthreshold voltage range, where other large, voltage-gated conductances are not active (Chandler & Meves, 1966; Crill, 1996). There is a wealth of studies in different types of neurons demonstrating consistently that INaP is markedly reduced from functional loss of Nav1.6 in mice, which supports that Nav1.6 channels carry most subthreshold INaP and are crucial for repetitive firing (Raman et al. 1997; Maurice et al. 2001; Enomoto et al. 2007; Royeck et al. 2008; Osorio et al. 2010).
Among the nine sodium channels, Nav1.1, Nav1.2 and Nav1.6 are the primary subtypes abundantly expressed in the adult brain (Beckh et al. 1989; Schaller et al. 1995; Trimmer & Rhodes, 2004; Wimmer et al. 2010). Although each has been directly implicated in human genetic epilepsy (e.g. see Meisler et al. 2010; Wimmer et al. 2010), Nav1.1 is not a likely candidate for direct involvement in Celf4-mediated convulsive seizures because its expression is primarily in GABAergic interneurons (Ogiwara et al. 2007; Lorincz & Nusser, 2008, 2010). Nav1.6 and Nav1.2 are better candidates not only because of their role in INaP generation but because of their cell type expression and enrichment at the AIS. Numerous studies revealed that both Nav1.6 and Nav1.2 are concentrated along the length of the AIS, but Nav1.6 accumulates nearer the distal AIS where APs initiate (Van Wart et al. 2007; Hu et al. 2009; Wimmer et al. 2010). Using an improved imaging technique, Nav1.6 channels were also localized in soma and proximal dendritic plasma membrane in hippocampal pyramidal neurons (Lorincz & Nusser, 2010), but somatodendritic Nav1.2 was not detected in the same study. Developmentally, Nav1.2 channels start to express in the embryonic brain and are the main sodium channel subunits at AIS of excitatory neurons during embryonic and early postnatal period. In contrast, Nav1.6 expression increases soon after birth. While Nav1.2 is thought to be important in back-propagation of AP, Nav1.6 is thought to be the critical sodium channel subtype for AP initiation and is of direct relevance in our mice (Hu et al. 2009). For all these reasons, on balance we think that increase of Nav1.6 is the most plausible explanation for increased intrinsic excitability in Celf4 cortical pyramidal neurons.
Increase in INaP is causally associated with seizure disorders in human and in animal models (Kearney et al. 2001; Liao et al. 2010; Veeramah et al. 2012). The transgenic Q54 mouse is a gain-of-function mutation in Scn2a that leads to an elevated INaP and a dominant seizure disorder (Meisler et al. 2001). Similarly in humans, a de novo mutation (p.Ala263Val) in SCN2A was found to cause a pronounced gain-of-function, in particular an increased INaP, and causes neonatal-onset dominant seizures (Liao et al. 2010). Both mutations alter the biophysical properties of Nav1.2 channels. Very recently, a gain-of-function SCN8A mutation was found in a 15-year-old patient, for the first time directly connecting mutant Nav1.6 with human epilepsy (Veeramah et al. 2012). The biophysical properties of the mutant channel reveal a dramatic increase in INaP and incomplete channel inactivation. Current-clamp analysis revealed increased spontaneous firing, paroxysmal-depolarizing-shift-like complexes and an increased firing frequency, consistent with a gain-of-function phenotype in the heterozygous patient. These effects are different from those of all Nav1.6 mutations previously reported, and are more like those observed in Celf4 mice.
Nav1.6 in genetic seizure disorders and a paradox
Several independent mutations in Scn8a have been studied extensively, with generally strong phenotypic concordance among them (Burgess et al. 1995; Meisler et al. 2004; Trudeau et al. 2006; Sharkey et al. 2009). All homozygous mutant Scn8a mice show severe motor impairment, whereas heterozygotes appear grossly normal (although at least one allele confers more intricate abnormal behavior; McKinney et al. 2008). In the first direct tie between Scn8a mutation and seizures, it was found that Scn8a gene dosage actually elevates seizure threshold (i.e. is protective) and also diminishes severe spontaneous seizures when crossed to an Scn1a heterozygous null model of Dravet syndrome – suggesting that SCN8A may be a modifier gene in some human epilepsies, or at least those caused by sodium channel mutations (Martin et al. 2007). Consistent with those studies, we have also found that a normal amount of Scn8a is necessary for seizure susceptibility of Celf4 mutants (Supplemental Fig. 1). Intriguingly, however, at least three murine Scn8a alleles, including the one studied here, while dramatically convulsive seizure resistant, confer absence seizures when heterozygous and these are genetic-background dependent (Papale et al. 2009). Although such opposite effects –Scn8a loss-of-function alleles that suppress convulsive seizures but enhance absence seizures – might appear to present a paradox, the cellular etiology of Nav1.6 mutation in these seizure types has not yet been explored. It may be informative to determine this, as generalized spike-wave activity was also observed in the most recently described human SCN8A gain-of-function mutation (Veeramah et al. 2012).
CELF4 and genetically complex neurological disease
Ion channels are obvious candidates for IE, and it is therefore not a surprise that most of the known human mutations reside in ion channel or transporter genes. However, these were ascertained mostly from Mendelian families, and represent only a fraction of the vast heritability of epilepsy – most of which is not yet defined (Ottman, 2005). Interestingly, although many potentially deleterious, non-synonymous variants were identified in a recent DNA sequencing study of 237 ion channel genes in 152 patients with IE and 139 controls, there was little evidence for an overall correlation with disease (Klassen et al. 2011). Mouse genetics offers a parallel means of peering into the genetic complexity of IEs. Of the ∼16,000 genes for which there has been the opportunity to observe mutant phenotypes in mice (and most of these are loss-of-function including knockout), at least 100 cause spontaneous seizures and a much smaller fraction encode ion channels (Frankel, 2009; also see http://www.informatics.jax.org/phenotypes.shtml). As appears to be the case with Celf4, however, they may themselves affect channel expression or function.
Human CELF4 sits in a region of Chr 18q that has strong evidence for linkage with adolescent-onset idiopathic generalized epilepsy (IGE; Durner et al. 2001). Although the gene encoding malic enzyme 2 was proposed as a candidate (Greenberg et al. 2005), this is only by association; no causal variants were identified. CELF4 may still be a candidate gene for this Chr 18q IGE locus.
However, Celf4-deficient mice have neurological abnormalities that extend beyond seizures; it is likely that CELF4 haploinsufficient patients would exhibit broader symptoms. In this respect, it is quite intriguing that patients with del(18q) syndrome – caused by deletion in the long arm of Chr 18 at an estimated incidence of 1 in 40,000 live births – often suffer from complex seizure disorder. These patients also have moderate to severe intellectual disability, developmental and speech delay, and various behavioral abnormalities, including autism spectrum disorder, hyperactivity and aggressiveness. After reviewing the most recent clinical case reports of proximal del(18q) syndrome with precisely refined breakpoints, we noted in nine cases where deletions did not include CELF4, only one patient suffered seizures (Cody et al. 2007; Buysse et al. 2008; Bouquillon et al. 2011; Filges et al. 2011; Marseglia et al. 2012). Accordingly, the remaining six patients all had seizures and all had deletions that span CELF4 (Gribble et al. 2005; Kotzot et al. 2005; Cody et al. 2007; Feenstra et al. 2007; Bouquillon et al. 2011). Very recently, a case report described a male patient with similar symptoms carrying a de novo translocation involving the CELF4 gene itself (Halgren et al. 2012), thus solidifying the causal relationship between CELF4 and major features of proximal del(18q) syndrome. Although it is not yet known whether more subtle CELF4 variants are associated with more common neurological disease, we expect that further intragenic CELF4 mutations will eventually arise, adding to our understanding of its function.
Despite its compelling role in hyperexcitability of Celf4-deficient mice, in a related study we find that Scn8a is just one of over 2000 mRNAs directly targeted by CELF4 in the brain, including many others with well-appreciated roles in neuronal function (Wagnon et al., in press). We have validated Celf4-dependent alterations in whole-cell, steady-state abundance of several mRNA targets – some are increased like Scn8a, and others are decreased (also see Yang et al. 2007). While most of these changes are individually modest in nature, like that of Scn8a, we find that abundance of many CELF4 mRNA targets is shifted translationally (e.g. from monosome to polysome fractions in sucrose gradients of brain extracts) and subcellularly (e.g. from cell body to neuropil of dissected CA1 tissue), and that CELF4 itself is most tightly associated with very dense gradient fractions where a variety of RNA granules are known to reside. Together with its 3′-UTR binding specificity, at least for synaptically localized targets, these results suggest a mechanism whereby CELF4 regulates expression by sequestering target mRNAs to very large particles until needed for translation. While we may presume this is the case for Scn8a, however, more extensive studies are required, especially when the effect is local to the AIS, making translational profiling more challenging.
Although we suspect it is a key feature, increased intrinsic cellular excitability associated with excessive Nav1.6 in the AIS may explain only part of the CELF4 neurological disease. In our aforementioned related studies, functional annotation clustering revealed that the subset of CELF4 target mRNAs that shifts from cell body to neuropil is highly enriched for genes known to be important for regulation of synaptic plasticity. Together with the data herein, this suggests a model whereby a fairly modest local increase in ion channel function leads to disease in part because it occurs in the context of an impaired synaptic response. While this is indeed a natural way to think about complex phenotypes that result from primary genetic deficiencies of transcriptional and post-transcriptional regulatory proteins with multiple targets (e.g. FMRP, RBFOX1 and MECP2 among others (Huber et al. 2002; Deng et al. 2011; Gehman et al. 2011; Liu-Yesucevitz et al. 2011; Na et al. 2012; Qiu et al. 2012), one can appreciate that more subtle functional variation in these and other synaptic function regulators may, along with coding variants in individual targets (e.g. ion channels; Klassen et al. 2011), together comprise the vast and heretofore impenetrable genetic landscape of the most common neurological diseases, such as IE.
Acknowledgments
The authors thank Zhong-wei Zhang and Verity Letts for helpful advice and suggestions. This work was supported by the National Institutes of Health (NS061971) and a generous gift from the Relf family. The Jackson Laboratory's Imaging Sciences service was subsidized by an NCI core grant (5 P30 CA034196-27).
Glossary
- ACSF
artificial cerebrospinal fluid
- AIS
axon initial segment
- AnkG
AnkyrinG
- AP
action potential
- ECT
electroconvulsive threshold
- IE
idiopathic epilepsy
- IGE
idiopathic generalized epilepsy
- RBP
RNA-binding protein
- TEA
tetraethylammonium
Author contributions
W.S. designed the experiments, and performed electrophysiological studies and analysed the data. J.L.W. and M.B. performed the gene expression analysis and RNA-binding experiments, respectively, and J.U. supervised M.B.'s work. J.L.W. and C.L.M. performed immunostaining experiments. W.N.F. supervised the work, and helped to design and analyse experiments. All authors contributed to the preparation of the manuscript.
Author's present address
W. Sun: Janelia Farm Research Campus, Howard Hughes Medical Institute, 19700 Helix Dr., Ashburn, VA 20147, USA.
Supplementary material
Supplemental Fig. 1
References
- Beckh S, Noda M, Lubbert H, Numa S. Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 1989;8:3611–3616. doi: 10.1002/j.1460-2075.1989.tb08534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquillon S, Andrieux J, Landais E, Duban-Bedu B, Boidein F, Lenne B, Vallee L, Leal T, Doco-Fenzy M, Delobel B. A 5.3Mb deletion in chromosome 18q12.3 as the smallest region of overlap in two patients with expressive speech delay. Eur J Med Gen. 2011;54:194–197. doi: 10.1016/j.ejmg.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant 'motor endplate disease'. Nat Gen. 1995;10:461–465. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- Buysse K, Menten B, Oostra A, Tavernier S, Mortier GR, Speleman F. Delineation of a critical region on chromosome 18 for the del(18)(q12.2q21.1) syndrome. Am J Med Gen A. 2008;146A:1330–1334. doi: 10.1002/ajmg.a.32267. [DOI] [PubMed] [Google Scholar]
- Chandler WK, Meves H. Incomplete sodium inactivation in internally perfused giant axons from Loligo forbesi. J Physiol. 1966;186:121P–122P. [PubMed] [Google Scholar]
- Chang YT, Chen PC, Tsai IJ, Sung FC, Chin ZN, Kuo HT, Tsai CH, Chou IC. Bidirectional relation between schizophrenia and epilepsy: a population-based retrospective cohort study. Epilepsia. 2011;52:2036–2042. doi: 10.1111/j.1528-1167.2011.03268.x. [DOI] [PubMed] [Google Scholar]
- Cody JD, Sebold C, Malik A, Heard P, Carter E, Crandall A, Soileau B, Semrud-Clikeman M, Cody CM, Hardies LJ, Li J, Lancaster J, Fox PT, Stratton RF, Perry B, Hale DE. Recurrent interstitial deletions of proximal 18q: a new syndrome involving expressive speech delay. Am J Med Gen A. 2007;143A:1181–1190. doi: 10.1002/ajmg.a.31729. [DOI] [PubMed] [Google Scholar]
- Congdon E, Poldrack RA, Freimer NB. Neurocognitive phenotypes and genetic dissection of disorders of brain and behaviour. Neuron. 2010;68:218–230. doi: 10.1016/j.neuron.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Ann Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Dasgupta T, Ladd AN. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip Rev RNA. 2012;3:104–121. doi: 10.1002/wrna.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Sojka D, Klyachko VA. Abnormal presynaptic short-term plasticity and information processing in a mouse model of fragile X syndrome. J Neurosci. 2011;31:10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durner M, Keddache MA, Tomasini L, Shinnar S, Resor SR, Cohen J, Harden C, Moshe SL, Rosenbaum D, Kang H, Ballaban-Gil K, Hertz S, Labar DR, Luciano D, Wallace S, Yohai D, Klotz I, Dicker E, Greenberg DA. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol. 2001;49:328–335. [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Chandler SH. Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J Neurophysiol. 2007;98:710–719. doi: 10.1152/jn.00292.2007. [DOI] [PubMed] [Google Scholar]
- Feenstra I, Vissers LE, Orsel M, van Kessel AG, Brunner HG, Veltman JA, van Ravenswaaij-Arts CM. Genotype-phenotype mapping of chromosome 18q deletions by high-resolution array CGH: an update of the phenotypic map. Am J Med Gen A. 2007;143:1858–1867. doi: 10.1002/ajmg.a.31850. [DOI] [PubMed] [Google Scholar]
- Filges I, Shimojima K, Okamoto N, Rothlisberger B, Weber P, Huber AR, Nishizawa T, Datta AN, Miny P, Yamamoto T. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J Med Gen. 2011;48:117–122. doi: 10.1136/jmg.2010.084582. [DOI] [PubMed] [Google Scholar]
- Frankel WN. Genetics of complex neurological disease: challenges and opportunities for modelling epilepsy in mice and rats. Trends Gen. 2009;25:361–367. doi: 10.1016/j.tig.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel WN, Taylor L, Beyer B, Tempel BL, White HS. Electroconvulsive thresholds of inbred mouse strains. Genomics. 2001;74:306–312. doi: 10.1006/geno.2001.6564. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M, Jr, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Gen. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–1986. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg DA, Cayanis E, Strug L, Marathe S, Durner M, Pal DK, Alvin GB, Klotz I, Dicker E, Shinnar S, Bromfield EB, Resor S, Cohen J, Moshe SL, Harden C, Kang H. Malic enzyme 2 may underlie susceptibility to adolescent-onset idiopathic generalized epilepsy. Am J Hum Gen. 2005;76:139–146. doi: 10.1086/426735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, Fiegler H, Carr P, Kalaitzopoulos D, Clegg S, Sandstrom R, Temple IK, Youings SA, Thomas NS, Dennis NR, Jacobs PA, Crolla JA, Carter NP. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Gen. 2005;42:8–16. doi: 10.1136/jmg.2004.024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren C, Bache I, Bak M, Myatt MW, Anderson CM, Brondum-Nielsen K, Tommerup N. Haploinsufficiency of CELF4 at 18q12.2 is associated with developmental and behavioral disorders, seizures, eye manifestations, and obesity. Eur J Hum Gen. 2012 doi: 10.1038/ejhg.2012.92. (in press: doi: 10.1038/ejhg.2012.1092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesdorffer DC, Ludvigsson P, Olafsson E, Gudmundsson G, Kjartansson O, Hauser WA. ADHD as a risk factor for incident unprovoked seizures and epilepsy in children. Arch Gen Psych. 2004;61:731–736. doi: 10.1001/archpsyc.61.7.731. [DOI] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R, Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohane IS, McMurry A, Weber G, MacFadden D, Rappaport L, Kunkel L, Bickel J, Wattanasin N, Spence S, Murphy S, Churchill S. The co-morbidity burden of children and young adults with autism spectrum disorders. PLoS One. 2012;7:e33224. doi: 10.1371/journal.pone.0033224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP-transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp. 2011;50:e2638. doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzot D, Haberlandt E, Fauth C, Baumgartner S, Scholl-Burgi S, Utermann G. Del(18)(q12.2q21.1) caused by a paternal sister chromatid rearrangement in a developmentally delayed girl. Am J Med Gen A. 2005;135:304–307. doi: 10.1002/ajmg.a.30727. [DOI] [PubMed] [Google Scholar]
- Kumada T, Ito M, Miyajima T, Fujii T, Okuno T, Go T, Hattori H, Yoshioka M, Kobayashi K, Kanazawa O, Tohyama J, Akasaka N, Kamimura T, Sasagawa M, Amagane H, Mutoh K, Yamori Y, Kanda T, Yoshida N, Hirota H, Tanaka R, Hamada Y. Multi-institutional study on the correlation between chromosomal abnormalities and epilepsy. Brain Dev. 2005;27:127–134. doi: 10.1016/j.braindev.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278:27333–27339. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- Liao Y, Anttonen A-K, Liukkonen E, Gaily E, Maljevic S, Schubert S, Bellan-Koch A, Petrou S, Ahonen VE, Lerche H, Lehesjoki A-E. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology. 2010;75:1454–1458. doi: 10.1212/WNL.0b013e3181f8812e. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria PM, Duke A, Rand JB, Hobert O. Two neuronal, nuclear-localized RNA binding proteins involved in synaptic transmission. Curr Biol. 2003;13:1317–1323. doi: 10.1016/s0960-9822(03)00532-3. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Molecular identity of dendritic voltage-gated sodium channels. Science. 2010;328:906–909. doi: 10.1126/science.1187958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Gen. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- McKinney BC, Chow CY, Meisler MH, Murphy GG. Exaggerated emotional behaviour in mice heterozygous null for the sodium channel Scn8a (Nav1.6) Genes Brain Behav. 2008;7:629–638. doi: 10.1111/j.1601-183X.2008.00399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Joerges J, Huguenard JR, Sejnowski TJ. A model of spike initiation in neocortical pyramidal neurons. Neuron. 1995;15:1427–1439. doi: 10.1016/0896-6273(95)90020-9. [DOI] [PubMed] [Google Scholar]
- Marseglia G, Scordo MR, Pescucci C, Nannetti G, Biagini E, Scandurra V, Gerundino F, Magi A, Benelli M, Torricelli F. 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur J Med Gen. 2012;55:216–221. doi: 10.1016/j.ejmg.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Gen. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci. 2001;21:2268–2277. doi: 10.1523/JNEUROSCI.21-07-02268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney J, Ottman R, Escayg A. Identification of epilepsy genes in human and mouse. Ann Rev Gen. 2001;35:567–588. doi: 10.1146/annurev.genet.35.102401.091142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, O'Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588:1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Plummer NW, Burgess DL, Buchner DA, Sprunger LK. Allelic mutations of the sodium channel SCN8A reveal multiple cellular and physiological functions. Genetica. 2004;122:37–45. doi: 10.1007/s10709-004-1441-9. [DOI] [PubMed] [Google Scholar]
- Moore JW, Stockbridge N, Westerfield M. On the site of impulse initiation in a neurone. J Physiol. 1983;336:301–311. doi: 10.1113/jphysiol.1983.sp014582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Hecquet S, McLellann A, Ville D, Grid D, Picard F, Moulard B, Asherson P, Makoff AJ, McCormick D, Nashef L, Froguel P, Arzimanoglou A, LeGuern E, Bailleul B. Polymorphism analysis of JRK/JH8, the human homologue of mouse jerky, and description of a rare mutation in a case of CAE evolving to JME. Epilepsy Res. 2001;46:157–167. doi: 10.1016/s0920-1211(01)00275-3. [DOI] [PubMed] [Google Scholar]
- Morgan CL, Baxter H, Kerr MP. Prevalence of epilepsy and associated health service utilization and mortality among patients with intellectual disability. Am J Ment Retard. 2003;108:293–300. doi: 10.1352/0895-8017(2003)108<293:POEAAH>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Na ES, Nelson ED, Adachi M, Autry AE, Mahgoub MA, Kavalali ET, Monteggia LM. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci. 2012;32:3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicita F, De Liso P, Danti FR, Papetti L, Ursitti F, Castronovo A, Allemand F, Gennaro E, Zara F, Striano P, Spalice A. The genetics of monogenic idiopathic epilepsies and epileptic encephalopathies. Seizure. 2012;21:3–11. doi: 10.1016/j.seizure.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio N, Cathala L, Meisler MH, Crest M, Magistretti J, Delmas P. Persistent Nav1.6 current at axon initial segments tunes spike timing of cerebellar granule cells. J Physiol. 2010;588:651–670. doi: 10.1113/jphysiol.2010.183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman R. Analysis of genetically complex epilepsies. Epilepsia. 2005;46(Suppl 10):7–14. doi: 10.1111/j.1528-1167.2005.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, Letts VA, Meisler MH, Frankel WN, Escayg A. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum Mol Gen. 2009;18:1633–1641. doi: 10.1093/hmg/ddp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett syndrome protein MeCP2 regulates synaptic scaling. J Neurosci. 2012;32:989–994. doi: 10.1523/JNEUROSCI.0175-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Rapp M, Yarom Y, Segev I. Modeling back propagating action potential in weakly excitable dendrites of neocortical pyramidal cells. Proc Natl Acad Sci U S A. 1996;93:11985–11990. doi: 10.1073/pnas.93.21.11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royeck M, Horstmann MT, Remy S, Reitze M, Yaari Y, Beck H. Role of axonal NaV1.6 sodium channels in action potential initiation of CA1 pyramidal neurons. J Neurophysiol. 2008;100:2361–2380. doi: 10.1152/jn.90332.2008. [DOI] [PubMed] [Google Scholar]
- Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH. A novel, abundant sodium channel expressed in neurons and glia. J Neurosci. 1995;15:3231–3242. doi: 10.1523/JNEUROSCI.15-05-03231.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharkey LM, Cheng X, Drews V, Buchner DA, Jones JM, Justice MJ, Waxman SG, Dib-Hajj SD, Meisler MH. The ataxia3 mutation in the N-terminal cytoplasmic domain of sodium channel Nav1.6 disrupts intracellular trafficking. J Neurosci. 2009;29:2733–2741. doi: 10.1523/JNEUROSCI.6026-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahata E, Iwasaki H, Takagi M, Lin C, Bennett V, Okamura Y, Hayasaka K. Ankyrin-G regulates inactivation gating of the neuronal sodium channel, Nav1.6. J Neurophysiol. 2006;96:1347–1357. doi: 10.1152/jn.01264.2005. [DOI] [PubMed] [Google Scholar]
- Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr Res. 2009;65:599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan Y, Elmer L, Davis J, Bennett V, Angelides K. Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature. 1988;333:177–180. doi: 10.1038/333177a0. [DOI] [PubMed] [Google Scholar]
- Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Ann Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LPW, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. J Med Gen. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J. Ribonucleoprotein complexes in neurologic diseases. Curr Opin Neurobiol. 2008;18:516–523. doi: 10.1016/j.conb.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500:339–352. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- Veeramah KrishnaR, O'Brien JanelleE, Meisler MiriamH, Cheng X, Dib-Hajj SulaymanD, Waxman StephenG, Talwar D, Girirajan S, Eichler EvanE, Restifo LindaL, Erickson RobertP, Hammer MichaelF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Gen. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Briese M, Sun W, Mahaffey CL, Curk T, Rot G, Ule J, Frankel WN. CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic plasticity. PLoS Genet. 2012;8(11):e1003067. doi: 10.1371/journal.pgen.1003067. doi: 10.1371/journal.pgen.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Mahaffey CL, Sun W, Yang Y, Chao HT, Frankel WN. Etiology of a genetically complex seizure disorder in Celf4 mutant mice. Genes Brain Behav. 2011;10:765–777. doi: 10.1111/j.1601-183X.2011.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer VC, Reid CA, So EY-W, Berkovic SF, Petrou S. Axon initial segment dysfunction in epilepsy. J Physiol. 2010;588:1829–1840. doi: 10.1113/jphysiol.2010.188417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski KE, French JH, Fernando S, Brown WT, Jenkins EC, Friedman E, Hill AL, Miezejeski CM. Fragile X syndrome: associated neurological abnormalities and developmental disabilities. Ann Neurol. 1985;18:665–669. doi: 10.1002/ana.410180607. [DOI] [PubMed] [Google Scholar]
- Yang Y, Mahaffey CL, Berube N, Maddatu TP, Cox GA, Frankel WN. Complex seizure disorder caused by Brunol4 deficiency in mice. PLoS Genet. 2007;3:e124. doi: 10.1371/journal.pgen.0030124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.