

Key Points
miR-155 tg mice have increased NK-cell number, enhanced NK-cell survival, excess immature CD11blowCD27high NK cells, and an activated phenotype.
miR-155 tg NK cells exhibit enhanced expansion, interferon-γ production, AKT and ERK activation, and killing of lymphoma in vivo.
Abstract
It is known that microRNAs (miRs) are involved in lymphocyte development, homeostasis, activation, and occasionally malignant transformation. In this study, a miR-155 transgene (tg) was driven to be overexpressed off of the lck promoter in order to assess its effects on natural killer (NK) cell biology in vivo. miR-155 tg mice have an increase in NK-cell number with an excess of the CD11blowCD27high NK subset, indicative of a halt in terminal NK-cell differentiation that proved to be intrinsic to the cell itself. The increase in NK cells results, in part, from improved survival in medium alone and enhanced expansion with endogenous or exogenous interleukin 15. Phenotypic and functional data from miR-155 tg NK cells showed constitutive activation and enhanced target cell conjugation, resulting in more potent antitumor activity in vitro and improved survival of lymphoma-bearing mice in vivo when compared with wild type NK cells. The enhanced NK-cell survival, expansion, activation, and tumor control that result from overexpression of miR-155 in NK cells could be explained, in part, via diminished expression of the inositol phosphatase SHIP1 and increased activation of ERK and AKT kinases. Thus, the regulation of miR-155 is important for NK-cell development, homeostasis, and activation.
Introduction
Natural killer (NK) cells directly kill pathogen-infected and tumor cells and regulate the immune system via production of cytokines and chemokines.1 During maturation, NK cells acquire cytokine receptors, activating and inhibitory receptors, adhesion molecules, and effector functions.2-4 The committed NK-cell precursors (NKPs) in mice express the common β chain receptor (R) for interleukin 2 (IL-2) and IL-15 (CD122), IL-7Rα (CD127), and c-kit (CD117). NKPs then acquire an immature phenotype with the surface expression of NK1.1, CD94, the TNFR superfamily member CD27, the integrin CD11b, and Ly49 receptors.2 Additionally, during terminal maturation, NK cells downregulate CD27 and acquire high surface density expression of CD11b.5,6
Acquisition of lytic functions and interferon γ protein (IFN-γ) production in NK cells depends on complex interactions that involve signaling molecules, transcription factors, and microRNAs (miRs).7-10 miRs are small noncoding RNAs that modulate posttranscriptional gene expression of multiple targets and are implicated in regulating several cellular and developmental processes.11 miRs regulate gene expression by binding to the 3′ untranslated region (UTR) and inducing either suppression of mRNA translation or mRNA degradation.
miR-155 plays a protective role in immunity when its expression is tightly regulated.12 Reportedly, miR-155 controls the development and functions of different immune cells, including T, B, and dendritic cells.13,14 In human NK cells, the constitutive expression of miR-155 is different in CD56bright and CD56dim subsets, which represent stages 4 and 5 of NK-cell development and is also upregulated during human NK-cell activation. In particular, the induction of miR-155 expression depends on IL-18 or CD16 stimulation and can be synergistically induced by the combination of these stimuli with IL-12.15 miR-155 inhibits the expression of SH2 containing 5′ inositol phosphatase (SHIP1) inositol phosphatase in human NK cells, which contributes, at least in part, to its regulation of IFN-γ production.15
To further understand the role of miR-155 in regulating NK-cell development and function, we assessed NK cells in mice genetically modified to overexpress miR-155 driven off the lck promoter. Our results show that miR-155 is important for NK-cell development, homeostasis, and the regulation of several intrinsic NK cellular functions.
Methods
Mice
The lck-miR-155 C57BL/6 (B6) tg mouse was described16 as was the IL-15 B6 tg mouse.17 Littermates aged 8.5 to 14 weeks were used for experiments. Double tg mice for miR-155 and IL-15 were generated from a single cross. Rag2/II2rg double knockout (Rag2−/−xII2rg−/−)18 mice were from Taconic. The Ohio State University Animal Care and Use Committee approved animal work.
NK-cell preparations
All experiments were performed with purified NK cells via microbead selection (Miltenyi Biotech Inc.) followed by fluorescence-activated cell sorter (FACS) giving >99.5% purity. Cultured NK cells were in RPMI-1640 (Invitrogen) containing 900 IU/mL of human IL-2 (Hoffman-LaRoche Inc.) and 55 μM β-mercaptoethanol (Gibco).
Cell lines
The YAC-1 mouse T-lymphoma cell line, RMA mouse T-cell lymphoma tumor cells, and Rae1β-transduced RMA tumor cells were maintained in RPMI-1640, and the murine mastocytoma P815 cells were maintained in Dulbecco’s modified Eagle medium, both with 10% fetal bovine serum (FBS; Invitrogen) and 2 mM L-glutamine (Gibco).
Flow cytometry analysis
Splenocytes were stained with antibodies (Abs; clones) from BD Biosciences reactive against: NK1.1 (PK136), CD3ε (145-2C11), CD117 (ACK45), CD27 (LG.3A10), CD11b (M1/70), CD49b (DX5), CD122 (TM-beta1), Ly-49C/I (5E6), Ly-49I (YLI-90), Ly-49A (A1), CD16/32 (2.4G2), Ly-49D (4E5), and Ly-49G2 (4D11). Rat Abs to mouse CD94 (18d3), CD69 (H1.2F3), NKG2D (CX5), NKG2ACE (20d5), 2B4 (eBio244F4), NKp46 (29A1.4), and Ly-49C/I/F/H (14B11) were from eBioscience. NK1.1+CD3− cells were gated for FACS analysis of these antigens and analyzed with FlowJo v7.6.1 software (TreeStar).
Survival assay
Fresh splenocytes were cultured in 24-well plates at 1 million/mL in RPMI-1640 with 10% FBS and 55 μM β−mercaptoethanol for 24 hours. NK1.1+CD3− NK cells were labeled with annexin V and 7-AAD (BD Bioscience) and analyzed by FACS within 1 hour of staining.
In vivo BrdU incorporation assay
Mice were given 5-bromo-2′-deoxyuridine (BrdU) daily in drinking water (0.8 mg/mL) containing 1% glucose. All mice were injected intraperitoneally once with 1.5 mg of BrdU in phosphate-buffered saline (PBS) on day 1. Ten days later, splenocytes were stained for FITC-CD3, PE-NK1.1, 7-AAD, and APC-BrdU following the manufacturer’s protocol.
Assessment of in vitro NK-cell expansion
Sorted NK cells were cultured (2 × 104) in triplicate wells of 200 μL of RPMI-1640, 10% FBS, IL-15 (100 ng/mL), and 55 μM β-mercaptoethanol. After 2 to 7 days, viable cells were enumerated by trypan blue exclusion. To measure NK-cell proliferation, NK1.1+CD3−–sorted NK cells labeled with the cell fluorescence dye eFluor 670 (eBioscience) were cultured in the presence of IL-15 for 11 days, followed by flow cytometric analysis to quantify serial dilution of fluorescence intensity while gated on live cells.
Transfer experiment
CD11blowCD27high splenic NK cells from CD45.2+ wild type (wt) and miR-155 tg mice were sorted to >98% purity and injected intravenously into sublethally irradiated or nonirradiated CD45.1+ wt littermate recipient mice. CD27 and CD11b expression were analyzed by FACS of NK-cell–enriched splenocytes after 16 days by gating on CD45.2+ NK1.1+CD3− cells.
Cell stimulation
Next, wt and miR-155 tg NK cells were stimulated with IL-2 (90 ng/mL), IL-15 (100 ng/mL; Amgen), or IL-12 (20 ng/mL; Genetics Institute Inc.) plus IL-18 (10 ng/mL; R&D Systems) for indicated times. For stimulation with YAC-1 tumor cells, NK cells were expanded in IL-2 for >8 days, starved from IL-2 for 2 hours on ice, and then mixed with paraformaldehyde-fixed YAC-1 cells at a 5:1 ratio and stimulated for the indicated times.
NK cell IFN-γ production
Sorted subsets from wt and miR-155 tg NK1.1+CD3− NK cells were untreated or stimulated and analyzed as previously described.19 Results are shown as the means of triplicate wells ± standard error of the mean (SEM).
Cytotoxicity assays
YAC-1, RMA, RMA-Rae-1β cells, or P815 cells coated with an anti-mouse lymphocyte rabbit Ab (Accurate Chemical) were used as targets in a 4-hour 51Cr release assay20 with fresh NK cells or NK cells cultured in IL-2 90 ng/mL for 8 days.
Conjugate formation
Immune complex formations among NK cells and YAC-1 tumor cells were examined by flow cytometry. YAC-1 tumor cells were infected using the green fluorescent protein (GFP)-encoding lentivirus vector pCDH (System Biosciences) and sorted for GFP. For the conjugate assay, 2 × 105 PE-labeled NK1.1+CD3− NK cells were mixed with 2 × 105 GFP+ YAC-1 cells in 200 μL of cold RPMI-1640 with 10% FBS and centrifuged at 600 rpm for 1 minute. To induce the formation of immune complexes, cells were incubated at 37°C for 0 and 10 minutes. Reactions were stopped with ice-cold PBS. NK1.1+GFP+ conjugates were detected by FACS analysis as described.21
Immunoblot analysis
NK cells were harvested and lysed into RIPA buffer or directly lysed in Laemmli buffer (2 × 105 cells/20 μl) as described.22 Immunoblots were performed according to published protocols, and Ab-reactive proteins were detected as described.20 The Abs used included the following: the anti-SHIP1, anti-actin, and anti-granzyme M (Santa Cruz Biotechnology); anti-granzyme B, anti-perforin, anti-phospho-AKTSer473, and anti-phospho-extracellular signal-regulated kinaseThr202/Tyr204 (Cell Signaling Technology).
Real-time RT-PCR
RNA was extracted using Trizol (Invitrogen). Reverse transcription (RT) was performed with a Taqman MicroRNA reverse transcription kit and RT primers specific for miR-155 and U6 or 292 as control (Applied Biosystems). Real-time reverse-transcription polymerase chain reactions (RT-PCRs) were performed as described.15 Data were analyzed with the comparative CT method using internal control U6 or 292 RNA levels to normalize differences in sample loading. Results (mean ± SEM of triplicate reaction wells) represent the n-fold difference of transcript levels in a particular sample compared with samples of wt NK cells.
Tumor growth assay
For the tumor growth assay, 2 × 105 wt or miR-155 tg NK cells were mixed with 1 × 105 tumor cells and injected subcutaneously into the shaved flank of Rag2−/−xII2rg−/− recipient mice. As a positive control, 1 × 105 RMA-Rae-1β tumor cells were injected without NK cells. Tumor volumes [0.5 × (length) × (width)2] were measured every 2 days. Animals were considered tumor free when no tumor was found 40 days after inoculation. Mice were sacrificed when lymphoma tumor burden became excessive.
Statistics
Data were compared using the Student 2-tailed t test. A P value < .05 was considered significant. Survival data were analyzed using Kaplan-Meier and log-rank test methods (GraphPad Prism Version 5.0).
Results
Effect of miR-155 overexpression on NK-cell number
To investigate the effects of miR-155 overexpression on NK cells, we used lck-miR-155 tg mice that we previously generated.16 We quantified miR-155 expression on RNA obtained from NK1.1+CD3− wt and miR-155 tg NK cells, noting significantly higher values in the latter (average induction of 26.6 +/− 5.3-fold; Figure 1A; P < .0001, n = 6). Similar data were noted for T cells from miR-155 tg mice compared with wt mice (data not shown and16). MiR-155 tg mice also had a higher percentage of splenic NK1.1+CD3− NK cells compared with wt mice (Figure 1B; P < .0001, n = 16) and a higher absolute number of NK cells (Figure 1C; P < .0001, n = 13). Comparable changes were observed in bone marrow and blood (data not shown). On the other hand, we observed a clear reduction in the percentage and absolute number of splenic NK1.1+CD3+ NKT cells in miR-155 tg mice compared with wt mice (P < .0001; n = 12; supplemental Figure 1).
Figure 1.
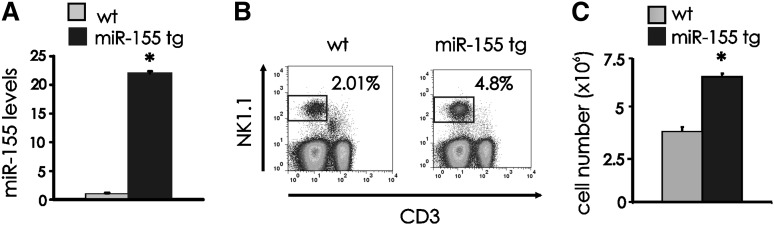
NK cell expansion in miR-155 tg mice. (A) NK1.1+CD3− FACS-sorted NK cells from spleen of wt and miR-155 tg mice were analyzed for miR-155 expression by real-time RT-PCR. This experiment is representative of 6 performed with similar results. Results are expressed as the mean ± SEM of triplicate wells. (B) Freshly isolated splenocytes of wt and miR-155 tg mice were stained with anti-NK1.1 and anti-CD3 Abs and analyzed by flow cytometry for percentage of NK1.1+CD3− NK cells. (C) The absolute number of NK cells was calculated in spleens of wt vs miR-155 tg mice. The graph summarizes data from 4 wt and 2 miR-155 tg littermate mice. *Statistically significant; see text for details.
Survival and proliferative capacities of miR-155 tg vs wt NK cells
To explain the higher number of NK cells in miR-155 tg mice, we investigated NK-cell proliferative and survival capacities. For in vivo proliferation studies we did not see a significant difference in the rate of BrdU incorporation between miR-155 tg and wt NK1.1+CD3− NK cells (Figure 2A; P = .17, n = 10). To determine if these miR-155 tg NK cells had the capacity for enhanced proliferation, parallel studies were performed with sorted NK1.1+CD3− NK cells cultured ex vivo in the presence of IL-15, the endogenous survival and growth factor for NK cells.23-25 In vitro, miR-155 tg NK cells showed significantly greater expansion when compared with wt NK cells in the presence of IL-15 (Figure 2B, left; P < .001, n = 6) and significantly greater proliferation, as indicated by serial dilution of the cell fluorescence dye eFluor 670 (Figure 2B, right). To determine if endogenous IL-15 cooperates with miR-155 in regulating NK-cell number in vivo, we crossed miR-155 tg B6 mice with IL-15 tg B6 mice.17,26 We observed that miR-155/IL-15 double tg mice had a significantly higher percentage and absolute number of NK1.1+CD3− splenic NK cells (Figure 2C, left and right, respectively; P < .01, n = 4) compared with either miR-155 tg or IL-15 tg mice alone.
Figure 2.

Proliferation and survival of miR-155 tg vs wt NK cells. (A) BrdU drinking water was administrated daily for 10 days to wt and miR-155 tg mice. BrdU incorporation in splenic NK cells was determined by surface staining of NK1.1 and CD3, followed by intracellular staining of BrdU. The histogram represents BrdU incorporation within a gated population of NK1.1+CD3− wt and miR-155 tg NK cells. Mean ±SEM of percentage of BrdU incorporation of wt and miR-155 NK cells from 10 mice is shown on graph (right panel). (B) Left: 2 × 104 FACS-sorted NK1.1+CD3− splenic NK cells were cultured in IL-15 (100 ng/mL) for 7 days. Viable cells were enumerated after culturing for 3, 5, and 7 days by trypan blue dye exclusion. Right: eFluor 670–labeled NK1.1+CD3− wt and miR-155 tg NK cells were cultured with IL-15 for 11 days. Viable cells were assessed for proliferation by measuring serial dilution of dye fluorescence. (C) Splenocytes of wt, miR-155 tg, IL-15 tg, or double miR-155/IL-15 tg mice were stained with anti-NK1.1 and anti-CD3 Abs and analyzed by flow cytometry for percentage of NK1.1+CD3− NK cells (left) and for absolute number of NK cells (right). (D) Freshly isolated splenocytes were cultured in medium without cytokines for 24 hours, followed by surface staining of NK1.1 and CD3 and labeling with 7-AAD and annexin V. Representative dot plots from 5 experiments show staining for 7-AAD and annexin V in NK1.1+CD3− NK cells. *Statistically significant; see text for details.
For survival studies, splenic NK cells were harvested and cultured in vitro for 24 hours without cytokines and then stained with annexin V and 7-amino-actinomycin D (7-AAD) to assess for cell death. Figure 2D is representative of 5 experiments. MiR-155 tg NK cells showed a significantly greater fraction of viable cells at 24 hours, as noted by a higher percentage of cells lacking an increase in annexin V and 7-AAD when compared with wt NK cells (P < .001). In contrast, wt NK cells had a greater fraction of 7-AAD+ and annexin V− cells consistent with necrosis at 24 hours when compared with miR-155 tg NK cells (P < .001). Both wt and miR-155 tg NK cells had a low fraction of 7-AAD- and annexin V+ NK cells (consistent with early apoptosis) at time zero, at 6 hours (not shown), and at 24 hours. No significant differences were observed in 7-AAD+ and annexin V+ between wt and miR-155 tg NK cells, which is indicative of the late stage of apoptosis (P = .18).
Collectively, these data indicate that miR-155 tg NK cells have an enhanced intrinsic ability for survival in the absence of cytokines, and this likely contributes to their in vivo increase in absolute cell number when compared with wt NK cells. In addition, their growth can be further enhanced over that seen with wt NK cells in the presence of abundant exogenous or endogenous IL-15.
NK-cell surface antigen expression in miR-155 tg mice
To determine whether overexpression of miR-155 could affect NK cell development and/or NK cell activation, we analyzed a variety of well-characterized cell surface antigens in miR-155 tg NK cells and compared their expression with wt mice. MiR-155 tg mice had a significantly higher percentage of splenic NK cells expressing CD94, CD117, CD27, CD69, NKG2D, Ly49G2, and NKG2ACE (Figure 3A; P < .01). There were no significant differences in the expression of NKp46, 2B4, DX5, CD122, Ly49CIFH, Ly49C/I, Ly49I, and Ly49A (Figure 3B; P > .06), while miR-155 tg NK cells had a lower percent expression of CD11b, CD16, and Ly49D compared with wt NK cells (Figure 3C; P < .01). These differences were then used to explore developmental and functional differences between miR-155 tg NK cells and their wt counterparts.
Figure 3.

Comparative analysis of surface antigen expression on NK cells from miR-155 tg vs wt mice. Antigen expression was performed on gated NK1.1+CD3− NK cells harvested from spleens of miR-155 tg and wt mice. The antigen of interest is labeled along the x-axis. (A) Antigens with significantly higher expression on miR-155 tg NK cells compared with wt NK cells; (B) antigens with no difference in expression; and (C) antigens with significantly lower expression on miR-155 tg NK cells compared with wt NK cells. The y-axis indicates the mean ± SEM percent of surface antigen expression on gated NK1.1+CD3− cells from at least 5 mice/group for each antigen. *Statistically significant; see text for details.
Effect of miR-155 overexpression on NK-cell development
The noted decrease in the percentage of miR-155 tg NK cells expressing the late maturation markers CD11b and CD16 and their increased expression of early maturation markers CD27 and CD117 suggested a block of terminal differentiation. To extend this analysis, we gated on NK1.1+CD3− NK cells and quantified each subset representing the 3 sequential stages of terminal NK mouse maturation5,6: CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low. miR-155 tg splenocytes showed an increase in the CD11blowCD27high NK subset (P < .001, n = 7) and a decrease in the CD11bhighCD27low NK subset (P < .001, n = 7) when compared with wt NK subsets (Figure 4A-B). No difference was observed in the CD11bhighCD27high subset in miR-155 tg vs wt NK cells (Figure 4A-B; P = .60, n = 7). These data are consistent with a block in NK-cell development at the earlier CD11blowCD27high stage of differentiation.
Figure 4.

Effect of miR-155 overexpression on terminal differentiation of NK cells. (A) Splenocytes from wt and miR-155 tg mice were stained with anti-NK1.1, anti-CD3, anti-CD27, and anti-CD11b Abs. Contour maps represent surface density expression of CD27 and CD11b on gated NK1.1+CD3− NK cells. (B) The graphs summarize mean percentage (left) and absolute number (right) of +/− SEM of CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low NK-cell subsets from 10 miR-155 tg and 4 wt mice. (C) 1 × 105 to 2 × 105 CD11blowCD27high NK cells collected from 2 CD45.2+ wt and 3 CD45.2+ miR-155 tg mice were intravenously injected into sublethally irradiated CD45.1+ recipient mice. After 16 days, splenic NK cells were harvested and the progeny of adoptively transferred CD11blowCD27high NK cells from wt and miR-155 tg mice were analyzed via FACS with a gate on CD45.2 expression and assessment for coexpression of CD27 and CD11b. With this, the frequency of CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low NK-cell subsets was quantified and graphed. (D) Total wt and miR-155 tg NK1.1+CD3− NK cells were each costimulated in vitro for 18 hours using IL-12 (20 ng/mL) and IL-18 (10 ng/mL) and analyzed for IFN-γ secretion by ELISA. (E) FACS-sorted CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low NK-cell subsets from wt and tg mice were stimulated for 18 hours in vitro with IL-12 (20 ng/mL) and IL-18 (10 ng/mL). Supernatants were then collected and analyzed for IFN-γ by ELISA. *Statistically significant; see text for details.
To determine whether the block in differentiation was due to an intrinsic defect, we purified the CD11blowCD27high CD45.2 NK subset from wt and miR-155 tg mice, verified the overexpression of miR-155 in the tg mice, and adoptively transferred this subset into CD45.1 wt recipient mice. After 16 days we analyzed the percentage of CD11blowCD27high, CD11bhighCD27high, and CD11bhighCD27low CD45.2+ NK subsets in spleen by FACS. We observed a higher percentage of miR-155 tg CD11blowCD27high immature NK cells in vivo relative their wt counterpart (Figure 4C; P < .02, n = 8) and a clear decrease in the percentage of miR-155 tg CD11bhighCD27low mature NK cells in vivo relative to their wt counterpart (P < .001, n = 8). Thus, the accumulation of the relatively immature CD11blowCD27high NK cells in miR-155 tg mice appears to be secondary to an intrinsic arrest in NK-cell maturation.
Effect of miR-155 overexpression on NK-cell IFN-γ production
The increase in CD69 and CD94 expression suggested that a majority of miR-155 tg NK cells were in a heightened state of activation.27 Indeed, miR-155 tg NK cells produced higher IFN-γ when compared with wt NK cells following in vitro stimulation with IL-12 plus IL-18 (Figure 4D; P < .001, n = 6). Further, each of the 3 CD27/CD11b stages of miR-155 tg NK-cell subsets produced higher IFN-γ than their comparable wt NK-cell subset (Figure 4E; P ≤ .02, n = 3). We also noted that the miR-155 tg CD11blowCD27high immature NK-cell subset produced more IFN-γ than the more mature miR-155 tg or wt CD11bhighCD27low NK-cell subset (Figure 4E). Finally, we confirmed results from a previously published report indicating that CD11bhighCD27high NK cells secrete a higher level of IFN-γ compared with the more terminally mature CD11bhighCD27low NK-cell subset.6
Effect of miR-155 overexpression on NK-cell lytic function
We compared NK1.1+CD3− NK cells from miR-155 tg and wt mice for their ex vivo ability to kill the prototypic tumor cell target YAC-1, as well as the RMA tumor cell line that has high surface density expression of the NKG2D ligand Rae1β (RMA-Rae1β).28 Fresh miR-155 tg NK cells lysed both YAC-1 (P < .01, n = 9) and RMA-Rae1β (P = .02, n = 5) lymphoma targets with significantly higher efficiency compared with wt NK cells (Figure 5A). Very low cytotoxicity was observed using control RMA target cells not expressing Rae1β (not shown). Following 8 days of culture in IL-2, these differences in spontaneous cytotoxic function between miR-155 tg NK cells and wt NK cells disappeared (data not shown), yet miR-155 tg NK cells had significantly higher antibody-dependent cellular cytotoxicity (ADCC) against P815 antibody–coated target cells when compared with wt NK cells when incubated with IL-2 (Figure 5B; P < .001, n = 5). NK-cell ADCC was low yet significantly higher in freshly isolated miR-155 tg NK cells compared with wt NK cells (not shown).
Figure 5.

Effect of miR-155 overexpression on NK-cell cytotoxic effector functions. (A) FACS-sorted NK1.1+CD3− NK cells from wt and miR-155 tg mice were used as effector cells in a 4-hour 51Cr release assay using YAC-1 and RMA-Rae1β tumor cells as targets. (B) Eight days after expansion in IL-2, sorted wt and miR-155 tg NK cells were assayed for ADCC against 51Cr-labeled P815 Ab-coated targets cells. (C) The wt and miR-155 tg NK cells were mixed with RMA-Rae1β tumor cells at a ratio of 2:1 and injected subcutaneously into the flank of Rag2−/−xII2rg−/− recipient mice (left). RMA-Rae1β tumor cells alone were injected as control. Tumor volumes were calculated every 2 days. The graph summarizes mean ± SEM data from 2 experiments with a total of 7 mice for the wt and the miR-155 tg groups. The percent survival of mice that had been inoculated with either wt or miR-155 tg NK cells in combination with RMA-Rae1β tumor cells at a ratio of 2:1 or RMA-Rae1β tumor cells alone as control is shown (right). Data from 3 independent experiments using 11 mice for each group are summarized in the Kaplan-Meier survival plots. (D) Resting (left) and IL-2–activated (8 days; right) NK1.1+CD3− wt and miR-155 tg NK cells were analyzed for granzyme B, granzyme M, perforin, and actin protein levels by immunoblot. (E) The wt and miR-155 tg NK cells labeled with PE-conjugated anti-NK1.1 Ab were incubated with GFP+ YAC-1 tumor cells (left). Conjugate formation was analyzed at time 0 and after 10 minutes of incubation by flow cytometry. NK-cell target cell conjugates were gated and identified as NK1.1+GFP+ cells. The percentages of conjugates are shown on top of the representative dot plots. The graph summarizes the data of conjugate formation obtained from 3 wt and 3 miR-155 tg NK cell samples coincubated with YAC-1 tumor cells (right). *Statistically significant; see text for details.
To assess cytotoxicity of lymphoma in vivo, FACS-sorted wt and miR-155 NK cells were co-injected with RMA-Rae1β cells subcutaneously into Rag2−/−xII2rg−/− mice. These mice do not have T cells or NK cells. miR-155 tg NK cells significantly inhibited the growth of RMA-Rae1β tumor when compared with wt NK cells in vivo (Figure 5C; P < .01, n = 7). Further, in 2 independent experiments, 63.6% of mice receiving RMA-Rae1β tumor cells co-injected with miR-155 tg NK cells survived, while only 27.2% survived when co-injected with the same tumor and wt NK cells (Figure 5C; P < .001, n = 11).
To investigate potential cellular mechanisms responsible for the significantly enhanced NK-cell cytolysis in miR-155 tg NK cells, we first analyzed protein levels of granzyme B, granzyme M, and perforin by immunoblot analysis. Of interest, granzyme B protein levels were significantly decreased in miR-155 tg NK cells compared with wt NK cells, yet this downregulation was completely reversed following activation in the presence of IL-2 (Figure 5D). No significant differences were otherwise observed in levels of granzyme M and perforin in miR-155 tg vs wt NK cells.
Effector-target conjugate formation is critical for NK-cell cytotoxicity. As shown in Figure 5E, miR-155 tg NK cells have a significantly enhanced ability to form conjugates with YAC-1 cells when compared with wt NK cells (Figure 5E; P < .01, n = 3). This can explain, at least in part, the enhanced ability of miR-155 NK cells to kill tumor cell targets over wt NK cells.
SHIP1 expression and phosphorylation of AKT and ERK in miR-155 tg NK cells
We have previously shown that SHIP1 is negatively regulated by miR-155 in human NK cells,15 and indeed, SHIP1 is downregulated in miR-155 tg mouse NK cells (Figure 6A). SHIP1 is a negative regulator of the PI-3K pathway of which AKT is a downstream target.29 Following activation by either IL-2 or IL-15 (and occasionally in medium alone), AKT was visibly more activated in miR-155 tg NK cells compared with wt NK cells (Figure 6B). ERK is also a downstream target of PI-3K in NK cells,10,30 and similar to AKT, we found its activation was visibly higher in miR-155 tg NK cells compared with wt NK cells cultured in IL-2 or IL-15 (and occasionally in medium alone; Figure 6B). Since ERK signaling regulates cytotoxicity in NK cells,10,20 we also quantified ERK phosphorylation after binding YAC-1 tumor target cells. miR-155 tg NK cells have higher levels of phospho-active ERK compared with wt NK cells after conjugation with YAC-1 tumor target cells (Figure 6C). Thus, at least following stimulation with cytokines or following interaction with tumor target cells, NK cells from miR-155 tg mice possess intrinsically enhanced activation of AKT and ERK when compared with wt NK cells.
Figure 6.

Effect of miR-155 overexpression on SHIP1 protein expression and AKT and ERK activation in NK cells. (A) NK1.1+CD3− NK cells from wt and miR-155 tg mice were analyzed for SHIP1 protein levels by immunoblot. Actin was assessed to ensure equal loading. (B) NK1.1+CD3− NK cells from wt and miR-155 tg mice were left untreated or stimulated with IL-2 (90 ng/mL) or IL-15 (100 ng/mL) for 10 minutes. Immunoblot analysis was performed on total lysates using anti-phospho-ERKThr202/Tyr204, anti-phospho-AKTSer473, and actin Abs. (C) NK1.1+CD3− NK cells from wt and miR-155 tg mice and paraformaldehyde-treated YAC-1 cells were mixed and incubated at a ratio of 5:1 for the indicated times. Lysates from NK-cell and YAC-1 samples were analyzed by immunoblot using anti-phospho-ERKThr202/Tyr204 and actin Abs. These blots are representative of at least 2 independent experiments.
Discussion
In our study, we used a tg mouse with overexpression of miR-155 driven off of the lck promoter to assess the relevance of this miR on the development, homeostasis, and function of NK cells. Our first in vivo observation was that overexpression of miR-155 increased the number of NK cells. This is reflective of altered homeostasis, a process that depends on a balance of NK-cell production, proliferation, survival, and cell death,31,32 each of which is dependent, at least in part, on endogenous IL-15.23-25 miR-155 tg NK cells have improved intrinsic survival in the absence of cytokines and improved expansion in the presence of exogenous IL-15. In support of this, we consistently found enhanced expression of phospho-AKT and phospho-ERK in IL-15–activated miR-155 tg NK cells compared with wt NK cells; both are important components of NK-cell survival and growth pathways, likely in response to endogenous IL-15.24 As a validation, we show that when endogenous IL-15 is increased, the in vivo expansion of NK cells in the double tg mice was significantly greater than the significant increase seen in either single tg model. If IL-15 does not directly control miR-155, then in vivo NK-cell homeostasis may also be regulated by the intrinsic expression of miR-155 in NK cells.
A clear and significant enrichment in the earliest stage of terminal NK-cell maturation, CD11blowCD27high, was discovered in miR-155 tg mice. Moreover, the excess persisted upon adoptive transfer into wt mice, consistent with a potential intrinsic block in the process of NK-cell differentiation being mediated by miR-155. The reason for this block is currently under investigation. Both SHIP1 knockout (KO) and the KO of the p110δ or the p110γ PI-3K isoforms result in altered NK-cell receptor expression.10,33 However, NK cells from the p110δ or p110γ PI-3K isoform KO mice have an immature CD11blowCD27high phenotype,10 suggesting that miR-155 overexpression, which decreases SHIP1 thereby enhancing PI-3K signaling, is mediating the halt in terminal NK-cell differentiation, at least in part, independently of the PI-3K pathway, possibly involving other molecules, for example, specific transcriptional factor(s) or even additional miRs.
Despite this partial block in terminal differentiation, miR-155 tg NK cells are fully functional in terms of cytokine production and cytotoxicity. The latter is, in part, mediated by NKG2D, one of the activating NK cell receptors involved in eliminating tumor and virus-infected cells.34 The enhanced killing by freshly harvested resting miR-155 tg NK cells clearly does not depend on enhanced granzyme B expression, which is curiously low in these cells at rest. A previous study regarding granzyme B–deficient NK cells indicated that granzyme B−/− NK cells are still able to lyse YAC-1 targets but less efficiently than wt NK cells in a chromium release assay.35 We therefore speculate that other granzymes may compensate for low expression of granzyme B in resting miR-155 tg NK cells. Alternatively, or additionally, we show enhanced activation of ERK upon effector–target conjugation, which could provide compensatory upregulation of granzyme B translation in miR-155 tg NK cells, similar to what was previously observed for IL-15–activated NK cells.36 In support of this, we show that granzyme B levels can be rescued by IL-2 activation in miR-155 tg NK cells. Thus, low expression of granzyme B may not depend on a direct effect of miR-155. To our knowledge, the 3′ UTR of granzyme B mRNA does not have binding sites for miR-155. miR-223 directly binds the 3′ UTR of granzyme B mRNA, and its expression is downregulated by IL-15 in NK cells.37 Whether miR-155 regulates granzyme B expression by altering this or other miRs remains to be investigated.
The enhanced killing of the lymphoma cell lines by miR-155 tg vs wt NK cells could be due to the significantly higher levels of NKG2D or CD94 receptors or the heightened state of activation (eg, CD69, higher ERK, and AKT activation) in miR-155 tg NK cells. Specifically, the enhanced killing can be explained by the witnessed increased activation of ERK that correlated with increased conjugate formation among miR-155 tg NK cells and tumor target cells.10,20 Indeed, the significantly greater number of NK tumor conjugates in miR-155 tg vs wt mice supports this notion. Our ability to translate these in vitro observations into successful control of lymphoma growth and improved survival in vivo by adoptively transferring miR-155 tg NK cells into immune-deficient hosts suggests that such adoptive therapy with NK cells engineered to overexpress miR-155 may have a role in diseases such as lymphoma or acute myeloid leukemia where NK-cell therapy has shown some efficacy.38
Despite lower CD16 expression, we show that ADCC is significantly enhanced in miR-155 tg NK cells compared with wt NK cells, possibly explained by data showing that direct downregulation of SHIP1 can enhance NK-cell ADCC.39 Likewise, the miR-155–mediated downregulation of SHIP1 can explain the enhanced induction of IFN-γ in miR-155 tg mice following monokine stimulation.15,19 In addition, STAT4, a direct transactivator of the interferon-γ gene promoter in NK cells after monokine stimulation,40 is higher in miR-155 NK cells compared with wt NK cells (not shown). It is currently unclear if STAT4 expression in miR-155 tg NK cells depends on reduced SHIP1 expression.
The downregulation of SHIP1 in miR-155 tg NK cells also correlates with an enhanced PI-3K and ERK pathway in these mice. Because SHIP1 normally targets the PI-3K lipid product phosphatidyl-inositol 3,4,5-trisphosphate (PI3,4,5P3),29 its suppression in miR-155 tg NK cells, in fact, leads to enhanced ERK activation.10,30,41 One aspect of miR-mediated gene regulation is that each specific miR is predicted to target hundreds of genes.42 As miR-155 has multiple targets, it is very likely that the mechanism and observed effects of miR-155 overexpression on NK-cell development, homeostasis, and function are not yet completely understood.
In summary, we show that the overexpression of miR-155 in NK cells positively regulates NK-cell expansion and effector functions despite an intrinsic block in terminal differentiation. The elucidation of factors that control these processes is important for host defense. The selective enhancement of miR-155 in NK cells via direct delivery of miR-155 oligonucleotides43 could improve NK-cell activation against challenging cancers or infections while avoiding the well-known toxicities that patients have encountered when undergoing systemic administration of NK-cell–activating cytokines such as IL-2 or IL-12.
Acknowledgments
The authors thank Paolo Neviani and Nicola Zanesi for useful discussions.
This work was supported, in part, by grants from the National Cancer Institute (CA124541, C.M.C.; CA16058, M.A.C.; CA95426, M.A.C.; and CA68458, M.A.C.). S.J. and E.L.B. have support from the Pelotonia Fellowship Program (Columbus, OH). B.L.M.-B. is supported by a National Institutes of Health training grant (T32-CA009338) (M.A.C). We thank Dr David H. Raulet (Berkeley, CA) for providing the RMA T-cell lymphoma tumor cells, and Rae1β-transduced RMA tumor cells.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.T. designed the study, performed research work, analyzed data, and wrote the manuscript; L.C., S.J., and D.C. performed experimental work and analyzed data. B.L.M.-B., A.M., and K.K.M. performed experiments. C.M. and E.L.B. performed cell-sorting experiments; L.Y. analyzed data; S.C. and C.M.C developed the miR-155 transgenic mice; and M.A.C. contributed to the design of the study and to the writing and editing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Rossana Trotta and Michael A. Caligiuri, The Ohio State University Comprehensive Cancer Center, 460 West 12th Ave, Columbus, OH 43210; e-mail: rossana.trotta@osumc.edu; michael.caligiuri@osumc.edu.
References
- 1.Caligiuri MA. Human natural killer cells. Blood. 2008;112(3):461–469. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7(9):703–714. doi: 10.1038/nri2154. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Iizuka K, Kang HS, Dokun A, French AR, Greco S, Yokoyama WM. In vivo developmental stages in murine natural killer cell maturation. Nat Immunol. 2002;3(6):523–528. doi: 10.1038/ni796. [DOI] [PubMed] [Google Scholar]
- 4.Vosshenrich CA, Samson-Villéger SI, Di Santo JP. Distinguishing features of developing natural killer cells. Curr Opin Immunol. 2005;17(2):151–158. doi: 10.1016/j.coi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113(22):5488–5496. doi: 10.1182/blood-2008-10-187179. [DOI] [PubMed] [Google Scholar]
- 6.Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol. 2006;176(3):1517–1524. doi: 10.4049/jimmunol.176.3.1517. [DOI] [PubMed] [Google Scholar]
- 7.Bezman NA, Cedars E, Steiner DF, Blelloch R, Hesslein DG, Lanier LL. Distinct requirements of microRNAs in NK cell activation, survival, and function. J Immunol. 2010;185(7):3835–3846. doi: 10.4049/jimmunol.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hesslein DG, Lanier LL. Transcriptional control of natural killer cell development and function. Adv Immunol. 2011;109:45–85. doi: 10.1016/B978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan RP, Leong JW, Schneider SE, Keppel CR, Germino E, French AR, Fehniger TA. MicroRNA-deficient NK cells exhibit decreased survival but enhanced function. J Immunol. 2012;188(7):3019–3030. doi: 10.4049/jimmunol.1102294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tassi I, Cella M, Gilfillan S, Turnbull I, Diacovo TG, Penninger JM, Colonna M. p110gamma and p110delta phosphoinositide 3-kinase signaling pathways synergize to control development and functions of murine NK cells. Immunity. 2007;27(2):214–227. doi: 10.1016/j.immuni.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 11.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol. 2008;9(8):839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 12.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28(5):264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 15.Trotta R, Chen L, Ciarlariello D, et al. miR-155 regulates IFN-γ production in natural killer cells. Blood. 2012;119(15):3478–3485. doi: 10.1182/blood-2011-12-398099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranganathan P, Heaphy CE, Costinean S, et al. Regulation of acute graft-versus-host disease by microRNA-155. Blood. 2012;119(20):4786–4797. doi: 10.1182/blood-2011-10-387522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaser BW, Schwind NR, Karol S, et al. Trans-presentation of donor-derived interleukin 15 is necessary for the rapid onset of acute graft-versus-host disease but not for graft-versus-tumor activity. Blood. 2006;108(7):2463–2469. doi: 10.1182/blood-2006-04-019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, Shores EW, Hu-Li J, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2(3):223–238. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 19.Trotta R, Parihar R, Yu J, et al. Differential expression of SHIP1 in CD56bright and CD56dim NK cells provides a molecular basis for distinct functional responses to monokine costimulation. Blood. 2005;105(8):3011–3018. doi: 10.1182/blood-2004-10-4072. [DOI] [PubMed] [Google Scholar]
- 20.Trotta R, Puorro KA, Paroli M, Azzoni L, Abebe B, Eisenlohr LC, Perussia B. Dependence of both spontaneous and antibody-dependent, granule exocytosis-mediated NK cell cytotoxicity on extracellular signal-regulated kinases. J Immunol. 1998;161(12):6648–6656. [PubMed] [Google Scholar]
- 21.Dong Z, Davidson D, Pérez-Quintero LA, Kurosaki T, Swat W, Veillette A. The adaptor SAP controls NK cell activation by regulating the enzymes Vav-1 and SHIP-1 and by enhancing conjugates with target cells. Immunity. 2012;36(6):974–985. doi: 10.1016/j.immuni.2012.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Trotta R, Vignudelli T, Candini O, et al. BCR/ABL activates mdm2 mRNA translation via the La antigen. Cancer Cell. 2003;3(2):145–160. doi: 10.1016/s1535-6108(03)00020-5. [DOI] [PubMed] [Google Scholar]
- 23.Cooper MA, Bush JE, Fehniger TA, et al. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood. 2002;100(10):3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- 24.Guimond M, Freud AG, Mao HC, et al. In vivo role of Flt3 ligand and dendritic cells in NK cell homeostasis. J Immunol. 2010;184(6):2769–2775. doi: 10.4049/jimmunol.0900685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy MK, Glaccum M, Brown SN, et al. Reversible defects in natural killer and memory CD8 T cell lineages in interleukin 15-deficient mice. J Exp Med. 2000;191(5):771–780. doi: 10.1084/jem.191.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fehniger TA, Suzuki K, Ponnappan A, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Wei M, Mao H, et al. CD94 defines phenotypically and functionally distinct mouse NK cell subsets. J Immunol. 2009;183(8):4968–4974. doi: 10.4049/jimmunol.0900907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohrschneider LR, Fuller JF, Wolf I, Liu Y, Lucas DM. Structure, function, and biology of SHIP proteins. Genes Dev. 2000;14(5):505–520. [PubMed] [Google Scholar]
- 30.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, Djeu JY. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol. 2000;1(5):419–425. doi: 10.1038/80859. [DOI] [PubMed] [Google Scholar]
- 31.Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. 2011;11(10):645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–429. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 33.Wang JW, Howson JM, Ghansah T, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295(5562):2094–2097. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- 34.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shresta S, MacIvor DM, Heusel JW, Russell JH, Ley TJ. Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc Natl Acad Sci USA. 1995;92(12):5679–5683. doi: 10.1073/pnas.92.12.5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity. 2007;26(6):798–811. doi: 10.1016/j.immuni.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Fehniger TA, Wylie T, Germino E, et al. Next-generation sequencing identifies the natural killer cell microRNA transcriptome. Genome Res. 2010;20(11):1590–1604. doi: 10.1101/gr.107995.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295(5562):2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 39.Galandrini R, Tassi I, Mattia G, Lenti L, Piccoli M, Frati L, Santoni A. SH2-containing inositol phosphatase (SHIP-1) transiently translocates to raft domains and modulates CD16-mediated cytotoxicity in human NK cells. Blood. 2002;100(13):4581–4589. doi: 10.1182/blood-2002-04-1058. [DOI] [PubMed] [Google Scholar]
- 40.Thierfelder WE, van Deursen JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382(6587):171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 41.Parihar R, Trotta R, Roda JM, Ferketich AK, Tridandapani S, Caligiuri MA, Carson WE., III Src homology 2-containing inositol 5′-phosphatase 1 negatively regulates IFN-gamma production by natural killer cells stimulated with antibody-coated tumor cells and interleukin-12. Cancer Res. 2005;65(19):9099–9107. doi: 10.1158/0008-5472.CAN-04-4424. [DOI] [PubMed] [Google Scholar]
- 42.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010;18(9):1650–1656. doi: 10.1038/mt.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]