

Background: Thrombin is generated from zymogen precursors with the assistance of cofactors.
Results: Thrombin precursors prethrombin-2 and prothrombin are capable of catalytic activity and autoactivate.
Conclusion: Conformational selection regulates activity in the mature protease and has the potential to unleash autoactivation in the zymogen.
Significance: The paradigm of the inactive zymogen to active protease conversion needs revision to account for conformational selection.
Keywords: Coagulation Factors, Protease, Site-directed Mutagenesis, Structural Biology, Thrombin, Argatroban, Conformational Selection
Abstract
Trypsin-like proteases are synthesized as inactive zymogens and convert to the mature form upon activation by specific enzymes, often assisted by cofactors. Central to this paradigm is that the zymogen does not convert spontaneously to the mature enzyme, which in turn does not feed back to activate its zymogen form. In the blood, the zymogens prothrombin and prethrombin-2 require the prothrombinase complex to be converted to the mature protease thrombin, which is unable to activate prothrombin or prethrombin-2. Here, we show that replacement of key residues within the activation domain causes these zymogens to spontaneously convert to thrombin. The conversion is started by the zymogen itself, which is capable of binding ligands at the active site, and is abrogated by inactivation of the catalytic residue Ser-195. The product of autoactivation is functionally and structurally equivalent to wild-type thrombin. Zymogen autoactivation is explained by conformational selection, a basic property of the trypsin fold uncovered by structural and rapid kinetics studies. Both the zymogen and protease undergo a pre-existing equilibrium between active and inactive forms. The equilibrium regulates catalytic activity in the protease and has the potential to unleash activity in the zymogen to produce autoactivation. A new strategy emerges for the facile production of enzymes through zymogen autoactivation that is broadly applicable to trypsin-like proteases of biotechnological and clinical interest.
Introduction
Four protease families account for >40% of all proteolytic enzymes in humans and mediate extracellular matrix remodeling, blood coagulation, immunity, development, protein processing, cell signaling, and apoptosis. Trypsin-like proteases constitute the largest and best characterized group (1) and utilize a catalytic triad for activity, composed of the highly conserved residues His-57, Asp-102, and Ser-195 (chymotrypsinogen numbering). Additional structural components required for substrate binding and catalysis are the oxyanion hole defined by the backbone nitrogens of Ser-195 and Gly-193, residue Asp-189 at the bottom of the primary specificity pocket conferring specificity toward an Arg side chain at the P1 position of substrate, and the 215–217 segment flanking the active site that engages the P3-P4 residues of substrate (1, 2). Activity in trypsin-like proteases ensues via a common mechanism that involves the irreversible processing of an inactive zymogen precursor. The zymogen is proteolytically cut at Arg-15 in nearly all members of the family to generate a new N terminus that ion-pairs with the highly conserved Asp-194 next to the catalytic Ser-195 and organizes both the oxyanion hole and primary specificity pocket for substrate binding and catalysis (3). Central to this paradigm is that the zymogen does not convert spontaneously to the mature enzyme, which in turn does not feed back to activate its zymogen form (4, 5). A safety mechanism is thereby established to guarantee stability of the zymogen until a biological signal triggers activation via a distinct protease, a trypsin-like enzyme with primary specificity toward Arg side chains, which often acts in tandem with a cofactor in the context of a proteolytic cascade (6). Factors that perturb activation of a zymogen or the ordered sequence of reactions in a proteolytic cascade result in serious disruptions of homeostasis, as illustrated by disorders of the immune response (7), coagulation (8), and acute and chronic ailments of the pancreas (9).
The widely accepted notion of the zymogen as an immature precursor of the mature protease is buttressed by the structural observation of incorrect architectures of the catalytic triad, oxyanion hole, and primary specificity pocket prior to the proteolytic cleavage at Arg-15 (2, 3). However, the validity of this paradigm is called into question by the autocatalytic activity of zymogens such as proprotein convertases furin and kexin type 9 (10–12), plasma hyaluronan-binding protein (13), recombinant factor VII (14), and the membrane-bound matriptases (9, 15). Inspection of the structural database casts additional doubt on the current view of the zymogen as an immature precursor of the protease. A number of zymogens crystallize in a conformation (E* form) with the active site occluded by collapse of the 215–217 segment, the oxyanion hole incorrectly formed, and residues of the catalytic triad not optimally aligned for H-bonding interaction. Other zymogens such as trypsinogen (3, 16), the zymogen of MASP-2 (17), chymotrypsinogen (18), coagulation factor XI (19), and complement profactor B (20) crystallize in an alternative conformation (E form) where the active site is fully accessible to substrate and organized as in the mature protease (21, 22). In the case of chymotrypsinogen (18) and prethrombin-2 (23), the immediate zymogen precursor of the clotting protease thrombin, alternative conformations of the 215–217 segment consistent with a pre-existing E*-E equilibrium in solution have been trapped in the same crystal structure or different crystals harvested from the same crystallization well. The equilibrium between active (E) and inactive (E*) forms is a basic property of the trypsin fold for both the protease and zymogen (21, 22) and provides a relevant example of conformational selection as the basis of functional and structural plasticity in proteins (24, 25). Conformational selection in terms of the E*-E equilibrium is the only ligand binding mechanism that can be assigned unambiguously from rapid kinetics data (26) and was originally reported for chymotrypsin >40 years ago (27). Similar kinetic evidence for the zymogen has been obtained recently for the first time in the case of prethrombin-2 (26), thereby proving that the E* and E forms also exist in the precursor of the active protease. Although the E*-E equilibrium explains activity and regulation in the protease, questions arise about the role of the active E form in the zymogen that is supposed to be inactive. Here, we show that the presence of the E form confers the zymogen catalytic activity that may be targeted toward the zymogen itself to produce autoactivation.
EXPERIMENTAL PROCEDURES
Numbering
To facilitate comparison among different trypsin-like proteases, it is customary to number residues relative to chymotrypsinogen (3). Insertions relative to chymotrypsinogen are labeled with a lowercase letter, which makes numbering somewhat cumbersome for residues of the mature A chain of thrombin. The corresponding prothrombin numbering for these residues is given in Fig. 1.
FIGURE 1.
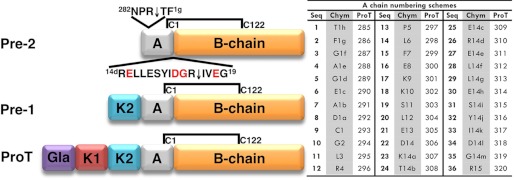
Schematic representation of prethrombin-2, prethrombin-1, and prothrombin, the zymogen precursors of thrombin. Prothrombin is composed of fragment 1 (residues 1–155), fragment 2 (residues 156–271) and a protease domain (residues 272–579). Fragment 1 contains a Gla domain and a kringle (K1), fragment 2 contains a second kringle (K2), and the protease domain contains the A chain (residues 272–320) and the catalytic B chain (residues 321–579). Prethrombin-2 is cleaved at Arg-15 (residue 320 of prothrombin) to separate the A and B chains and generate the mature protease thrombin. The A and B chain remain covalently attached after activation through the disulfide bond Cys-1-Cys-122. Cleavage at Arg-284 by thrombin itself reduces the length of the A chain to its final 36 amino acids composition (residues 285–320 of prothrombin). Residues within this final construct composed of a total of 295 amino acids (residues 285–579 of prothrombin) are numbered according to chymotrypsinogen, and insertions relative to chymotrypsinogen are labeled with a lowercase letter. The table lists residues of the mature A chain sequentially (Seq) and according to the prothrombin (ProT) and chymotrypsinogen (Chym) numbering. The sequence around the site of activation at Arg-15 shows residues subject to mutagenesis in red.
Materials
Prethrombin-2 wild-type and mutants S195A, E14eA, D14lA, G14mP, G14mP/S195A (GS), E18A, E14eA/D14lA (ED), E14eA/E18A (EE), D14lA/E18A (DE), E14eA/D14lA/E18A (EDE), E14eA/D14lA/E18A/S195A (EDES), and E14eA/D14lA/G14mP/E18A (EDGE) were expressed in Escherichia coli, refolded, and purified to homogeneity as described previously (23). Benzamidine was added to the refolding, dialysis, and purification buffers at a final concentration of 10–50 mm. Unlike all other autoactivating mutants, the conversion of EDGE to the mature protease could not be inhibited by addition of benzamidine up to 50 mm to the refolding buffer. Production of the mutant EDGE with the additional S195A substitution as a control and for crystallization failed repeatedly due to lack of refolding. Prethrombin-1 wild-type and mutants EDE and EDES were expressed in baby hamster kidney cells as described (28). Benzamidine was added after collecting the cell supernatant at a final concentration of 10 mm to prevent proteolysis. Gla domainless prothrombin wild-type and mutants EDE and EDES were expressed in baby hamster kidney cells (28) after introduction of an ApaI restriction site in the human prothrombin cDNA. Homogeneity and chemical identity of final preparations were verified by SDS-PAGE and by reverse phase HPLC mass spectrometry analysis, giving a purity of >98%.
Fluorescein Labeling Studies
Thrombin (10 μm), prethrombin-2 (24 μm), and prothrombin purified from plasma (28 μm, Enzyme Research Labs) were incubated with fluorescein-H-d-Phe-Pro-Arg-CH2Cl (fluorescein-PPACK,2 Hematologic Technologies) at concentrations of 20 μm (thrombin) or 500 μm (prethrombin-2, prothrombin), in the presence of 20 mm Tris, 4% dimethyl sulfoxide, 200 mm NaCl, pH 7.4, at room temperature for 2 h (thrombin) or 40 h (prethrombin-2 and prothrombin). Aliquots (4 μl) were quenched by the addition of 4× loading buffer (2 μl) at different time points and monitored by SDS-PAGE. SeeBlue Plus2 prestained standard (Invitrogen) was used as a molecular weight marker and produced the expected myoglobin red band at 17 kDa when the gel was irradiated with UV light. Incorporation of fluorescein-PPACK in thrombin, prethrombin-2, and prothrombin was quantified in terms of the absorbance ratio at 429/280 nm after removal of unreacted probe by G-25 gel filtration. PPACK binds irreversibly to the active site of thrombin (29, 30) and also prethrombin-2, as confirmed by a preliminary x-ray crystal structure of the complex (data not shown).
Autoactivation Studies
Autoactivation of prethrombin-2, prethrombin-1, and prothrombin was followed at room temperature at a final concentration of 1.1 mg/ml after removal of benzamidine by G-25 gel filtration. Autoactivation was confirmed by N-terminal sequencing. The reaction leading to generation of thrombin and depletion of the zymogen was quenched at different times with 10 μl of NuPAGE LDS sample buffer containing β-mercaptoethanol as the reducing agent. Samples were processed by electrophoresis using 4–12% NuPAGE gels and MES running buffer, stained with Coomassie Brilliant Blue R-250, and analyzed by quantitative densitometry. The activity of the autoactivated constructs was tested against the chromogenic substrate H-d-Phe-Pro-Arg-p-nitroanilide and physiological substrates fibrinogen, PAR1, and protein C in the presence of 100 nm thrombomodulin and 5 mm CaCl2 under experimental conditions of 5 mm Tris, pH 7.4, 0.1% PEG 8000, 145 mm NaCl, 37 °C, as detailed elsewhere (28). Proteolytically degraded β-thrombin and γ-thrombin were removed by hydrophobic interaction chromatography on a TSK-Gel Phenyl 5PW column (75 × 7.5 mm, 10 μm, Supleco). Cleavage of prethrombin-2 mutants S195A (6.8 μm) and EDES (6.8 μm) by thrombin (1.3 μm) was studied by following consumption of the zymogen and appearance of the band of the active enzyme at 29 kDa. Experiments were performed in 20 mm Tris, pH 7.4, at 25 °C, 145 mm NaCl, and 2 mm EDTA. Aliquots (16 μl) were quenched by the addition of 4× loading buffer (12 μl) at different time points and monitored by quantitative SDS-PAGE.
Isothermal Titration Calorimetry Studies
Binding of the active site inhibitor argatroban was studied by isothermal titration calorimetry under experimental conditions of 20 mm Tris, 0.1% PEG 6000, 200 mm NaCl, pH 7.4, at 25 °C using an iTC200 calorimeter (MicroCal Inc., Northampton, MA) with the sample cell containing the S195A mutant of thrombin (10 μm) or prethrombin-2 (54 μm) and the syringe injecting argatroban (100–575 μm). The concentrations of thrombin and prethrombin-2 were calculated at 280 nm from absorption coefficients of 66,390 m−1 cm−1 and 67,880 m−1 cm−1, respectively. The concentration of argatroban was calculated at 333 nm from an absorption coefficient of 5000 m−1 cm−1. Thermodynamic parameters were obtained by fitting the data, after subtraction of the base line, with a one-site binding model. Experiments were performed in triplicate.
X-ray Studies
Crystallization of prethrombin-2 mutants EDES and S195A (mixed 1:10 with argatroban) and thrombin mutants EDES and S195A (mixed 1:8 with argatroban) was achieved at 22 °C by the vapor diffusion technique using an Art Robbins Instruments Phoenix liquid handling robot and mixing equal volumes (0.3 μl) of protein and reservoir solution. Optimization of crystal growth was achieved by the hanging drop vapor diffusion method mixing 3 μl of protein (10 mg/ml) with equal volumes of reservoir solution (Table 1). Crystals grew in the P41212 space group for prethrombin-2 EDES and in the P21212 space group for thrombin EDES, with one molecule in the asymmetric unit in both cases. Complexes with argatroban grew in the P21 space group for prethrombin-2 S195A and in the P41 space group for thrombin S195A, with two molecules in the asymmetric unit in both cases. Diffraction quality crystals for all structures grew in ∼2 weeks and were cryoprotected in a solution similar to the mother liquor but containing 20% glycerol prior to flash-freezing. X-ray diffraction data were collected with a home source (Rigaku 1.2 kW MMX007 generator with VHF optics) Rigaku Raxis IV++ detector and were indexed, integrated, and scaled with the HKL2000 software package (31). Structures were determined by molecular replacement using MOLREP from the CCP4 suite (32) and Protein Data Bank codes 3SQH (23) for the prethrombin-2 structures and 1SHH (30) for the thrombin structures as search models. Refinement and electron density generation were performed with REFMAC5 from the CCP4 suite, and 5% of the reflections were randomly selected as a test set for cross-validation. Model building and analysis of the structures were conducted with COOT (33). In the final stages of refinement, translation/libration/screw tensors modeling rigid-body anisotropic temperature factors were calculated and applied to the model, except for the structure of prethrombin-2 S195A bound to argatroban. Ramachandran plots were calculated using PROCHECK (34). Statistics for data collection and refinement are summarized in Table 1. Atomic coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 4H6T for prethrombin-2 EDES, 4HFY for prethrombin-2 S195A bound to argatroban, 4H6S for thrombin EDES, and 4HFP for thrombin S195A bound to argatroban).
TABLE 1.
Crystallographic data for prethrombin-2 and thrombin mutants EDES (free) and S195A (bound to argatroban)
| Prethrombin-2 EDES | Thrombin EDES | Prethrombin-2 S195A/argatroban | Thrombin S195A/argatroban | |
|---|---|---|---|---|
| Buffer/salt | 200 mm KH2PO4 | 200 mm NH4Cl | 100 mm Tris, pH 8.5, 200 mm Li2SO4 | 100 mm sodium acetate, pH 4.6 |
| PEG | 3350 (15%) | 3350 (20%) | 3000 (30%) | 8000 (25%) |
| PDB code | 4H6T | 4H6S | 4HFY | 4HFP |
| Data collection | ||||
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Space group | P41212 | P21212 | P21 | P41 |
| Unit cell dimensions | a = 135.5, b = 135.5, and c = 42.5 Å | a = 63.9, b = 70.8, and c = 56.1 Å | a = 43.9, b = 81.8, and c = 77.8 Å; β = 100.6° | a = 77.2, b = 77.2, and c = 94.9 Å |
| Molecules/asymmetric unit | 1 | 1 | 2 | 2 |
| Resolution range (Å) | 40–2.4 | 40–2.2 | 40–3.0 | 40–2.4 |
| Observations | 139,696 | 48,768 | 36,961 | 135,941 |
| Unique observations | 16,004 | 13,133 | 10,552 | 21,611 |
| Completeness (%) | 98.8 (97.8) | 97.2 (97.0) | 94.6 (89.8) | 98.9 (91.5) |
| Rsym (%) | 7.0 (44.0) | 10.7 (44.4) | 15.6 (44.7) | 10.7 (36.2) |
| I/σ(I) | 21.3 (2.9) | 10.5 (2.5) | 5.9 (2.0) | 13.9 (2.8) |
| Refinement | ||||
| Resolution (Å) | 40–2.4 | 40–2.2 | 40–3.0 | 40–2.4 |
| Rcryst/Rfree | 0.212/0.253 | 0.196/0.259 | 0.248/0.343 | 0.167/0.238 |
| Reflections (working/test) | 14,274/802 | 12,369/646 | 9833/477 | 20,437/1105 |
| Protein atoms | 2366 | 2275 | 4610 | 4637 |
| Argatroban molecules | 2 | 2 | ||
| Na+ | 1 | 0 | 2 | |
| PO43− | 7 | |||
| Solvent molecules | 63 | 103 | 0 | 66 |
| r.m.s.d. bond lengthsa (Å) | 0.014 | 0.012 | 0.007 | 0.006 |
| r.m.s.d. anglesa | 1.5° | 1.5° | 1.1° | 1.0° |
| r.m.s.d. ΔB (Å2) (mm/ms/ss)b | 1.06/0.47/2.02 | 1.30/1.29/2.41 | 0.30/0.10/0.49 | 1.25/1.02/1.18 |
| 〈B〉 protein (Å2) | 47.5 | 41.0 | 30.0 | 34.0 |
| 〈B〉 argatroban (Å2) | 30.2 | 27.3 | ||
| 〈B〉 Na+ (Å2) | 27.7 | 27.0 | ||
| 〈B〉 PO43− (Å2) | 64.4 | |||
| 〈B〉 Solvent (Å2) | 50.9 | 41.1 | 33.5 | |
| Ramachandran plot | ||||
| Most favored (%) | 98.4 | 99.6 | 98.4 | 98.6 |
| Generously allowed (%) | 1.2 | 0.4 | 1.4 | 1.0 |
| Disallowed (%) | 0.4 | 0.0 | 0.2 | 0.4 |
a Root mean squared deviation (r.m.s.d.) from ideal bond lengths and angles and root mean square deviation in B-factors of bonded atoms.
b mm, main chain-main chain; ms, main chain-side chain; ss, side chain-side chain; PDB, Protein Data Bank.
RESULTS
In the blood, thrombin is generated from the zymogen prothrombin by the prothrombinase complex, composed of the trypsin-like protease factor Xa, the cofactor factor Va, phospholipid membranes, and Ca2+ (8). Prothrombin (Fig. 1) is a vitamin K-dependent protein composed of fragment 1 (residues 1–155), fragment 2 (residues 156–271), and a protease domain (residues 272–579). Fragment-1 contains a Gla domain and a kringle, fragment-2 contains a second kringle, and the protease domain contains the A chain (residues 272–320) and the catalytic B chain (residues 321–579). The prothrombinase complex converts prothrombin to thrombin by cleaving at Arg-271 and Arg-320. A single cleavage at Arg-320 (corresponding to Arg-15 in the chymotrypsinogen numbering, see Fig. 1) generates the active intermediate meizothrombin. Cleavage at Arg-271 generates the inactive precursor prethrombin-2 that differs from thrombin only in the intact Arg-15-Ile-16 peptide bond (Fig. 1). Rapid kinetics of substrate binding to the active site have recently shown that prethrombin-2 exists in equilibrium between the E* and E forms (26) as the mature enzyme and may therefore assume a conformation (E) where the active site is fully accessible. Indeed, prethrombin-2 interacts with the active site inhibitor argatroban with a significant affinity (Fig. 2). Direct measurements of the interaction by calorimetry yield a value of Kd = 8.0 ± 0.1 μm, corresponding to a significant ΔG = −7.0 ± 0.1 kcal/mol. The binding interaction is driven predominantly by enthalpic components (ΔH = −8.1 ± 0.3 kcal/mol) with a small entropy loss (TΔS = −1.1 ± 0.1 kcal/mol). The interaction of argatroban with thrombin under identical solution conditions (Fig. 2) gives a value of Kd = 42 ± 2 nm, corresponding to a free energy change ΔG = −10.1 ± 0.1 kcal/mol and is 200-fold tighter compared with the interaction with prethrombin-2. The interaction is again driven predominantly by enthalpic components (ΔH = −10.9 ± 0.2 kcal/mol) with a small entropy loss (TΔS = −0.8 ± 0.1 kcal/mol). Interestingly, the difference in binding free energy between prethrombin-2 and thrombin is almost entirely enthalpic and amounts to 3 kcal/mol, vouching for differences in molecular contacts between argatroban and the active site in the two cases. The lack of a significant difference in the entropy contribution to argatroban binding between prethrombin-2 and thrombin suggests a basic similarity in the conformational changes associated with the interaction for both the zymogen and protease.
FIGURE 2.

a and b, crystal structures of the mutant S195A of prethrombin-2 (a) and thrombin (b) in surface over ribbon representation (gold) bound to the active site inhibitor argatroban (green sticks). Relevant residues from the protein are depicted as yellow sticks and labeled. Argatroban binds to thrombin and prethrombin-2 in a similar orientation but with some important differences discussed in the text. The distinct orientation of Asp-189 in the primary specificity pocket between prethrombin-2 and thrombin accounts for the 200-fold difference in binding affinity between zymogen and protease (see c and d). c and d, isothermal titration calorimetry traces of argatroban binding to the mutants S195A of prethrombin-2 (c) and thrombin (d), used for x-ray crystal studies (a and b), under experimental conditions of 20 mm Tris, 0.1% PEG 8000, 200 mm NaCl, pH 7.4, at 25 °C. Argatroban binds to the prethrombin-2 mutant S195A with a value of Kd = 8.0 ± 0.1 μm, corresponding to ΔG = −7.0 ± 0.1 kcal/mol with thermodynamic components ΔH = −8.1 ± 0.3 kcal/mol and TΔS = −1.1 ± 0.1 kcal/mol. Binding of the inhibitor to the thrombin mutant S195A gives a value of Kd = 42 ± 2 nm, corresponding to ΔG = −10.1 ± 0.1 kcal/mol, and thermodynamic components ΔH = −10.9 ± 0.2 kcal/mol and TΔS = −0.8 ± 0.1 kcal/mol.
Independent support to the conclusions drawn from calorimetric measurements comes from the crystal structures of thrombin and prethrombin-2 bound to argatroban (Fig. 2). Although the inhibitor binds to the active site in exactly the same orientation for the zymogen and protease, there are small but significant differences. The Arg moiety of argatroban penetrates the primary specificity pocket but in prethrombin-2 the side chain of Asp-189 remains 6 Å away. The defect in the orientation of Asp-189 caused by the lack of cleavage at Arg-15 in the activation domain of the zymogen in the free form (23) is not corrected by binding of argatroban to the primary specificity pocket. Therefore, ligand recognition by the zymogen involves selection of the E form pre-existing with the E* form in the free zymogen (23, 26) rather than an induced fit. The carboxylate of the piperidine of argatroban contacts the Nϵ1 atom of Trp-148 in the autolysis loop of prethrombin-2 but not thrombin and in both cases packs against Trp-60d and Tyr-60a in the hydrophobic portion of the 60-loop. The quinoline is in hydrophobic interaction with Trp-148 in prethrombin-2 but not in thrombin and engages Trp-215 and Leu-99 in the aryl binding site in both cases. Overall, the lack of interaction of argatroban with Asp-189 in the primary specificity pocket of prethrombin-2 accounts for most of the 3 kcal/mol difference in binding free energy between the zymogen and protease because it is similar to the free energy penalty caused by the D189A substitution on substrate binding (35).
Having established ligand binding to the active site of prethrombin-2, we tested the possibility that the zymogen would also possess catalytic activity. Prethrombin-2 does not show any appreciable cleavage of synthetic and physiological substrates under several experimental conditions tested, but it does react with the irreversible inhibitor fluorescein-PPACK (Fig. 3), proving that the active site residues His-57 and Ser-195 in the zymogen are already in the optimal orientation for catalysis. The interaction is slower than that observed with thrombin but specific enough that PPACK is found bound to the active site of prethrombin-2 in a preliminary x-ray crystal structure (data not shown). Prothrombin also interacts with fluorescein-PPACK (Fig. 3) but incorporates the label with significantly lower yield (20%) compared with prethrombin-2 (50%). The presence of fragment-1 and fragment-2 in prothrombin likely affects the E*-E equilibrium by reducing the relative population of the active E form required for PPACK binding.
FIGURE 3.
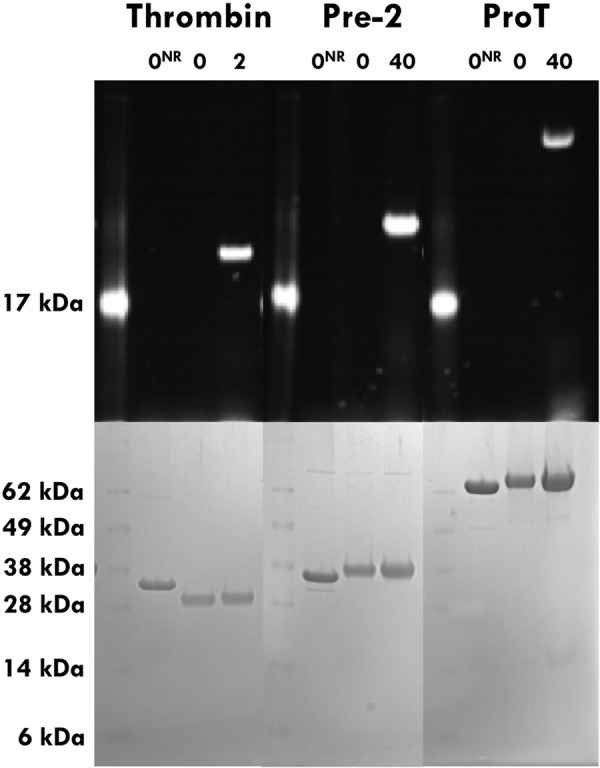
Prethrombin-2 (Pre2) and prothrombin (ProT) incorporate fluorescein-PPACK at the active site based on fluorescence (upper panel) and protein staining (lower panel) SDS gels under reducing and non-reducing (NR) conditions. Thrombin is shown as a positive control. SeeBlue Plus2 prestained standard (Invitrogen) was used as a molecular weight marker and produced the expected myoglobin red band at 17 kDa when the gel was irradiated with UV light. The standard is present at a concentration too low for detection in the SDS gels. Under reducing conditions, thrombin migrates as a doublet due to separation of the A (4 kDa) and B (29 kDa) chains, but the zymogen prethrombin-2 migrates as a single band (37 kDa). Incorporation of fluorescein-PPACK proves that the active site of prethrombin-2 and prothrombin may be catalytically competent.
If prethrombin-2 and prothrombin possess catalytic activity, what is their biological target? A recent crystal structure of prethrombin-2 reveals Arg-15 in the activation domain in ionic interactions with the acidic residues Glu-14e, Asp-14l, and Glu-18 instead of being exposed to solvent for proteolytic attack (23). When the acidic residues are mutated to Ala, the resulting prethrombin-2 triple mutant EDE slowly converts to the mature enzyme. The mutant EDES, carrying an additional Ala substitution of the catalytic residue Ser-195, does not autoactivate (23), implying that it is the zymogen itself that initiates the autocatalytic conversion to the mature protease. Thrombin is known to cleave prethrombin-2 at Arg-284 to reduce the A chain to its final length (Fig. 1) but has no specificity toward Arg-15 in the activation domain. Unlike wild-type, the prethrombin-2 mutant EDES is cleaved by thrombin at Arg-15 according to a single exponential with kobs = 0.030 ± 0.003 h−1 (Fig. 4), implying that Arg-15 has distinct solvent accessibility in the two proteins. The crystal structure of the prethrombin-2 mutant EDES (Fig. 4) shows an intact Arg-15-Ile-16 peptide bond and the segment around the cleavage site at Arg-15 significantly different from wild-type (23). Neutralization of the acidic residues Glu-14e, Asp-14l, and Glu-18 in the activation domain causes Arg-15 to swing out of its anionic cage and to direct its side chain to solvent for proteolytic attack. In this orientation, Arg-15 has an accessible surface area of 196 Å2 or 73% of that of an Arg residue in solution (268 Å2). Solvent exposure of Arg-15 in the wild-type is only 28% (23). The effect of the mutation is local on the architecture of the activation domain and does not propagate to other regions of the zymogen that assume a conformation similar to that of wild-type (23).
FIGURE 4.
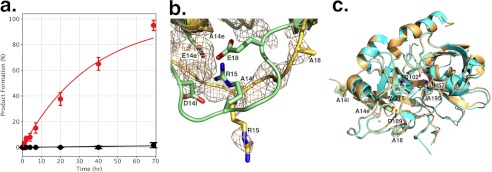
a, kinetics of activation of prethrombin-2 mutants S195A (6. 8 μm, black circles) and EDES (6.8 μm, red circles) by thrombin (1.3 μm), plotted as the percent of product formation generated from cleavage of the zymogen at Arg-15 as a function of time. No appreciable cleavage is detected for prethrombin-2 S195A up to 70 h. The prethrombin-2 mutant EDES is cleaved by thrombin according to a single exponential with kobs = 0.030 ± 0.003 h−1. b, overlay of the activation domains of prethrombin-2 S195A (green) as reported recently (23), and mutant EDES (yellow) reported in this study. The Arg-15-Ile-16 peptide bond is intact in both zymogens, but the organization of the segment around the cleavage site at Arg-15 is significantly different. Arg-15 is caged by Glu-14e, Asp-14l, and Glu-18 in the S195A mutant, but becomes exposed to solvent in the EDES mutant. The simulated annealing Fo − Fc omit map (beige mesh) for the mutant is contoured at 2σ. c, structure of the EDES mutant of thrombin (yellow) obtained from proteolytic cleavage of prethrombin-2 mutant EDES by the thrombin mutant EDE. The mutant (yellow) crystallizes in the active E form with Na+ (gold ball) bound to its site. The structure is very similar (root mean square deviation, 0.309 Å2) to that of the 1SG8 structure of wild-type (cyan, with Na+ as a cyan ball) (30). Sites of Ala substitution of the EDES mutant are clearly detected in the density map contoured at 1σ.
The EDE mutation also produces autoactivation of prethrombin-1 and prothrombin, and the additional S195A replacement abrogates it (Fig. 5). The thrombin mutant EDE generated by autoactivation crystallizes in the E form with Na+ bound to its site (Fig. 4) in a conformation that is practically identical to that of wild-type thrombin in the Na+-bound E form (30). The EDE mutation does not alter the fold of the molecule or the conformation of the active site and produces a construct that cleaves chromogenic substrates, fibrinogen, PAR1, and protein C with values of kcat/Km nearly identical to those of wild-type (Fig. 5). Therefore, lack of autoactivation of prethrombin-2, prethrombin-1, and prothrombin is not due to lack of catalytic activity in the zymogen but rather to the buried conformation of Arg-15 in the activation domain. A similar conclusion has recently emerged for the anticoagulant zymogen protein C, which shares sequence similarities with prethrombin-2 in the activation domain (36). Replacement of charged residues in the anionic cage produces a variant protein C that slowly autoactivates and is rapidly activated by thrombin without the need of thrombomodulin (36).
FIGURE 5.

a, the triple substitution EDE in the activation domain of Gla-domainless prothrombin (GD-ProT), prethrombin-1 (Pre-1), and prethrombin-2 (Pre-2) generates autoactivation. The reaction was monitored on SDS gels as the consumption of zymogen and the appearance of the band for the mature enzyme at 35 kDa for GD-ProT, 32 kDa, for Pre-1 and 29 kDa for Pre-2. N-terminal sequencing confirmed the sequence 16IVAGS20 of the B chain with the E18A substitution. The different electrophoretic mobility of the B chains is due to glycosylation of the prethrombin-1 and prothrombin constructs. Autoactivation proceeds through the expected intermediates and generates prethrombin-1 (49 kDa, with checked N-terminal sequence 156SEGSS160) and prethrombin-2 (38 kDa, with checked N-terminal sequence 285TFGSG289) in Gla-domainless prothrombin, and only prethrombin-2 (38 kDa, with checked N-terminal sequence 285TFGSG289) in prethrombin-1. Autoactivation was not detected in wild-type zymogens and was specifically abrogated by the additional S195A mutation in the EDES constructs. Optimization of autoactivation was achieved with the single mutant G14mP and the quadruple mutant EDGE that converts to thrombin rapidly upon refolding. Autoactivation in the G14mP construct is abrogated by the additional S195A mutation in the GS double mutant. b, kinetics of autoactivation of wild-type and mutant prethrombin-2 constructs measured as the extent of zymogen conversion to thrombin (A). The t½ for complete conversion of the zymogen to the mature enzyme is as follows: ∞ h (wild-type, E14eA), 220 ± 20 h (D14lA), 158 ± 6 h (ED), 124 ± 4 h (E18A), 116 ± 4 h (EE), 13 ± 2 h (EDE), 8 ± 1 h (G14mP), 5.0 ± 0.5 h (DE), 0 h (EDGE). Prethrombin-2 wild-type and mutant E14eA do not autoactivate. The single mutant G14mP and the double mutant DE autoactivate slightly faster than the triple mutant EDE, and the quadruple mutant EDGE fully converts to thrombin upon refolding (a). c, values of the log of the specificity constant s = kcat/Km (m−1 s−1) for the hydrolysis of synthetic (H-d-Phe-Pro-Arg-p-nitroanilide; FPR) and physiological (fibrinogen, PAR1, and protein C) substrates by thrombin mutants relative to wild-type under experimental conditions of 5 mm Tris, 0.1% PEG 8000, 145 mm NaCl, pH 7.4, at 37 °C. Abbreviations used for the mutants are listed under “Experimental Procedures.” The mutations afford minimal perturbations of substrate hydrolysis in all cases. The values of kcat/Km for wild-type are as follows: 39 ± 1 μm−1 s−1 (H-d-Phe-Pro-Arg-p-nitroanilide), 10 ± 1 μm−1 s−1 (fibrinogen), 14 ± 1 μm−1 s−1 (PAR1), and 0.33 ± 0.04 μm−1 s−1 (protein C).
Autoactivation of prethrombin-2 suggests a new method for thrombin production that obviates the need of activators such as prothrombinase or the snake venom metalloprotease ecarin. The autocatalytic conversion of the EDE mutant of prethrombin-2 to the mature enzyme is slow but can be optimized. Systematic Ala scanning mutagenesis was carried out on residues of the anionic cage to determine which was necessary and/or sufficient to shield Arg-15 from solvent. A total of 10 prethrombin-2 constructs (wild-type and nine mutants) were expressed, purified, and tested for autoactivation. Quantification and analysis was carried out as described previously from progress curves of zymogen conversion (23, 36). Of the three possible single mutants, D14lA and E18A but not E14eA produced autoactivation (Fig. 5). The three acidic residues are not functionally equivalent in caging Arg-15, even though they make significant ionic interactions with the guanidinium group (Fig. 4). The dispensable role of Glu-14e is confirmed by the two double mutants ED and EE, which autoactivate no faster than the single mutants D14lA and E18A, and by the triple mutant EDE, which autoactivates even slower than the double DE mutant (Fig. 5). Mutation of either Asp-14l or Glu-18 is sufficient yet not necessary to produce autoactivation, as directly demonstrated by mutation of Gly-14m at the P2 position of the activation domain (Fig. 1). Thrombin has a strong preference for Pro at this position, as documented by numerous studies on chromogenic substrates (37–39), and indeed, optimization of the activation domain of prethrombin-2 for thrombin cleavage has recently involved mutation of Gly-14m (40). Remarkably, the G14mP single mutant of prethrombin-2 autoactivates as rapidly as the DE double mutant and faster than the EDE triple mutant (Fig. 5). Again, autoactivation in the G14mP construct is abrogated by the additional S195A mutation (GS mutant). The side chain of Arg-15 may be constrained to point to the solvent by the single G14mP substitution, and the presence of Gly-14m in the activation domain of prehtrombin-2 may play the important role of providing the necessary backbone flexibility for Arg-15 to be sequestered within the anionic cage. In this scenario, G14m and residues within the anionic cage synergistically lock Arg-15 in a buried conformation. Consistent with this hypothesis, the quadruple mutant EDGE of prethrombin-2 is already fully converted to thrombin when harvested from the refolding column, and the product of autoactivation is functionally equivalent to wild-type (Fig. 5). A thrombin construct functionally equivalent to wild-type can be generated directly by autoactivation of its zymogen precursor prethrombin-2, without appreciable delay after refolding and without the need of activators such as prothrombinase or the snake venom ecarin.
DISCUSSION
Autoactivation of the zymogen has two necessary requirements: an intrinsic catalytic activity prior to conversion to the mature protease and specificity toward the site of cleavage around Arg-15 in the activation domain. The pre-existing equilibrium between fully open (E) and collapsed (E*) conformations of the active site is a key property of the trypsin fold and is supported by structural biology (21, 22) and rapid kinetics (26, 27, 41, 42). The E*-E equilibrium offers a simple framework to understand the molecular basis of activity and regulation in the mature protease, but its relevance extends to the zymogen. When the zymogen is in the E* form, no activity is possible, and the protein behaves as an immature precursor of the active protease. However, when the E form is significantly populated, the zymogen may acquire catalytic activity toward suitable targets, including the zymogen itself. This explains autocatalysis in proprotein convertases furin and kexin type 9 (10–12), plasma hyaluronan-binding protein (13), recombinant factor VII (14), and the membrane-bound matriptases (9, 15). The conceptual framework of the E*-E equilibrium extends to other classes of enzymes such as caspases and explains how small molecules can induce activation of the pro-enzyme (43) by stabilizing the E form. Prethrombin-2 exists in the E* and E forms (23, 26) and carries a P1–P4 sequence in the activation domain that closely resembles that of PAR1, the most specific substrate of thrombin (44). What prevents autoactivation in prethrombin-2 is burial of Arg-15 in the activation domain, an unanticipated feature uncovered when the structure of this zymogen was solved for the first time in the free form (23). Inspection of the structural database reveals Arg-15 in the activation domain of several zymogens either completely disordered or exposed to solvent, with prethrombin-2 being the only known exception (23). However, in the structure of prethrombin-1 (45) and many others, exposure of Arg-15 may be an artifact due to interaction of this residue with neighbor molecules in the crystal lattice. Therefore, the buried conformation of Arg-15 documented in prethrombin-2 may be more prevalent than currently suggested by the structural database. Burial of Arg-15 may be one of the most important factors controlling autoactivation in zymogens of the trypsin family. Prethrombin-2, prethrombin-1, and prothrombin become autocatalytic once Arg-15 is forced outside of its anionic cage with specific mutations, but it is conceivable that exposure of Arg-15 may be triggered in the wild-type by suitable effector molecules. In this case, thrombin would be generated in vivo directly from prothrombin through mechanisms that bypass the coagulation cascade.
Conformational selection in terms of the pre-existing E*-E equilibrium provides a more realistic paradigm for the function of the zymogen and protease and challenges the current dogma of the zymogen as an inactive precursor of the active protease (4, 5). The molecular basis of autoactivation in terms of conformational selection also defines a strategy for the facile production of trypsin-like proteases of clinical and biotechnological relevance. Tissue-type plasminogen activator, thrombin, and activated protein C are or have been on the market for the treatment of medically relevant conditions such as stroke, hemorragic complications, and sepsis (46). Thrombin variants engineered for anticoagulant and antithrombotic activity in vivo are in preclinical stage (47). A method of producing an active form of these and other proteases rapidly, in high yield and purity, without the need of additional enzymes, would be highly desirable and significant. Whether produced from plasma prothrombin (48–50) or made recombinant from prethrombin-1 (51, 52) or prethrombin-2 (23, 40, 53), thrombin must eventually separate from components of the prothrombinase complex or the snake venom metalloprotease ecarin used for activation, which adds regulatory hurdles and costs to large-scale production (54, 55). We have shown that the prethrombin-2 quadruple mutant EDGE obviates the use of activating enzymes because it converts to thrombin rapidly upon refolding and the product of autoactivation has functional properties toward synthetic and physiological substrates similar to those of wild-type. Recent studies show the feasibility of extending this strategy to production of activated protein C (36) and the anticoagulant thrombin mutant W215A/E217A (23) for clinical applications.
Acknowledgment
We are grateful to Tracey Baird for help with illustrations.
This work was supported in part by National Institutes of Health Research Grants HL49413, HL73813, and HL112303 (to E. D. C.).
The atomic coordinates and structure factors (codes 4H6T, 4HFY, 4H6S, and 4HFP) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PPACK
- H-d-Phe-Pro-Arg-CH2Cl.
REFERENCES
- 1. Page M. J., Di Cera E. (2008) Serine peptidases: classification, structure and function. Cell Mol. Life Sci. 65, 1220–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hedstrom L. (2002) Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524 [DOI] [PubMed] [Google Scholar]
- 3. Fehlhammer H., Bode W., Huber R. (1977) Crystal structure of bovine trypsinogen at 1–8 A resolution. II. Crystallographic refinement, refined crystal structure and comparison with bovine trypsin. J. Mol. Biol. 111, 415–438 [DOI] [PubMed] [Google Scholar]
- 4. Bode W., Schwager P., Huber R. (1978) The transition of bovine trypsinogen to a trypsin-like state upon strong ligand binding. The refined crystal structures of the bovine trypsinogen-pancreatic trypsin inhibitor complex and of its ternary complex with Ile-Val at 1.9 A resolution. J. Mol. Biol. 118, 99–112 [DOI] [PubMed] [Google Scholar]
- 5. Neurath H., Dixon G. H. (1957) Structure and activation of trypsinogen and chymotrypsinogen. Fed. Proc. 16, 791–801 [PubMed] [Google Scholar]
- 6. Krem M. M., Di Cera E. (2002) Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem. Sci. 27, 67–74 [DOI] [PubMed] [Google Scholar]
- 7. Carroll M. C., Isenman D. E. (2012) Regulation of humoral immunity by complement. Immunity 37, 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davie E. W., Fujikawa K., Kisiel W. (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 30, 10363–10370 [DOI] [PubMed] [Google Scholar]
- 9. Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K., Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 [DOI] [PubMed] [Google Scholar]
- 10. Gawlik K., Shiryaev S. A., Zhu W., Motamedchaboki K., Desjardins R., Day R., Remacle A. G., Stec B., Strongin A. Y. (2009) Autocatalytic activation of the furin zymogen requires removal of the emerging enzyme's N-terminus from the active site. PLoS One 4, e5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Artenstein A. W., Opal S. M. (2011) Proprotein convertases in health and disease. N. Engl. J. Med. 365, 2507–2518 [DOI] [PubMed] [Google Scholar]
- 12. Piper D. E., Jackson S., Liu Q., Romanow W. G., Shetterly S., Thibault S. T., Shan B., Walker N. P. (2007) The crystal structure of PCSK9: a regulator of plasma LDL-cholesterol. Structure 15, 545–552 [DOI] [PubMed] [Google Scholar]
- 13. Yamamoto E., Kitano Y., Hasumi K. (2011) Elucidation of crucial structures for a catechol-based inhibitor of plasma hyaluronan-binding protein (factor VII activating protease) autoactivation. Biosci. Biotechnol. Biochem. 75, 2070–2072 [DOI] [PubMed] [Google Scholar]
- 14. Sichler K., Banner D. W., D'Arcy A., Hopfner K. P., Huber R., Bode W., Kresse G. B., Kopetzki E., Brandstetter H. (2002) Crystal structures of uninhibited factor VIIa link its cofactor and substrate-assisted activation to specific interactions. J. Mol. Biol. 322, 591–603 [DOI] [PubMed] [Google Scholar]
- 15. Stirnberg M., Maurer E., Horstmeyer A., Kolp S., Frank S., Bald T., Arenz K., Janzer A., Prager K., Wunderlich P., Walter J., Gütschow M. (2010) Proteolytic processing of the serine protease matriptase-2: identification of the cleavage sites required for its autocatalytic release from the cell surface. Biochem. J. 430, 87–95 [DOI] [PubMed] [Google Scholar]
- 16. Kossiakoff A. A., Chambers J. L., Kay L. M., Stroud R. M. (1977) Structure of bovine trypsinogen at 1.9 A resolution. Biochemistry 16, 654–664 [DOI] [PubMed] [Google Scholar]
- 17. Gál P., Harmat V., Kocsis A., Bián T., Barna L., Ambrus G., Végh B., Balczer J., Sim R. B., Náray-Szabó G., Závodszky P. (2005) A true autoactivating enzyme. Structural insight into mannose-binding lectin-associated serine protease-2 activations. J. Biol. Chem. 280, 33435–33444 [DOI] [PubMed] [Google Scholar]
- 18. Wang D., Bode W., Huber R. (1985) Bovine chymotrypsinogen A X-ray crystal structure analysis and refinement of a new crystal form at 1.8 A resolution. J. Mol. Biol. 185, 595–624 [DOI] [PubMed] [Google Scholar]
- 19. Papagrigoriou E., McEwan P. A., Walsh P. N., Emsley J. (2006) Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat. Struct. Mol. Biol. 13, 557–558 [DOI] [PubMed] [Google Scholar]
- 20. Milder F. J., Gomes L., Schouten A., Janssen B. J., Huizinga E. G., Romijn R. A., Hemrika W., Roos A., Daha M. R., Gros P. (2007) Factor B structure provides insights into activation of the central protease of the complement system. Nat. Struct. Mol. Biol. 14, 224–228 [DOI] [PubMed] [Google Scholar]
- 21. Gohara D. W., Di Cera E. (2011) Allostery in trypsin-like proteases suggests new therapeutic strategies. Trends Biotechnol. 29, 577–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pozzi N., Vogt A. D., Gohara D. W., Di Cera E. (2012) Conformational selection in trypsin-like proteases. Curr. Opin. Struct. Biol. 22, 421–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pozzi N., Chen Z., Zapata F., Pelc L. A., Barranco-Medina S., Di Cera E. (2011) Crystal structures of prethrombin-2 reveal alternative conformations under identical solution conditions and the mechanism of zymogen activation. Biochemistry 50, 10195–10202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boehr D. D., Nussinov R., Wright P. E. (2009) The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. James L. C., Tawfik D. S. (2003) Conformational diversity and protein evolution–a 60-year-old hypothesis revisited. Trends Biochem. Sci. 28, 361–368 [DOI] [PubMed] [Google Scholar]
- 26. Vogt A. D., Di Cera E. (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fersht A. R., Requena Y. (1971) Equilibrium and rate constants for the interconversion of two conformations of -chymotrypsin. The existence of a catalytically inactive conformation at neutral p H. J. Mol. Biol. 60, 279–290 [DOI] [PubMed] [Google Scholar]
- 28. Marino F., Pelc L. A., Vogt A., Gandhi P. S., Di Cera E. (2010) Engineering thrombin for selective specificity toward protein C and PAR1. J. Biol. Chem. 285, 19145–19152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bode W., Turk D., Karshikov A. (1992) The refined 1.9-A x-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human α-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein Sci. 1, 426–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pineda A. O., Carrell C. J., Bush L. A., Prasad S., Caccia S., Chen Z. W., Mathews F. S., Di Cera E. (2004) Molecular dissection of Na+ binding to thrombin. J. Biol. Chem. 279, 31842–31853 [DOI] [PubMed] [Google Scholar]
- 31. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected by oscillation methods. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 32. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Morris A. L., MacArthur M. W., Hutchinson E. G., Thornton J. M. (1992) Stereochemical quality of protein structure coordinates. Proteins 12, 345–364 [DOI] [PubMed] [Google Scholar]
- 35. Prasad S., Cantwell A. M., Bush L. A., Shih P., Xu H., Di Cera E. (2004) Residue Asp-189 controls both substrate binding and the monovalent cation specificity of thrombin. J. Biol. Chem. 279, 10103–10108 [DOI] [PubMed] [Google Scholar]
- 36. Pozzi N., Barranco-Medina S., Chen Z., Di Cera E. (2012) Exposure of R169 controls protein C activation and autoactivation. Blood 120, 664–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vindigni A., Dang Q. D., Di Cera E. (1997) Site-specific dissection of substrate recognition by thrombin. Nat. Biotechnol. 15, 891–895 [DOI] [PubMed] [Google Scholar]
- 38. Lottenberg R., Hall J. A., Blinder M., Binder E. P., Jackson C. M. (1983) The action of thrombin on peptide p-nitroanilide substrates. Substrate selectivity and examination of hydrolysis under different reaction conditions. Biochim. Biophys. Acta 742, 539–557 [DOI] [PubMed] [Google Scholar]
- 39. Claeson G. (1994) Synthetic peptides and peptidomimetics as substrates and inhibitors of thrombin and other proteases in the blood coagulation system. Blood Coagul. Fibrinolysis 5, 411–436 [PubMed] [Google Scholar]
- 40. Virel A., Saa L., Pavlov V. (2012) Quantification of prothrombin in human plasma amplified by autocatalytic reaction. Anal. Chem. 84, 2380–2387 [DOI] [PubMed] [Google Scholar]
- 41. Fersht A. R. (1999) Enzyme Structure and Mechanism, Freeman, New York, NY [Google Scholar]
- 42. Niu W., Chen Z., Gandhi P. S., Vogt A. D., Pozzi N., Pelc L. A., Zapata F., Di Cera E. (2011) Crystallographic and kinetic evidence of allostery in a trypsin-like protease. Biochemistry 50, 6301–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wolan D. W., Zorn J. A., Gray D. C., Wells J. A. (2009) Small-molecule activators of a proenzyme. Science 326, 853–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gandhi P. S., Chen Z., Di Cera E. (2010) Crystal structure of thrombin bound to the uncleaved extracellular fragment of PAR1. J. Biol. Chem. 285, 15393–15398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Z., Pelc L. A., Di Cera E. (2010) Crystal structure of prethrombin-1. Proc. Natl. Acad. Sci. U.S.A. 107, 19278–19283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Craik C. S., Page M. J., Madison E. L. (2011) Proteases as therapeutics. Biochem. J. 435, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Di Cera E. (2011) Thrombin as an anticoagulant. Prog. Mol. Biol. Transl. Sci. 99, 145–184 [DOI] [PubMed] [Google Scholar]
- 48. Fenton J. W., 2nd, Fasco M. J., Stackrow A. B. (1977) Human thrombins. Production, evaluation, and properties of α-thrombin. J. Biol. Chem. 252, 3587–3598 [PubMed] [Google Scholar]
- 49. Bajaj S. P., Mann K. G. (1973) Simultaneous purification of bovine prothrombin and factor X. Activation of prothrombin by trypsin-activated factor X. J. Biol. Chem. 248, 7729–7741 [PubMed] [Google Scholar]
- 50. Ngai P. K., Chang J. Y. (1991) A novel one-step purification of human α-thrombin after direct activation of crude prothrombin enriched from plasma. Biochem. J. 280, 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Le Bonniec B. F., Esmon C. T. (1991) Glu-192—-Gln substitution in thrombin mimics the catalytic switch induced by thrombomodulin. Proc. Natl. Acad. Sci. U.S.A. 88, 7371–7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu Q. Y., Sheehan J. P., Tsiang M., Lentz S. R., Birktoft J. J., Sadler J. E. (1991) Single amino acid substitutions dissociate fibrinogen-clotting and thrombomodulin-binding activities of human thrombin. Proc. Natl. Acad. Sci. U.S.A. 88, 6775–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. DiBella E. E., Maurer M. C., Scheraga H. A. (1995) Expression and folding of recombinant bovine prethrombin-2 and its activation to thrombin. J. Biol. Chem. 270, 163–169 [DOI] [PubMed] [Google Scholar]
- 54. Aizawa P., Winge S., Karlsson G. (2008) Large-scale preparation of thrombin from human plasma. Thromb. Res. 122, 560–567 [DOI] [PubMed] [Google Scholar]
- 55. Heffernan J. K., Ponce R. A., Zuckerman L. A., Volpone J. P., Visich J., Giste E. E., Jenkins N., Boster D., Pederson S., Knitter G., Palmer T., Wills M., Early R. J., Rogge M. C. (2007) Preclinical safety of recombinant human thrombin. Regul. Toxicol. Pharmacol. 47, 48–58 [DOI] [PubMed] [Google Scholar]