

Key Points
A new molecular pathway involving the MASL1 gene during erythroid differentiation has been identified.
Abstract
Human erythropoiesis is a dynamic and complex multistep process involving differentiation of early erythroid progenitors into enucleated RBCs. The mechanisms underlying erythropoiesis still remain incompletely understood. We previously demonstrated that erythropoietin-stimulated clone-1, which is selectively expressed in normal human erythroid-lineage cells, shares 99.5% identity with malignant fibrous histiocytoma-amplified sequences with leucine-rich tandem repeats 1 (MASL1). In this study, we hypothesized that the MASL1 gene plays a role in erythroid differentiation, and used a human erythroid cell culture system to explore this concept. MASL1 mRNA and protein expression levels were significantly increased during the erythroid differentiation of CD34+ cells following erythropoietin (EPO) treatment. Conversely, MASL1 knockdown reduced erythroid differentiation in EPO-treated CD34+ cells. In addition, MASL1 knockdown interrupted the Raf/MEK/ERK signaling pathway in CD34+ cells. MASL1 mutant-transfected CD34+ cells also showed decreased erythroid differentiation. Furthermore, inhibition of the SH3 domain of Son of Sevenless, which is an upstream adapter protein in EPO-induced erythroid differentiation, also reduced MASL1 expression and phosphorylation of Raf/MEK/ERK kinases that consequently reduced erythroid differentiation of EPO-induced CD34+ cells. Importantly, we also demonstrated that MASL1 interacts physically with Raf1. Taken together, our data provide novel insights into MASL1 regulation of erythropoiesis through the Raf/MEK/ERK pathway.
Introduction
Differentiation of hematopoietic stem cells into mature blood cells involves lineage-specific activation and restriction of gene expression.1 Lineage-specific transcription factors play essential roles in RBC development. The zinc-finger transcription factor GATA-1, a central mediator of erythroid gene expression, interacts with multiple proteins, including Friend of GATA 1, Erythroid Krüppel-like Factor, SP1, CREB binding protein/E1A binding protein p300, and PU.1.2 The mechanisms by which these interactions influence GATA-1 function, as well as any possible relationships between these seemingly disparate complexes, remain incompletely understood. However, several new findings have provided further insight into their role in erythropoiesis.
The Ras/Raf/MEK/ERK signaling cascade is one of the key signaling pathways involved in erythropoiesis.3,4 In addition, oncogenic Ras leads to the constitutive activation of its downstream signaling pathways, a severe block of terminal erythroid differentiation, and cytokine-independent growth of primary erythroid progenitors.5 Deregulated erythropoiesis in polycythemia vera involves erythropoietin (EPO) hypersensitivity and apoptosis resistance of erythroid precursor cells, both of which are associated with abnormally increased activation of the Ras-ERK and phosphatidylinositol 3-kinase (PI3K)-AKT pathways.6 However, the role of Ras-GTPases in hematopoiesis and leukemogenesis is not completely known.
We have previously identified some potentially novel genes associated with hematopoietic-lineage commitment and differentiation.7 One of these, erythropoietin-stimulated clone-1, is selectively expressed in normal erythroid-lineage cells and shares 99.5% identity with malignant fibrous histiocytoma-amplified sequences with leucine-rich tandem repeats 1 (MASL1 or MFHAS1). This novel MASL1 gene was identified as a candidate oncogene from the genomic amplification at 8p23.1 observed in malignant fibrous histiocytoma.8 Amplification of 8p23 has also been found in a few solid tumors, such as gastric cancer,9 whereas genomic loss of chromosomal region 8p23 occurs frequently in leukemic mantle cell lymphoma.10 The primary structure of its deduced products shows a Ras-like GTPase, 3 leucine zipper domains, and a leucine-rich tandem repeat. These domains are all important structural or functional elements for interactions among proteins related to the cell cycle. Because of a lack of knowledge about the function of MASL1, the role and mechanisms of MASL1 in erythropoiesis still remain unknown. Here, we investigated the role of MASL1 in normal erythroid differentiation of human hematopoietic progenitor cells (CD34+ cells). Our data provide evidence for a novel mechanism of MASL1 action in erythropoiesis in which it activates erythroid differentiation through the Raf/MEK/ERK pathway.
Materials and methods
Cell culture and transfection
Primary human CD34+ cells were isolated by positive immunoselection from peripheral blood mononuclear cells harvested by leukapheresis after recombinant human granulocyte colony-stimulating factor injection under a protocol approved by the National Institute of Diabetes and Digestive and Kidney Diseases Institutional Review Board. All human participants provided written informed consent in accordance with the Declaration of Helsinki.11,12 In some instances, primary human CD34+ cells were obtained from commercial sources (Lonza, Walkerville, MD, or AllCells, Emeryville, CA). Cells were thawed and washed into StemSpan serum-free expansion medium (SFEM) (StemCell Technologies, Vancouver, BC, Canada) and then seeded in StemSpan SFEM containing 1× CC100 cytokine mix (StemCell Technologies) and 2% penicillin/streptomycin (Invitrogen, Carlsbad, CA). Cells were maintained in this expansion medium at a density of 0.1 to 1 × 106 cells/mL in a 5% CO2 atmosphere at 37°C for 6 days and then were induced with 4 U/mL EPO (Amgen, Thousand Oaks, CA) for 14 days. Cells were harvested at day 3, 5, 7, 10, and 14 of differentiation for MASL1 mRNA and protein expression profile studies.
For transfection studies, CD34+ cells were transfected with 10 nM of MASL1 siRNA (Dharmacon, Chicago, IL) using HiPerFect Transfection Reagent (Qiagen, Valencia, CA) or 1 µg of MASL1 shRNA or control shRNA plasmid DNA (Santa Cruz Biotechnology, Santa Cruz, CA) using Effectene Transfection Reagent (Qiagen) according to the manufacturer’s protocol after a 6-day period of expansion. In mock transfections, no siRNA or shRNA plasmid was included in the transfection reaction. Following transfection, cells were maintained in α Minimum Essential Medium (Sigma-Aldrich, St. Louis, MO) supplemented with 30% fetal bovine serum (Invitrogen), 1% bovine serum albumin (Sigma-Aldrich), 10 µM β-mercaptoethanol (Sigma-Aldrich), 1 µM dexamethasone (Sigma-Aldrich), 0.3 mg/mL holotransferrin (Sigma-Aldrich), 1× penicillin/streptomycin/glutamine (Invitrogen), and 4 U/mL EPO for an additional 14 days. Twenty-four hours after transfection (day 1 of differentiation), 0.5 µg/mL puromycin (Santa Cruz Biotechnology) was added to the medium for stable transfection. Cells were collected at day 3, 5, 7, 10, and 14 of differentiation for further analysis.
Cell counts and morphology
Cell counts were determined on an ICS standard 25 microscope (Zeiss, Thornwood, NY) equipped with a hemacytometer. Cells were spun for 2 minutes at 2000 rpm onto glass slides using a cytospin apparatus (Thermo Scientific, Waltham, MA). After air-drying for 1 minute, slides were fixed in methanol for 5 minutes and were stained with May-Grünwald and Giemsa stains (Sigma-Aldrich) following the manufacturer’s instructions. Stained cells were viewed with an Olympus BX51 microscope (20×) and QCapture Pro 6.0 (Olympus America Inc., Center Valley, PA).
Flow cytometry and cell-cycle, apoptosis, and cell-proliferation analyses
To evaluate transferrin receptor (CD71) and glycophorin A (GPA) expression, 5 × 105 CD34+ cells were fixed in 0.3% formaldehyde for 15 minutes and then were labeled with fluorescein isothiocyanate (FITC)-conjugated anti-CD71 and phycoerythrin (PE)-conjugated anti-GPA antibodies (BD Bioscience/Pharmingen, San Diego, CA) for 30 minutes. For cell-cycle analysis, cells were fixed in 70% ethanol in PBS and were then stained with 20 µg/mL propidium iodide (PI; Sigma-Aldrich) in 0.1% Triton X-100 and 200 µg/mL DNase-free RNase A (Sigma-Aldrich) for 30 minutes at room temperature. For apoptosis analysis, cells were costained with FITC-annexin V (BD Bioscience/Pharmingen) and PI (BD Bioscience/Pharmingen) according to the manufacturer’s protocol. Labeled cells were analyzed for fluorescence emission using a FACScalibur (BD Bioscience) and Cell Quest Pro software (BD Bioscience/Pharmingen) for acquisition and analysis. Unstained fixed cells and FITC- and PE-conjugated mouse IgG isotype controls were used as negative controls. Cell proliferation was analyzed using the 3-(4,5-dimethythizolyl-2)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to the manufacturer’s protocol (ATCC, Manassas, VA).
7-amino-actinomycin D (7-AAD) staining
Aliquots of 1 × 106 CD34+ cells were stained with FITC-conjugated anti-CD71 and PE-conjugated anti-GPA for 30 minutes on ice. After washing, the cells were further stained with 7-AAD solution (BD Bioscience/Pharmingen) according to the manufacturer’s protocol. The cells were then washed and resuspended in 500 μL 2% paraformaldehyde solution (Sigma-Aldrich). Unstained fixed cells and FITC- and PE-conjugated mouse IgG isotype controls were used as negative controls. Samples were analyzed by flow cytometry on 50 000 events using 5 parameters: forward scatter, right-angle side scatter, and triple-color immunofluorescence from FITC, PE, and 7-AAD by a FACScalibur within 30 minutes of fixation.
Glutathione-S-transferase (GST) pull-down assays
293T cells (ATCC) were grown in 10-cm-diameter culture dishes and were then transfected with 24 μg of myc-DDK-tagged MASL1-pCMV6 Entry plasmid or control pCMV6 Entry plasmid (Origene Technologies, Rockville, MD) with Lipofectamine 2000 (Invitrogen) by following the manufacturer’s protocol. In mock transfections, no plasmid was included in the transfection reaction. Cells were rinsed once with ice-cold TBS and then were lysed 48 hours after transfection on the culture plates with 500 μL of lysis/binding/wash buffer (25 mM Tris-HCl pH 6.8, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 5% glycerol) supplemented with protease inhibitor cocktail (Roche Applied Science). GST pull-down assays were performed using the Active Ras Pull-down and Detection kit (Thermo Scientific). Total protein lysates (500 μg) were incubated with 80 μg of GST-Raf1-RBD coupled to glutathione resin overnight at 4°C. GDP- or GTPγS-treated lysates were incubated with GST-Raf1-RBD and glutathione resin as a negative control for inactivated-MASL1 and a positive control for activated-MASL1, respectively. GTPγS-treated lysate was also incubated with GST alone in the presence of glutathione resin as an additional negative control. For each sample, the resins were centrifuged at 6000 × g for 30 seconds in a microcentrifuge at 4°C and the supernatants removed. The resins were washed 3 times with 500 μL of lysis/binding/wash buffer, resuspended in 50 μL of 2× reducing sodium dodecyl sulfate sample buffer, and boiled for 5 minutes. Each elution was analyzed by 4% to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting using anti-MASL1 antibody. Pull-down experiments were also performed using total lysates of CD34+ cells treated with 4 U/mL EPO or 50 ng/mL granulocyte-colony stimulating factor (G-CSF) (PeproTech, Rocky Hill, NJ) for 14 days.13
Statistical analysis
Data were expressed as mean ± standard deviation (SD). Results were analyzed by Student t test for comparison between 2 independent groups. P < .05 was considered to be statistically significant.
Results
MASL1 expression is upregulated during primary human CD34+ cell erythroid differentiation and is an abundant protein in human RBCs
Previously, we demonstrated that MASL1 is selectively expressed in the erythroid-lineage cell population.7 To determine how MASL1 is expressed during normal erythroid differentiation, we first analyzed MASL1 expression patterns in K562 cells that were differentiated by Ara-C treatment. The expression of MASL1 mRNA levels was significantly increased in K562 cells after 4 days of Ara-C treatment (P = .009) (data not shown). Next, we employed a serum-free in vitro erythrocytic differentiation protocol for primary human hematopoietic progenitor cells (CD34+ cells).14 Primary human CD34+ cells were expanded for 6 days and then induced to differentiate by EPO treatment during a 14-day period. The harvested cell pellet showed a red color, and the cells’ morphology was consistent with that of mature erythrocytes (data not shown). MASL1 mRNA levels were detectable starting at day 5 of differentiation in EPO-treated CD34+ cells and then exponentially rose from day 7 through 14 of differentiation by semiquantitative RT-PCR and qRT-PCR (Figures 1A-B). Western-blot analysis demonstrated that MASL1 protein levels increased during EPO-induced CD34+ cell differentiation through day 14 in parallel to its mRNA expression pattern. The increase in the expression of MASL1 mirrored the rise in hemoglobin-α accumulation and GPA protein levels (Figure 1C). Furthermore, we found significantly increased levels of MASL1 in human RBCs compared to EPO-induced CD34+ cells (Figures 1D-E). Taken together, these data indicate that MASL1 expression is upregulated during normal erythroid differentiation and is abundantly expressed in human RBCs.
Figure 1.

MASL1 expression is upregulated during primary human CD34+ cell erythroid differentiation and is an abundant protein in human red blood cells (RBCs). Human peripheral blood CD34+ cells were expanded for 6 days and were then induced to differentiate by EPO treatment during a 14-day period. (A) MASL1 gene expression was examined in CD34+ cells at day 0, 3, 5, 7, 10, and 14 after EPO treatment by semiquantitative RT-PCR. GAPDH was used as an internal control. (B) Mean relative MASL1 expression levels shown as fold induction compared with levels in CD34+ cells at day 0 by qRT-PCR. Values were normalized to the expression level of the housekeeping gene GAPDH. Error bars represent the SD from 3 individual experiments; *P < .05; **P < .01. (C) Western-blot analysis of protein lysates of EPO-induced CD34+ cells using anti-MASL1, hemoglobin-α, and GPA antibodies. β-actin was used as an internal control. (D) Western-blot analysis of protein lysates prepared from CD34+ cells at day 14 of EPO-induced differentiation (EPO Day 14) and human RBCs. GAPDH was used as an internal control. (E) Mean relative MASL1 protein expression level normalized to GAPDH. Error bars represent the SD from 3 individual experiments; *P < .05.
Knockdown of MASL1 markedly reduces erythroid differentiation in human erythroid progenitor CD34+ cells but does not affect granulocytic differentiation
After demonstrating that MASL1 was induced during CD34+ cell differentiation after EPO treatment, we next asked whether the downregulation of MASL1 could reduce the differentiation of erythroid cells. To assess this, we expanded CD34+ cells in cytokine cocktail-containing SFEM for 6 days, then transfected them with a MASL1 siRNA or shRNA plasmid to knock down MASL1 expression and examined their differentiation profiles. Fourteen days after induction with EPO, the cell pellet of MASL1-knockdown CD34+ cells did not demonstrate a red color when compared to mock- or control shRNA-transfected CD34+ cells (Figure 2A). The total cell counts remained relatively low during the 14-day differentiation period in MASL1-knockdown CD34+ cells. In contrast, we observed a more rapid, significant increase in cell numbers on day 14 of differentiation in mock- and control shRNA-transfected CD34+ cells (Figure 2B). Semiquantitative RT-PCR and qRT-PCR of CD34+ cells after 14 days of EPO induction demonstrated that the expression levels of MASL1 mRNA were significantly suppressed by MASL1 siRNA or shRNA knockdown (Figures 2C-D). Western-blot analysis demonstrated that MASL1 protein levels in CD34+ cells at day 14 of EPO induction were also dramatically decreased after MASL1 knockdown. Similarly, MASL1-knockdown CD34+ cells showed decreased levels of hemoglobin protein, indicating that erythroid differentiation was reduced (Figure 2E).
Figure 2.

Knockdown of MASL1 reduces erythroid differentiation in human erythroid progenitor CD34+ cells. (A) Cell pellets at day 14 of EPO-induced erythroid differentiation for mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells. Induced erythroid differentiation is evident by the pink-red cell pellets. (B) Cell counts per mL in culture at day 0, 7, and 14 of EPO-induced erythroid differentiation for mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells. Error bars represent the SD from 3 individual experiments; *P < .05; **P < .01. (C) Semiquantitative RT-PCR of MASL1 mRNA expression in mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells at day 14 of EPO-induced differentiation. GAPDH was used as an internal control. (D) qRT-PCR analysis of MASL1 gene expression in mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells. Mean relative MASL1 expression levels shown as fold induction compared with levels in mock-transfected CD34+ cells at day 14 of EPO-induced differentiation. Values were normalized to the expression level of the housekeeping gene GAPDH. Error bars represent the SD from 3 individual experiments; *P < .05; **P < .01. (E) Western-blot analysis of protein lysates prepared from mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells at day 14 of EPO-induced differentiation. β-actin was used as an internal control. (F) Flow cytometry analysis of CD71+ and GPA+ expression in mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells at day 14 of EPO-induced differentiation. Error bars represent the SD from 3 individual experiments; *P < .05; **P < .01. (G) (Top), representative density plots for data presented in F. CD34+ cells were stained with FITC-conjugated anti-CD71 and PE-conjugated anti-GPA monoclonal antibodies. The percent of CD71+ and GPA+ cells is labeled on each density plot. (Bottom), morphology of cells corresponding to flow-cytometric analysis obtained by May-Grünwald-Giemsa staining (original magnification ×20). Results are representative of 3 independent experiments.
Having demonstrated that MASL1 knockdown is probably involved in the reduction of erythroid differentiation, we next measured the erythroid differentiation marker, CD71 and GPA, in CD34+ cells with and without MASL1 knockdown treated with EPO by flow cytometry analysis. Knockdown of MASL1 expression exerted a significant inhibitory effect on erythroid differentiation; only 24.1 ± 14.7% and 11.5 ± 5.7% of MASL1 siRNA and shRNA-transfected CD34+ cells at day 14 of EPO induction were CD71+ GPA+, respectively. In contrast, mock- and control shRNA-transfected CD34+ cells were 70.0 ± 6.0% and 65.4 ± 8.2% double positive for CD71 and GPA expression, respectively (Figures 2F-G, upper panels). In addition, May-Grünwald-Giemsa staining confirmed that after 14 days of culture in the presence of EPO, most MASL1-knockdown CD34+ cells were nonerythroid or proerythroblast, whereas mock- or control shRNA-transfected CD34+ cells were orthochromatic erythroblasts (Figure 2G, lower panels). Furthermore, nonerythroid cells were also increased in MASL1-knockdown CD34+ cells at day 3 (macrophage precursor and/or myeloid precursor cells), day 5 (macrophage precursor and/or myeloid precursor cells), day 7 (early monocytic or myeloid cells), and day 14 (monocytes and/or macrophages and/or myeloid cells) of EPO-induced differentiation by differential counting (Figure 3).
Figure 3.
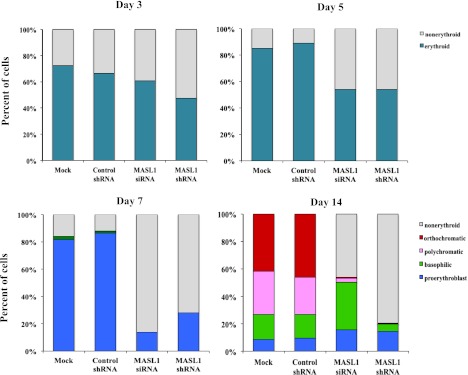
Nonerythroid cells accumulate in MASL1-knockdown CD34+ cells following EPO-induced erythroid differentiation. A hematopathologist, blinded to experimental details, reviewed all coded slides and quantitated myeloid and erythroid cells at day 3, 5, 7, and 14 of EPO-induced erythroid differentiation in mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD34+ cells. More than 200 cells were scored for each sample in triplicate individual experiments. The data represent the mean values of 3 independent experiments.
We also found that erythroid differentiation was reduced in MASL1-knockdown CD34+ cells at day 3 of EPO induction (supplemental Figure 1). To investigate further whether MASL1 is lineage-specific for erythroid differentiation, we also performed MASL1 knockdown in CD34+ cells treated with G-CSF to examine whether MASL1 may be involved in granulocytic differentiation. MASL1 knockdown did not significantly alter cell proliferation during granulocytic differentiation (supplemental Figure 2A). Notably, MASL1-knockdown CD34+ cells and mock- and control shRNA-transfected CD34+ cells were not significantly different in the percentage of CD13+ CD66b+ cells and apoptotic cells observed during granulocytic maturation (supplemental Figures 2B-C). Consistently, the phenotype of MASL1-knockdown CD34+ cells did not differ substantially from that of mock- or control shRNA-transfected CD34+ cells at day 3 and 7 of G-CSF-induced differentiation (supplemental Figure 2D). In addition, there was no change in phosphorylation of Raf/MEK/ERK signaling pathway in MASL1-knockdown CD34+ cells treated with G-CSF (supplemental Figure 2E). Based on these collective data, we conclude that knockdown of MASL1 causes a reduction of erythroid differentiation in CD34+ cells but had no effect on granulocytic differentiation.
Knockdown of MASL1 reduces cell proliferation and induces cell-cycle arrest in CD34+ cells but does not alter cell survival in CD71+ GPA- or CD71+ GPA+ subpopulations of CD34+ cells during erythroid differentiation
The underlying mechanisms responsible for reduced cell expansion in MASL1-knockdown CD34+ cells can be reduced proliferation and/or accelerated cell death. We first determined the cell proliferation rate of MASL1-knockdown CD34+ cells by the MTT assay. We found a significant reduction in proliferation for the MASL1-knockdown CD34+ cells compared with mock- and control shRNA-transfected cells at day 7 of EPO-induced differentiation (supplemental Figure 3A). No significant differences in the proliferation rate were observed at day 10 and 14. Next, we assessed induction of apoptosis by measuring annexin V binding and uptake of PI in MASL1-knockdown CD34+ cells undergoing EPO-induced erythroid differentiation in total CD34+ cell populations. We found an increase in early apoptotic cells (annexin V-positive/PI-negative), starting on day 3 (22.0 ± 7.3% for MASL1 siRNA and 24.9 ± 8.8% for MASL1 shRNA) and increasing further on day 5 (37.4 ± 1.2% for MASL1 siRNA and 37.5 ± 1.9% for MASL1 shRNA), in MASL1-knockdown CD34+ cells when compared with mock- (18.4 ± 2.2% for day 3 and 10.1 ± 1.3% for day 5) or control shRNA-transfected (19.2 ± 4.0% for day 3 and 10.2 ± 1.6% for day 5) CD34+ cells. Consistently, a significant increase in the percentage of cells that were annexin V-positive/PI-negative (early apoptotic) was observed for MASL1-knockdown CD34+ cells compared with control-transfected CD34+ cells at day 7 of differentiation (35.0 ± 1.0% in MASL1 siRNA- and 36.0 ± 2.1% in MASL1 shRNA-transfected cells vs 11.7 ± 3.5% in mock- and 11.3 ± 2.4% in control shRNA-transfected cells). However, compared to control-transfected cells, the percentage of cells that were annexin V-positive/PI-positive (late apoptotic) was not significantly increased in MASL1-knockdown CD34+ cells at day 7 of erythroid differentiation (17.1 ± 7.0% in MASL1 siRNA- and 18.4 ± 6.4% in MASL1 shRNA-transfected cells vs 7.6 ± 1.9% in mock- and 7.2 ± 2.1% in control shRNA-transfected cells), which suggests that apoptosis was increased by MASL1 knockdown (supplemental Figures 3B-C). At day 14 of EPO-induced differentiation, no significant difference in annexin V-positive/PI-negative (early apoptotic) or annexin V-positive/PI-positive (late apoptotic) levels was observed in MASL1-knockdown CD34+ cells compared with mock- or control shRNA-transfected CD34+ cells (P = .32) (supplemental Figure 3C).
To determine whether increased apoptosis is characterized in the total CD34+ cell population or is a feature of EPO-dependent erythroid cells, we further evaluated the survival characteristic of CD71+ GPA- cells at day 3 or CD71+ GPA+ cells at day 7 and 14 of differentiation using 7-AAD staining. Interestingly, MASL1-knockdown and mock- and control shRNA-transfected CD34+ cells were not significantly different in the percentage of apoptotic cells detected within the CD71+ GPA- cell fraction at day 3 (4.9 ± 6.4% in MASL1 siRNA- and 4.9 ± 6.3% in MASL1 shRNA-transfected cells vs 6.3 ± 7.1% in mock- and 7.4 ± 9.4% in control shRNA-transfected cells) (Figures 4A-C), or within the CD71+ GPA+ cell fraction at day 7 (P = .19) and 14 (P = .22) of differentiation (data not shown).
Figure 4.

MASL1 knockdown in CD34+ cells does not alter cell survival within the CD71+ GPA- subpopulation at day 3 of EPO-induced erythroid differentiation but causes cell-cycle arrest in all CD34+ cells during erythroid differentiation. (A) Mean percentages from flow cytometry analysis of 7-AAD- (viable), 7-AADdim (early apoptotic), and 7-AADbright (late apoptotic) in mock-, control shRNA-, MASL1 siRNA-, or MASL1 shRNA-transfected CD71+GPA- subpopulation cells at day 3 of EPO-induced differentiation. Error bars represent the SD from 3 individual experiments. (B) Scattergram of forward scatter (FSC) vs right-angle side scatter (SSC), to allow gating on CD34+ cells by excluding cell debris (R1). (C) Representative scattergram for mock-, control shRNA-, and MASL1-knockdown CD34+ cells of SSC vs anti-CD71 fluorescence gated on R1 to allow gating on CD71- (R2) or CD71+ (R3). (Middle) representative scattergram for mock-, control shRNA-, and MASL1-knockdown CD34+ cells of SSC vs anti-GPA fluorescence gated on R3 to allow gating on GPA- (R4) or GPA+ (R5) and lower panel is representative scattergram of FSC vs 7-AAD fluorescence gated on R4, showing 7-AADbright (late apoptotic), 7-AADdim (early apoptotic), and 7-AAD- (viable) within CD71+ GPA- cells on day 3 of differentiation. (D) Cell-cycle distribution of mock-, control shRNA-, and MASL1-knockdown CD34+ cells at day 3, 5, 7, and 14 of EPO-induced differentiation analyzed by propidium iodide staining and flow cytometry. The data are expressed as mean percentage of sub G0/G1, G0/G1, S, and G2/M phase cells. Error bars represent the SD from 3 independent experiments; *P < .05. (E) Representative histogram for data presented in D.
Cell-cycle analyses indicated that MASL1-knockdown CD34+ cells significantly accumulated in the G0/G1 phase of the cell cycle starting at day 3 and then began decreasing on day 7 of EPO-induced differentiation in total CD34+ cell populations. In addition, the percentage of MASL1-knockdown CD34+ cells in S-phase was significantly decreased at day 7 compared with mock- and control shRNA-transfected CD34+ cells (7.8 ± 2.5% in MASL1 siRNA- and 8.2 ± 1.7% in MASL1 shRNA-transfected cells vs 34.6 ± 4.2% in mock- and 33.8 ± 4.1% in control shRNA-transfected cells). The substantial population of control-transfected CD34+ cells in S-phase at day 7 of differentiation indicates that these cells continue to cycle (Figures 4D-E). The significant increase in the percentage of MASL1-knockdown CD34+ cells in sub G0/G1 phase compared with mock- and control shRNA-transfected cells correlated with the apoptotic analysis. No significant effect of MASL1 knockdown on cell cycle at day 14 of erythroid differentiation was observed (data not shown). These results suggest that downregulation of MASL1 expression reduced cell proliferation and caused cell-cycle arrest in total CD34+ cell populations, but did not alter cell survival in EPO-dependent erythroid cells, during erythroid differentiation.
MASL1 activates erythroid differentiation through the Raf/MEK/ERK pathway and Erk1/2 phosphorylation in MASL1-knockdown CD34+ cells is diminished in the absence of EPO and is induced by restimulation with EPO
It has been reported that several signaling pathways, including JAK/STAT, PI3K/AKT, and Ras/Raf/MEK/ERK, are involved in EPO-induced erythropoiesis.15 One of the important domains in the MASL1 protein structure that has recently been described is the Ras-like GTPase and C-terminal of Roc (ROCO).16 Therefore, we hypothesized that MASL1 could induce erythroid differentiation via the Raf/MEK/ERK pathway similar to Ras-GTPase proteins. To test this hypothesis, we transfected CD34+ cells with MASL1 siRNA or shRNA plasmid and then grew the cells in EPO-containing medium. Cells were harvested at various times, and lysates were subjected to western blotting. Interestingly, the phosphorylation of Raf/MEK/ERK signal effectors, including Raf1, Mek1/2, Erk1/2, and Elk1, was decreased in MASL1-knockdown CD34+ cells compared with mock- or control shRNA-transfected CD34+ cells at day 3 (supplemental Figure 1A) as well as at day 14 of differentiation (Figure 2E). In addition, we also detected a substantial decrease in hemoglobin-α protein levels in MASL1-knockdown CD34+ cells (supplemental Figures 1A and 2E). In contrast, we failed to detect any change in JAK/STAT or PI3K/AKT phosphorylation at day 14 of differentiation in MASL1-knockdown CD34+ cells (supplemental Figure 4).
Finally, we found that phosphorylation of Erk1/2 was further reduced when MASL1-knockdown CD34+ cells were EPO-starved for 4 hours; however, hyperstimulation of these cells with 10 U/mL EPO for 10 minutes was sufficient to restore the level of Erk1/2 phosphorylation to that observed before starvation (supplemental Figure 5A). There were no changes in phosphorylation of Akt or Stat5 level in MASL1-knockdown CD34+ cells compared to control groups after EPO starvation and restimulation with EPO (supplemental Figure 5A). To explore further the effects of EPO on Erk1/2, Akt, and Stat5 phosphorylation in CD34+ cells, we transfected CD34+ cells with control shRNA, MASL1 siRNA, or MASL1 shRNA in medium without EPO and grew the cells for 2 days in the absence of EPO. We then stimulated the cells with EPO for 15 or 30 minutes and examined the phosphorylation of Erk1/2, Akt, and Stat5. Akt and Stat5 phosphorylation pattern in this model was similar for MASL1-knockdown CD34+ cells and mock- or control shRNA-transfected CD34+ cells, whereas phosphorylation of Erk1/2 following EPO stimulation was decreased for MASL1-knockdown CD34+ cells compared to mock- or control-transfected CD34+ cells (supplemental Figure 5B). These observations indicate that MASL1 may affect erythroid differentiation through the Raf/MEK/ERK pathway and that this effect may depend on EPO induction.
Alteration of Ras-like GTPase region in MASL1 reduces erythroid differentiation in CD34+ cells
To verify that MASL1 is involved in erythroid differentiation through the Raf/MEK/ERK pathway via its Ras-like GTPase region, we generated a point mutation (MASL1 S450A) and a deletion of the Ras-like GTPase region of MASL1 (MASL1 Δ414-556) in myc-DDK-tagged MASL1 pCMV6-Entry plasmid vector using site-directed mutagenesis (supplemental Figure 6A), and then we transfected individually these 2 constructs into CD34+ cells to determine their effects on erythroid differentiation. Interestingly, we found smaller pink cell pellets in myc-DDK-tagged MASL1 S450A- or MASL1 Δ414-556-transfected CD34+ cells when compared with mock-, control vector-, or myc-DDK-tagged MASL1-transfected CD34+ cells (supplemental Figure 6B). Myc-DDK-tagged MASL1 S450A- or MASL1 Δ414-556-transfected CD34+ cells were only 50.6 ± 2.4% and 52.7 ± 3.6% double positive for CD71 and GPA expression using flow cytometry, respectively (supplemental Figures 6C-D, upper panels). Furthermore, May-Grünwald-Giemsa staining confirmed that more of the myc-DDK-tagged MASL1 S450A- or MASL1 Δ414-556-transfected CD34+ cells were pro- or basophilic erythroblasts compared to the mock-, control vector-, or myc-DDK-tagged MASL1-transfected CD34+ cells, where most cells were polychromatic or orthochromatic erythroblasts (supplemental Figure 6D, lower panels). In addition, the phosphorylation of Raf/MEK/ERK signaling effectors was diminished in myc-DDK-tagged MASL1 S450A- or MASL1 Δ414-556-transfected CD34+ cells (supplemental Figure 6E). These data confirm that MASL1 is involved in erythroid differentiation through the Raf/MEK/ERK pathway.
Inhibition of SOS activity reduces MASL1 expression and diminishes erythroid differentiation
Son of Sevenless (SOS) is an adapter protein that binds to a growth factor receptor bound protein-2 (GRB2) to activate the exchange of GDP to GTP on Ras guanine nucleotide-binding protein after EPO interacts with EPO receptor in the Ras/Raf/MEK/ERK pathway involving erythropoiesis.4,17 Selective disruption of the GRB2/SOS association has been considered an attractive mechanism for upstream control of the Ras/Raf/MEK/ERK pathway.18 To assess further the role of MASL1 in erythroid differentiation via the Raf/MEK/ERK pathway, we inhibited GRB2/SOS interaction in CD34+ cells at day 0 of EPO-induced differentiation by adding an SOS SH3 domain inhibitor (and replacing the inhibitor every 48 hours). CD34+ cells treated with Jak2 or PI3K inhibitors were used as internal controls. As expected, semiquantitative RT-PCR and Western-blot analysis demonstrated that SOS mRNA and protein levels were reduced at day 7 of EPO-induced erythroid differentiation in the presence of SOS SH3 domain inhibitor (supplemental Figures 7A-C). These results implied that the SOS SH3 domain inhibitor inhibited GRB2/SOS interaction. Interestingly, we also observed a significant decrease in MASL1 mRNA and protein levels in EPO-induced CD34+ cells treated with Jak2 or SOS SH3 domain inhibitor, but little or no change in MASL1 expression in EPO-induced CD34+ cells treated with PI3K inhibitors (supplemental Figures 7A-C). Moreover, we also detected a reduction of the phosphorylation of Raf1, Mek 1/2 and Erk 1/2. (supplemental Figure 7C).
Next, we investigated further the effects of SOS SH3 domain inhibitor on EPO-dependent differentiation in CD34+ cells. According to Wojda et al (2002),19 the S-phase of the cell cycle peaks on day 8 during the highly proliferative stage of erythroid differentiation. Therefore, we examined the cell-cycle, erythroid-differentiation, and cell-morphology profiles at day 7 of EPO-induced differentiation in CD34+ cells. The percent of CD34+ cells cultured in the presence of the SOS SH3 domain inhibitor and EPO that were in S-phase was significantly less than that of CD34+ cells treated with EPO only (P = .04) (supplemental Figure 7D). Only 25.7 ± 13.8% of the CD34+ cells treated with the SOS SH3 domain inhibitor and EPO were CD71+ GPA+, compared with 50.2 ± 10.3% of CD34+ cells treated with EPO alone (supplemental Figures 7E-F, upper panels). Most of the CD34+ cells treated with EPO and SOS SH3 domain inhibitor displayed a morphology characteristic of undifferentiated erythroid cells by May-Grünwald-Giemsa staining, similar to the CD34+ cells without EPO treatment. In contrast, we observed some proerythroblasts and a few basophilic normoblasts in EPO-induced CD34+ cells (supplemental Figure 7F, lower panels). Compared to CD34+ cells treated with EPO alone, we also detected significantly lower percentages of S-phase (P = .04) and CD71+ GPA+ cells (P = .01) in Jak2 inhibitor- or PI3K inhibitor-treated CD34+ cells grown in the presence of EPO for 7 days, which corresponded to their undifferentiated erythroid phenotype observed by May-Grünwald-Giemsa staining (supplemental Figures 7D-F). Taken together, the results of the SOS-inhibitor studies are consistent with the finding that MASL1 stimulates erythroid differentiation through the Raf/MEK/ERK pathway.
MASL1 interacts physically with Raf1
The reduction of the Raf/MEK/ERK pathway in MASL1-knockdown or MASL1 mutant-transfected CD34+ cells during EPO-induced differentiation suggests that MASL1 may function in regulating erythroid differentiation through the Raf/MEK/ERK pathway. To explore this possibility further, we ectopically expressed myc-DDK-tagged MASL1 in 293T cells followed by pull-down experiments with GST-Raf1-RBD protein. Strikingly, MASL1 was precipitated by GST-Raf1-RBD only in 239T cells transfected with myc-DDK-tagged MASL1; mock- or control vector-transfected 293T cells did not show the MASL1/GST-Raf1-RBD interaction (Figure 5A). Similarly, GST-Raf1-RBD immunoprecipitated endogenous MASL1 protein in CD34+ cells treated with EPO that was only visible at day 14 of differentiation (Figure 5B). In addition, we observed the association between MASL1 and Raf1 in erythroid cells regulated by EPO (Figure 5C). A slight amount of MASL1 protein was also detected in G-CSF-induced CD34+ cells at day 14 of differentiation (Figure 5C, lower panel), similar to the findings of Sakabe et al (1999)8 who also detected MASL1 expression in peripheral blood leukocytes. Therefore, these data demonstrate that MASL1 interacts physically with Raf1, confirming that MASL1 induces erythroid differentiation through the Raf/MEK/ERK pathway.
Figure 5.

MASL1 physically interacts with Raf1. (A) 239T cells were transfected with myc-DDK-tagged MASL1-pCMV6-Entry plasmid and collected for protein lysates 48 hours after transfection. The protein lysates were subjected to pull-down with GST-Raf1-RBD coupled to glutathione resin and the bound proteins were analyzed by western blot using anti-MASL1 antibody (top). (Lane 1), mock-transfected 293T cell lysate pulled-down with GST-Raf1-RBD (Mock). (Lane 2), control vector-transfected 293T cell lysate pulled-down with GST-Raf1-RBD (Control pCMV6). (Lane 3), myc-DDK-tagged MASL1-transfected 293T cell lysate pulled-down with GST-Raf1-RBD (MASL1 pCMV6). (Lane 4), GTPγS-treated myc-DDK-tagged MASL1-transfected 293T cell lysate pulled-down with GST alone (negative control) (GST). (Lane 5), GDP-treated myc-DDK-tagged MASL1-transfected 293T cell lysate pull-down (inactivated-MASL1 control). (Lane 6), GTPγS-treated myc-DDK-tagged MASL1-transfected 293T cell lysate pull-down (activated-MASL1 control). (Bottom 2 panels), expression levels of MASL1 and β-actin (as an internal control) in 10% of input samples. (B) CD34+ cells were induced to differentiate by EPO treatment of 14 days. At day 7 (Lane 1) and 14 (Lane 2) of differentiation, protein lysates were subjected to pull-down with GST-Raf1-RBD and then western blot with anti-MASL1 antibody. (Lane 3), EPO-induced CD34+ cells at day 14 of differentiation protein lysate pulled-down with GST alone (negative control). (Lane 4), GDP-treated EPO-induced CD34+ cells at day 14 of differentiation protein lysate pull-down (inactivated-MASL1 control). (Lane 5), GTPγS-treated EPO-induced CD34+ cells at day 14 of differentiation protein lysate pull-down (activated-MASL1 control). (Bottom 2 panels), expression levels of MASL1 and β-actin (as an internal control) in 10% of input samples. (C) CD34+ cells were induced to differentiate by G-CSF (Lane 1) or EPO (Lane 2) treatment of 14 days. The protein lysates were subjected to pull-down with GST-Raf1-RBD and then western blot with anti-MASL1 antibody. (Lane 3), EPO-induced CD34+ cells at day 14 of differentiation protein lysate pulled-down with GST alone (negative control). (Lane 4), GDP-treated EPO-induced CD34+ cells at day 14 of differentiation protein lysate pull-down (inactivated-MASL1 control). (Lane 5), GTPγS-treated EPO-induced CD34+ cells at day 14 of differentiation protein lysate pull-down (activated-MASL1 control). The lower 2 panels show expression levels of MASL1 and β-actin (as an internal control) in 10% of input samples. Minus (−) and Plus (+) indicate the addition of GDP or GTPγS, respectively.
Discussion
Erythroid differentiation proceeds in response to specific signaling pathways and according to a transcriptional program that is regulated by DNA binding and lineage-restricted transcription factors.2,20 Many questions remain unanswered regarding the underlying molecular mechanisms that control lineage-specific erythroid cell differentiation. However, we previously demonstrated that MASL1 is selectively expressed in erythroid-lineage cells.7 Here, we illustrate for the first time that MASL1 could induce erythroid differentiation through the Raf/MEK/ERK pathway but not specifically at any stage of erythroid differentiation (Figure 6).
Figure 6.

Proposed model for MASL1 involvement in the Raf/MEK/ERK pathway during erythropoiesis. MASL1 induces erythroid differentiation through the Raf/MEK/ERK pathway. EPO, erythropoietin; SCF, stem cell factor.
MASL1 is a novel gene on chromosome 8p23.1 that was identified following the amplification of genes in DNA from malignant fibrous histiocytoma tumors.8 It also has been identified as an important oncogene in gastric cancer and hematologic malignancies.9,21 In addition, genomic loss of MASL1 occurs frequently in leukemic mantle cell lymphoma and squamous cell carcinoma of the vulva or larynx and pharynx.10,22,23 MASL1 has also been reported to be tumorigenic in nude mice.21 However, no studies have described MASL1 function in erythropoiesis. Here, we demonstrate that MASL1 is highly expressed during erythroid differentiation starting at the early stage of erythroid development, and it is also an abundant protein in human RBCs. The knockdown of MASL1 expression produced antidifferentiation effects in erythroid cells and led to a shift from erythroid to granulocytic-monocytic populations. Our findings are consistent with the possibility of lineage-dependent maintenance or suppression.24 MASL1-knockdown erythroid cells were arrested at G0/G1 cell-cycle phase, and apoptosis was induced at an early stage of erythroid differentiation, with an additional reduction in the proliferation rate. Moreover, a reduction of erythroid differentiation was observed when a mutation or deletion in the Ras-like GTPase region of MASL1 was made. Our findings represent a new role for MASL1 in erythroid differentiation.
Loss of chromosome 8p23.1 has been found to be associated with some myelodysplastic syndromes (MDSs), a heterogeneous group of clonal hematological disorders characterized by ineffective hematopoiesis, dysplastic morphology of blood cells, and substantial risk for progression to acute myeloid leukemia.25-27 MDS clones are thought to arise from the clonal acquisition of mutations in primitive CD34+ hematopoietic cells,28,29 but the array of molecular abnormalities underlying these disorders has not been completely delineated. MASL1 also maps to the chromosome 8p23.1 genomic segment, and the current study demonstrates a role of MASL1 in erythropoiesis. It is therefore interesting to speculate that MASL1 could be a candidate gene involved in the molecular pathogenesis of some forms of MDS. However, this hypothesis will require formal validation in clinical studies involving genotype/phenotype analysis.
The mechanisms that are involved in EPO-induced erythropoiesis have been intensively investigated. Several pathways, including the JAK/STAT, PI3K/Akt, and Ras/Raf/MEK/ERK kinase cascades, promote cell survival, proliferation, and differentiation. Numerous other adapters and signaling intermediates are involved in EPO-receptor signal transduction. Sakabe et al (1999) demonstrated that the MASL1 protein has several domains: Ras; 3 leucine zippers; an ATP/GTP-binding site; and a leucine-rich repeat (LRR) motif.8 Recently, the LRR-containing protein MASL1 has been reported in the regulation of Toll-like receptor-dependent signal transduction.30 Given that a Ras-like GTPase region is found in the MASL1 protein, we propose that this region may participate in the molecular mechanisms by which MASL1 is involved in erythroid differentiation similar to the Ras-GTPase protein. The Ras-like GTPases perform central regulatory functions in cellular processes, such as cell division, differentiation, cell-cell adhesion, growth, and apoptosis. The Ras-like domain contains GTPase activity similar to Ras. Acting as molecular switches, the Ras-like GTPases respond to external signals by exchanging GTP for bound GDP, and the GTP-bound active protein interacts with specific downstream effectors, thereby triggering intracellular signaling cascades through their interaction with a variety of target proteins.31 Four distinct Ras proteins, including H-Ras, K-Ras4A, K-Ras4B, and N-Ras, have been identified in mammals.32 Several reports have also documented that the 4 Ras proteins have unique biological functions and participate in different signaling cascades.33-35 Ras signaling has also been shown essential for the development of erythroid progenitors.36 In contrast, H-Ras−/−, N-Ras−/−, and H-Ras−/− N-Ras−/− double-knockout mice had no apparent hematopoietic abnormality, indicating that Ras is dispensable for normal erythropoiesis.37,38 Regarding the roles of oncogenic Ras in erythropoiesis, it has been shown that oncogenic H-Ras blocks terminal erythroid differentiation and that enforced expression of an active mutant of N-Ras in primitive hematopoietic cells inhibits proliferation of erythroid cells.39,40 MASL1 is one of the human ROCO proteins, but its functions in general as well as its specific roles are still unknown. Here, we illustrated further insights into the role of MASL1 in erythroid differentiation beyond a candidate oncogene.
Among various signaling molecules downstream of Ras, the Raf/MEK/ERK pathway mainly promotes cell growth and differentiation and prevents apoptosis of hematopoietic cells.41 Our results reveal that MASL1 shRNA-mediated knockdown of MASL1 reduced downstream effectors of the Raf/MEK/ERK pathway. Moreover, Erk1/2 phosphorylation was further inhibited in MASL1-knockdown CD34+ cells after EPO starvation, but hyperstimulation of EPO-starved cells with 10 U/mL EPO was sufficient to restore the level of Erk1/2 phosphorylation to that observed before starvation. Consistently, an SOS SH3 domain inhibitor that we used to inhibit GRB2/SOS interaction, which is an upstream control element of the Ras/Raf/MEK/ERK pathway, greatly reduced MASL1 expression and consequently inhibited erythroid differentiation as well as the downstream effectors in the Raf/MEK/ERK pathway. MASL1 mRNA levels were also reduced when cells were treated with SOS inhibitor, suggesting the possibility of negative feedback. Furthermore, our data demonstrate that MASL1 levels were also decreased when Jak2, a molecule interacting with the EPO receptor, was inhibited. Strikingly, we found that MASL1 interacts physically with Raf1. Therefore, our findings suggest that MASL1 activates erythroid differentiation through the Raf/MEK/ERK pathway.
A negative network of feedback regulatory factors, including interleukin-6 (IL-6), transforming growth factor-β, tumor necrosis factor-α, and interferon-γ, appears to function negatively by inhibiting erythropoiesis and promoting cell death of erythroblasts.42-45 IL-6 induces an upregulation of hepcidin expression, which inhibits iron export from macrophages by binding to the iron exporter ferroportin and inducing its degradation, thereby blocking availability of iron for erythropoiesis.46 A study has demonstrated that downregulated MASL1 expression in macrophages strongly enhanced IL-6 production following lipopolysaccharide or polyinosinic-polycytidylic acid (polyIC) stimulation.30 Taken together, these studies indirectly support our findings that MASL1 is involved in erythropoiesis. However, further studies are required to investigate how MASL1 interacts with IL-6 in erythropoiesis.
In conclusion, we have established a new functional and molecular pathway for MASL1 during erythroid differentiation. These findings could be fundamental to the development of novel genetic and cellular therapies for RBC disorders.
Supplementary Material
Acknowledgment
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: C.K. designed the research, performed the experiments, analyzed the results, created the figures, and wrote the manuscript; W.A., W.L., J.Z., N.U., and M.M.H. participated in performing the research; R.K. performed differential counting for coded sample slides; J.F.T. commented and wrote the manuscript; G.P.R. designed the research, analyzed and interpreted the results, wrote the manuscript, and gave final approval of the manuscript; all authors checked and read the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Griffin P. Rodgers, Molecular and Clinical Hematology Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD 20892; e-mail:griffinr@extra.niddk.nih.gov
References
- 1.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet. 2000;1(1):57–64. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- 2.Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002;21(21):3368–3376. doi: 10.1038/sj.onc.1205326. [DOI] [PubMed] [Google Scholar]
- 3.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11(19):2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Lodish HF. Constitutive activation of the MEK/ERK pathway mediates all effects of oncogenic H-ras expression in primary erythroid progenitors. Blood. 2004;104(6):1679–1687. doi: 10.1182/blood-2004-04-1362. [DOI] [PubMed] [Google Scholar]
- 5.Zhang J, Liu Y, Beard C, et al. Expression of oncogenic K-ras from its endogenous promoter leads to a partial block of erythroid differentiation and hyperactivation of cytokine-dependent signaling pathways. Blood. 2007;109(12):5238–5241. doi: 10.1182/blood-2006-09-047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laubach JP, Fu P, Jiang X, et al. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp Hematol. 2009;37(12):1411–1422. doi: 10.1016/j.exphem.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumkhaek C, Liu W, Rodgers GP. Identification and characterization of novel full-length cDNAs expressed during hematopoietic lineage-specific differentiation of cultured human peripheral blood mononuclear cells. Blood Cells Mol Dis. Prepublished on December 21, 2012, as DOI pii: S1079-9796(12)00232-X. [DOI] [PubMed]
- 8.Sakabe T, Shinomiya T, Mori T, et al. Identification of a novel gene, MASL1, within an amplicon at 8p23.1 detected in malignant fibrous histiocytomas by comparative genomic hybridization. Cancer Res. 1999;59(3):511–515. [PubMed] [Google Scholar]
- 9.Sakakura C, Mori T, Sakabe T, et al. Gains, losses, and amplifications of genomic materials in primary gastric cancers analyzed by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;24(4):299–305. doi: 10.1002/(sici)1098-2264(199904)24:4<299::aid-gcc2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Climent JA, Vizcarra E, Sanchez D, et al. Loss of a novel tumor suppressor gene locus at chromosome 8p is associated with leukemic mantle cell lymphoma. Blood. 2001;98(12):3479–3482. doi: 10.1182/blood.v98.12.3479. [DOI] [PubMed] [Google Scholar]
- 11.Donahue RE, Kirby MR, Metzger ME, et al. Peripheral blood CD34+ cells differ from bone marrow CD34+ cells in Thy-1 expression and cell cycle status in nonhuman primates mobilized or not mobilized with granulocyte colony-stimulating factor and/or stem cell factor. Blood. 1996;87(4):1644–1653. [PubMed] [Google Scholar]
- 12.Hematti P, Tuchman S, Larochelle A, et al. Comparison of retroviral transduction efficiency in CD34+ cells derived from bone marrow versus G-CSF-mobilized or G-CSF plus stem cell factor-mobilized peripheral blood in nonhuman primates. Stem Cells. 2004;22(6):1062–1069. doi: 10.1634/stemcells.22-6-1062. [DOI] [PubMed] [Google Scholar]
- 13.Chen L, Zhang J, Tang DC, et al. Influence of lineage-specific cytokines on commitment and asymmetric cell division of haematopoietic progenitor cells. Br J Haematol. 2002;118(3):847–857. doi: 10.1046/j.1365-2141.2002.03638.x. [DOI] [PubMed] [Google Scholar]
- 14.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 15.Ingley E, Tilbrook PA, Klinken SP. New insights into the regulation of erythroid cells. IUBMB Life. 2004;56(4):177–184. doi: 10.1080/15216540410001703956. [DOI] [PubMed] [Google Scholar]
- 16.Lewis PA. The function of ROCO proteins in health and disease. Biol Cell. 2009;101(3):183–191. doi: 10.1042/BC20080053. [DOI] [PubMed] [Google Scholar]
- 17.McCormick F. Signal transduction. How receptors turn Ras on. Nature. 1993;363(6424):15–16. doi: 10.1038/363015a0. [DOI] [PubMed] [Google Scholar]
- 18.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 19.Wojda U, Noel P, Miller JL. Fetal and adult hemoglobin production during adult erythropoiesis: coordinate expression correlates with cell proliferation. Blood. 2002;99(8):3005–3013. [PubMed] [Google Scholar]
- 20.Tsiftsoglou AS, Vizirianakis IS, Strouboulis J. Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life. 2009;61(8):800–830. doi: 10.1002/iub.226. [DOI] [PubMed] [Google Scholar]
- 21.Tagawa H, Karnan S, Kasugai Y, et al. MASL1, a candidate oncogene found in amplification at 8p23.1, is translocated in immunoblastic B-cell lymphoma cell line OCI-LY8. Oncogene. 2004;23(14):2576–2581. doi: 10.1038/sj.onc.1207352. [DOI] [PubMed] [Google Scholar]
- 22.Stephen JK, Chen KM, Raitanen M, et al. DNA hypermethylation profiles in squamous cell carcinoma of the vulva. Int J Gynecol Pathol. 2009;28(1):63–75. doi: 10.1097/PGP.0b013e31817d9c61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alonso Guervós M, Alvarez Marcos C, Llorente JL, et al. Genetic differences between primary larynx and pharynx carcinomas and their matched lymph node metastases by multiplex ligation-dependent probe amplification. Oral Oncol. 2009;45(7):600–604. doi: 10.1016/j.oraloncology.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Sposi NM, Zon LI, Carè A, et al. Cell cycle-dependent initiation and lineage-dependent abrogation of GATA-1 expression in pure differentiating hematopoietic progenitors. Proc Natl Acad Sci USA. 1992;89(14):6353–6357. doi: 10.1073/pnas.89.14.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thiel A, Beier M, Ingenhag D, et al. Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance. Leukemia. 2011;25(3):387–399. doi: 10.1038/leu.2010.293. [DOI] [PubMed] [Google Scholar]
- 26.Starczynowski DT, Vercauteren S, Sung S, et al. Copy number alterations at polymorphic loci may be acquired somatically in patients with myelodysplastic syndromes. Leuk Res. 2011;35(4):444–447. doi: 10.1016/j.leukres.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Vercauteren SM, Sung S, Starczynowski DT, et al. Array comparative genomic hybridization of peripheral blood granulocytes of patients with myelodysplastic syndrome detects karyotypic abnormalities. Am J Clin Pathol. 2010;134(1):119–126. doi: 10.1309/AJCPH27ZIZEJLORF. [DOI] [PubMed] [Google Scholar]
- 28.Corey SJ, Minden MD, Barber DL, et al. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer. 2007;7(2):118–129. doi: 10.1038/nrc2047. [DOI] [PubMed] [Google Scholar]
- 29.Nilsson L, Astrand-Grundström I, Anderson K, et al. Involvement and functional impairment of the CD34(+)CD38(-)Thy-1(+) hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood. 2002;100(1):259–267. doi: 10.1182/blood-2001-12-0188. [DOI] [PubMed] [Google Scholar]
- 30.Ng AC, Eisenberg JM, Heath RJ, et al. Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4631–4638. doi: 10.1073/pnas.1000093107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell SL, Khosravi-Far R, Rossman KL, et al. Increasing complexity of Ras signaling. Oncogene. 1998;17(11 Reviews):1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 32.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 33.Prior IA, Harding A, Yan J, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3(4):368–375. doi: 10.1038/35070050. [DOI] [PubMed] [Google Scholar]
- 34.Rocks O, Peyker A, Kahms M, et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307(5716):1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 35.Matallanas D, Arozarena I, Berciano MT, et al. Differences on the inhibitory specificities of H-Ras, K-Ras, and N-Ras (N17) dominant negative mutants are related to their membrane microlocalization. J Biol Chem. 2003;278(7):4572–4581. doi: 10.1074/jbc.M209807200. [DOI] [PubMed] [Google Scholar]
- 36.Khalaf WF, White H, Wenning MJ, et al. K-Ras is essential for normal fetal liver erythropoiesis. Blood. 2005;105(9):3538–3541. doi: 10.1182/blood-2004-05-2021. [DOI] [PubMed] [Google Scholar]
- 37.Esteban LM, Vicario-Abejón C, Fernández-Salguero P, et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol Cell Biol. 2001;21(5):1444–1452. doi: 10.1128/MCB.21.5.1444-1452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umanoff H, Edelmann W, Pellicer A, et al. The murine N-ras gene is not essential for growth and development. Proc Natl Acad Sci USA. 1995;92(5):1709–1713. doi: 10.1073/pnas.92.5.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J, Socolovsky M, Gross AW, et al. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102(12):3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 40.Darley RL, Pearn L, Omidvar N, et al. Protein kinase C mediates mutant N-Ras-induced developmental abnormalities in normal human erythroid cells. Blood. 2002;100(12):4185–4192. doi: 10.1182/blood-2002-05-1358. [DOI] [PubMed] [Google Scholar]
- 41.Platanias LC. Map kinase signaling pathways and hematologic malignancies. Blood. 2003;101(12):4667–4679. doi: 10.1182/blood-2002-12-3647. [DOI] [PubMed] [Google Scholar]
- 42.Dai C, Chung IJ, Jiang S, et al. Reduction of cell cycle progression in human erythroid progenitor cells treated with tumour necrosis factor alpha occurs with reduced CDK6 and is partially reversed by CDK6 transduction. Br J Haematol. 2003;121(6):919–927. doi: 10.1046/j.1365-2141.2003.04367.x. [DOI] [PubMed] [Google Scholar]
- 43.Zermati Y, Fichelson S, Valensi F, et al. Transforming growth factor inhibits erythropoiesis by blocking proliferation and accelerating differentiation of erythroid progenitors. Exp Hematol. 2000;28(8):885–894. doi: 10.1016/s0301-472x(00)00488-4. [DOI] [PubMed] [Google Scholar]
- 44.Secchiero P, Melloni E, Heikinheimo M, et al. TRAIL regulates normal erythroid maturation through an ERK-dependent pathway. Blood. 2004;103(2):517–522. doi: 10.1182/blood-2003-06-2137. [DOI] [PubMed] [Google Scholar]
- 45.Chasis JA, Mohandas N. Erythroblastic islands: niches for erythropoiesis. Blood. 2008;112(3):470–478. doi: 10.1182/blood-2008-03-077883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.