

Abstract
Macroautophagy mediates the bulk degradation of cytoplasmic components. It accounts for the degradation of most long-lived proteins: cytoplasmic constituents, including organelles, are sequestered into autophagosomes, which subsequently fuse with lysosomes, where degradation occurs. Although the possible involvement of autophagy in homeostasis, development, cell death, and pathogenesis has been repeatedly pointed out, systematic in vivo analysis has not been performed in mammals, mainly because of a limitation of monitoring methods. To understand where and when autophagy occurs in vivo, we have generated transgenic mice systemically expressing GFP fused to LC3, which is a mammalian homologue of yeast Atg8 (Aut7/Apg8) and serves as a marker protein for autophagosomes. Fluorescence microscopic analyses revealed that autophagy is differently induced by nutrient starvation in most tissues. In some tissues, autophagy even occurs actively without starvation treatments. Our results suggest that the regulation of autophagy is organ dependent and the role of autophagy is not restricted to the starvation response. This transgenic mouse model is a useful tool to study mammalian autophagy.
INTRODUCTION
Eukaryotes have two major protein degradation systems within cells. One is the ubiquitin-proteasome system, which accounts for the selective degradation of most short-lived proteins (Hochstrasser, 1996; Hershko and Ciechanover, 1998). The other is the lysosomal system. Proteins from both inside and outside of the cell are delivered to the lytic compartment. Degradation of exogenous materials and plasma membrane proteins is mediated by the process of endocytosis/phagocytosis, whereas degradation of cytoplasmic components is achieved by autophagy (also known as autophagocytosis). Three types of autophagy have been proposed: macroautophagy, microautophagy, and chaperon-mediated autophagy (Seglen and Bohley, 1992; Dunn, 1994; Blommaart et al., 1997). Macroautophagy is thought to be responsible for the majority of the intracellular protein degradation in mammalian cells, particularly during starvation-induced proteolysis (Mortimore and Pösö, 1987).
Macroautophagy (simply referred to as autophagy hereafter) is mediated by a unique organelle termed the autophagosome. A membrane cisterna called the isolation membrane (also known as phagophore) encloses a portion of cytoplasm, resulting in the formation of the autophagosome. The autophagosome is a double-membrane structure containing undigested cytoplasmic materials including organelles. The sequestration step is generally thought to be nonselective. Next, the outer membrane of the autophagosome fuses with the lysosome membrane. Various hydrolytic enzymes are supplied to the autophagosome and the cytoplasm-derived contents are degraded together with the inner membrane of the autophagosome. This degrading structure is termed the autolysosome/autophagolysosome.
Autophagy is thought to be required for normal turnover of cellular components particularly in response to starvation (Mortimore and Pösö, 1987). Autophagy-defective yeast cells die quickly during starvation (Tsukada and Ohsumi, 1993). Autophagy also plays an important role in some types of differentiation/development: ATG genes (described below) are essential for spore formation in yeast (Tsukada and Ohsumi, 1993), and the development of Drosophila melanogaster (Juhasz et al., 2003), Dictyostelium discoideum (Otto et al., 2003), and Caenorhabditis elegans (Melendez et al., 2003). Plants deficient for autophagy genes show acceleration of senescence (Doelling et al., 2002; Hanaoka et al., 2002). In contrast, the precise roles of autophagy in mammals are not known, although a growing number of studies have suggested that autophagy might be important for cell death during embryogenesis (Clarke, 1990) and pathogenesis (Liang et al., 1999; Nishino et al., 2000). In addition, systematic analysis describing where and when autophagy occurs has not been performed. This is largely due to a lack of good diagnostic methods. To date, electron microscopy has been the only method to monitor autophagy. Unfortunately, this is a method requiring many skills and much time, and sometimes it is difficult to distinguish autophagic vacuoles from other structures just by morphology. Although an elegant transgenic mouse model was recently generated to monitor the ubiquitin/proteasome system (Lindsten et al., 2003), we have not had such in vivo assay systems for autophagy.
We have dissected the autophagic pathway at the molecular level using both yeast and mammalian cells. In the yeast, Saccharomyces cerevisiae, at least 16 of APG and AUT genes have been identified to be required for autophagosome formation (Klionsky and Ohsumi, 1999; Ohsumi, 2001; Mizushima et al., 2002a). The nomenclature of these autophagy-related genes were recently unified as ATG (Klionsky et al., 2003). We have found two novel ubiquitylation-like conjugation systems: one mediates conjugation of Atg12 to Atg5 (Mizushima et al., 1998a) and the other mediates a covalent linkage between Atg8 (Aut7/Atg8) and phosphatidylethanolamine (PE; Ichimura et al., 2000). The resulting conjugates, Atg12-Atg5 and Atg8-PE, function in autophagosome formation (Suzuki et al., 2001; Noda et al., 2002). These two conjugation systems are highly conserved in mammals (Mizushima et al., 1998b, 2002b; Kabeya et al., 2000; Tanida et al., 2001, 2002). Most Atg12-Atg5 conjugate exists in the cytosol as a complex with Atg16L (Mizushima et al., 1999, 2003; Kuma et al., 2002). Only a small fraction of the Atg12-Atg5·Atg16L complex localizes to the autophagic isolation membranes throughout its elongation process, and the complex dissociates from the membrane when autophagosome formation is completed (Mizushima et al., 2001). We have demonstrated that mouse Atg12-Atg5 conjugate is indeed essential for the membrane elongation process by generating ATG5-/- embryonic stem (ES) cells (Mizushima et al., 2001). LC3, one of the mammalian homologues of Atg8, also targets the isolation membrane in an Atg5-dependent manner. However, LC3 remains on the membrane even after spherical autophagosomes are completely formed (Kabeya et al., 2000; Mizushima et al., 2001). When we label autophagosomes with green fluorescent protein (GFP)-LC3, we are able to see them by fluorescence microscopy as ring-shaped structures (Mizushima et al., 2003), or dots in the case of small autophagosomes (Kabeya et al., 2000). Because autolysosomes have less LC3 on the membrane than autophagosomes, the fluorescent signal of autolysosomes is weaker. On the basis of these observations, we propose that LC3 could serve as a good molecular marker for isolation membranes, autophagosomes, and some autolysosomes (Kabeya et al., 2000).
We have applied this method to in vivo studies to monitor autophagy simply and accurately. We generated GFP-LC3 transgenic mice and examined the occurrence of autophagy in various tissues. Using this novel assay system, we performed comprehensive and quantitative analysis on the autophagic response during starvation.
MATERIALS AND METHODS
Antibodies
An antibody against recombinant rat LC3 has been described previously (Kabeya et al., 2000). Polyclonal anti-GFP antibody (for immunoblotting) and Alexa Fluor 568 goat anti-rabbit IgG (H+L) antibody were purchased from Molecular Probes (Eugene, OR). Polyclonal anti-GFP antibody (for immuno-electron microscopy) was purchased from Clontech (Palo Alto, CA). Rabbit antiporcine elastase antibody (Rockland), rabbit antibovine carboxypeptidase-A antibody (Rockland), rabbit antiserum to trypsin (Nordic Immunological Labs, Westbury, NY) and sheep anti-human amylase (pancreatic) antibody (The Binding Site Limited, San Diego, CA) were used for immunoblotting. Rabbit anti-mouse keratin antibody was purchased from LSL Co., Ltd. (Princeton, NJ).
Construction of Plasmids
A 1.8-kbp DNA fragment containing the rat LC3 cDNA fused to EGFP at the N-terminus (GFP-LC3) and the SV40 late polyadenylation signal was excised from pEGFP-LC3 (Kabeya et al., 2000) and inserted into the SmaI site of pK-CA6 (kindly provided by Dr. Zubair) downstream of the cytomegalovirus immediate-early (CMVie) enhancer and chicken β-actin (CAG) promotor (Niwa et al., 1991) to generate pCAG-GFP-LC3.
Generation of F9 Stable Transformants
F9 teratocarcinoma cells were cultured on gelatinized dishes in DMEM containing 10% fetal calf serum (FCS), 5 U/ml penicillin, and 50 μg/ml streptomycin. To obtain stable transformants, 5 × 106 F9 cells were electroporated with 10 μg of linearized pEGFP-LC3 (950 μF, 220 V) and selected in the presence of 0.5 mg/ml geneticin (Invitrogen, Carlsbad, CA). A clone (GLF3) overexpressing GFP-LC3 was used in this study. For amino acid starvation, cells were cultured in Hanks' solution containing 10 mM HEPES, pH 7.5 (without amino acids and FCS) for 2 h.
Generation of Transgenic Mice
The 3.4-kbp XhoI and SpeI fragment was isolated from pCAG-GFP-LC3 and microinjected into C57BL/6N Crj × BDF1 fertilized eggs. Microinjected eggs were transferred into the oviducts of pseudopregnant foster females. Sixty mice were screened for incorporation of the transgene by PCR using primers GFP1 (5′-TCCTGCTGGAGTTCGTGACCG-3′) and LC3* (5′-TTGCGAATTCTCAGC-CGTCTTCATCTCTCTCGC-3′). Another primer sets (mLC3ex3GT: 5′-TGAGC-GAGCTCATCAAGATAATCAGGT-3′ and mLC3ex4AG: 5′-GTTAGCATT-GAGCTGCAAGCGCCGTCT-3′) amplifying the third intron of the LC3 genome as an internal control was used. Eight of the mice were positive for the transgene. Positive mice were crossed with C57BL/6N Crj and maintained as heterozygotes for the GFP-LC3 transgene. One of the transgenic lines, GFP-LC3#53 was used in this study. Genotyping was carried out by PCR analysis or examination of the GFP signal by GFP macroscopy (Biological Laboratory Equipment, Maintenance, and Service Ltd., Budapest, Hungary). The lighting regimen for the mice was: light period 7 a.m. to 7 p.m. and dark period 7 p.m. to 7 a.m. GFP-LC3 mice aged from 6 to 8 weeks were used in this study. For studies of effects of starvation, mice were deprived of food for 24 or 48 h (10 a.m. to 10 a.m.). These mice had free access to drinking water.
Immunoblotting
F9 cell lysates were prepared with lysis buffer (2% NP-40, 0.2% SDS in PBS supplemented with protease inhibitors). Mouse tissues were homogenized in nine volumes of ice-cold PBS supplemented with protease inhibitors. The homogenates were centrifuged at 500 × g at 4°C for 10 min. Protein extracts, 50 μg, were subjected to SDS-PAGE and immunoblotting as previously described (Mizushima et al., 1998b). Quantification of intensities of the immunoreactive bands were carried out using LAS-1000 chemiluminescence imaging system (Fujifilm, Tokyo, Japan).
Fluorescence Microscopy
F9 cells expressing GFP-LC3 were directly observed by a fluorescence microscope (Olympus IX81, Tokyo, Japan) equipped with a CCD camera (ORCA ER, Hamamatsu Photonics, Hamamatsu, Japan). U-MGFPHQ mirror unit was used for GFP observation and U-MWIG2 mirror unit was used to check autofluorescence.
Tissue samples for GFP observation were prepared as follows. To prevent induction of autophagy during tissues preparation, mice were anesthetized with diethyl ether and immediately fixed by perfusion through the left ventricle with 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (PB; pH 7.4). Tissues were harvested and further fixed with the same fixative for at least 4 h, followed by treatment with 15% sucrose in PBS for 4 h and then with 30% sucrose solution overnight. Tissue samples were embedded in Tissue-Tek OCT compound (Sakura Finetechnical Co., Ltd., Tokyo, Japan) and stored at -70°C. The samples were sectioned at 5–7-μm thickness with cryostat (CM3050 S, Leica, Deerfield, IL), air-dried for 30 min, and stored at -20°C until use.
For immunofluorescence microscopy, mice were perfused with 2% PFA for 10 min, and tissues were removed and immediately immersed in sucrose solution. Cryosections were prepared as described above. After blocking with 4% skim milk in PBS, sections were incubated with rabbit anti-mouse keratin antibody for 1 h, followed by 1-h incubation of Alexa fluor 568 anti-rabbit IgG antibody (1:200 dilution). Samples were mounted using SlowFade Light Antifade Kit (Molecular Probes).
Quantitative Analysis of GFP-LC3 Dots
The number of GFP-LC3 dots in each organ was counted in five independent visual fields from at least five independent mice. The results were expressed as the mean ± SD. The probability of statistical differences between experimental groups was determined by the Student's t test.
Electron Microscopy and Immunoelectron Microscopy
Conventional electron microscopy of F9 cells was performed as described previously (Yoshimori et al., 2000), except that F9 cells grown on gelatinized plastic coverslips were prefixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4, for 2 h. Morphometric analysis was performed as previously described (Mizushima et al., 2001).
For immunoelectron microscopy, the preembedding silver enhancement immunogold method was performed as previously described (Yoshimori et al., 2000), with a slight modification. Mouse tissues were fixed in 4% paraformaldehyde in 0.1 M PB, pH 7.4, for 4 h. Cryosections, 6 μm, were prepared and incubated in PB containing 0.005% saponin, 10% bovine serum albumin, 10% normal goat serum, and 0.1% cold water fish skin gelatin for blocking for 30 min. Tissues sections were then treated with rabbit IgG against GFP (diluted 500×) in the blocking solution, overnight. Then, the sections were washed in PB containing 0.005% saponin for 10 min six times and incubated with goat anti-rabbit IgG conjugated to colloidal gold (1.4 nm diameter) in the blocking solution for 2 h. The samples were washed with PB for 10 min six times and fixed with 1% glutaraldehyde in PB for 10 min. After washing, the gold labeling was intensified by using a silver enhancement kit for 6 min at 20°C in the dark. After washing in distilled water, cells were postfixed in 0.5% OsO4 for 90 min at 4°C, washed in distilled water, incubated with 50% ethanol for 10 min, and stained with 2% uranyl acetate in 70% ethanol for 2 h. The samples were further dehydrated with a graded series of ethanol and embedded in epoxy resin. Ultrathin sections were doubly stained with uranyl acetate and lead citrate.
RESULTS
GFP-LC3 as a Marker for Autophagic Membranes
We have previously demonstrated that GFP-LC3 specifically labels autophagic isolation membranes and autophagosomes (Kabeya et al., 2000). Although some of the GFP-LC3 dissociates from the membrane after fusion with the lysosome, GFP-LC3 still weakly labels autolysosomes. Thus, GFP-LC3 serves as a specific marker for autophagic membranes.
It is a concern that exogenously overexpressed GFP-LC3 may inhibit or activate autophagic activity in the expressing cells. We tested this possibility using culture cells. We generated F9 teratocarcinoma cells stably overexpressing GFP-LC3 (Figure 1). The level of overexpressed GFP-LC3 was more than 10 times the level of endogenous LC3 (Figure 1B). After 2-h amino acid starvation, many ring-shaped GFP-LC3 structures were observed in these cells, confirming that GFP-LC3 functions as an autophagosome marker even when it is overexpressed (Figure 1A). We have shown that endogenous LC3 is present in two different forms (Kabeya et al., 2000). When autophagy is induced, cytosolic LC3-I is converted to the second form, LC3-II, which associates with autophagosomes and is likely conjugated to phosphatidylethanolamine (PE; Kabeya et al., unpublished results). These two forms of LC3 can be clearly distinguished by SDS-PAGE and the amount of LC3-II is highly correlated with the number of autophagosomes (Kabeya et al., 2000). Using the F9 stable cell line, we confirmed that overexpression of GFP-LC3 does not affect the endogenous LC3-II/LC3-I ratio when cells are cultured in nutrient-rich medium (Figure 1B). The conversion of LC3-I to LC3-II is also normal in this cell line after 2-h starvation. We next examined these cells by electron microscopy and morphometric analyses (Figure 1C). Very few autophagic vacuoles (autophagosomes and autolysosomes) were observed in wild-type F9 cells and F9 cells overexpressing GFP-LC3 cultured in nutrient-rich medium. However, after amino acid starvation, the volume of autophagic vacuoles increased to ∼2% of the total cytoplasm in both cell lines, and there were no significant differences. These results suggest that overexpression of GFP-LC3 does not affect autophagic activity.
Figure 1.

Overexpression of GFP-LC3 does not affect the autophagic status of F9 cells. (A) F9 cells overexpressing GFP-LC3 were incubated in Hanks' solution for the indicated time and directly observed by fluorescence microscopy. Bar, 10 μm. (B) Total cell lysates were prepared from wild-type F9 cells and F9 cells overexpressing GFP-LC3 before or after 2-h amino acid starvation and analyzed by immunoblotting using anti-LC3 antibody. The band intensities of endogenous LC3-I and LC3-II were quantified. The LC3-II/LC3–1 ratio was shown. (C) Wild-type F9 cells and F9 cells overexpressing GFP-LC3 before or after 2-h amino acid starvation were examined by electron microscopy and morphometric analysis. The ratio of the total area of autophagic vacuoles to the total cytoplasmic area is shown.
Generation of GFP-LC3 Transgenic Mice
Because exogenously expressed GFP-LC3 serves as an ideal molecular marker for autophagic membranes, we generated transgenic mouse lines expressing GFP-LC3 under the control of the constitutive CAG promoter. Eight transgenic mouse lines were obtained, and one line, GFP-LC3#53, was used in this study. This transgenic line shows no abnormality. GFP-LC3 was expressed during prenatal stages as in the case in previously reported green mice (Okabe et al., 1997). In adult mice, GFP-LC3 was expressed in almost all tissues we examined (Figure 2). The level of GFP-LC3 expression was comparable to the level of endogenous LC3 expression in the brain, whereas GFP-LC3 was overexpressed in other tissues. In particular, the level of GFP-LC3 expression was much higher than that of endogenous LC3 in the heart, liver, pancreas, and skeletal muscle. Such overexpression was comparable to that in the F9 GFP-LC3 stable transformants (Figure 1B). Various organs were removed from 4% PFA-perfused mice aged from 6 to 8 weeks. Conventional cryosections were prepared and examined.
Figure 2.

Expression of GFP-LC3 in the GFP-LC3 transgenic mice. Twenty-five milligrams of each tissue homogenate was subjected to SDS-PAGE and analyzed by immunoblotting with anti-GFP and anti-LC3 antibodies.
Liver
We first examined the liver because autophagy has been analyzed most frequently in this tissue. In the hepatocytes of fed mice, the GFP-LC3 signal was detected diffusely in the cytoplasm with few punctate dots (Figure 3A). The number of GFP-LC3 dots generally increased after 24-h starvation, and most of them were detected as cup-shaped and ring-shaped structures (Figure 3, B and C). To confirm that these GFP-positive structures indeed represented isolation membranes and autophagosomes, we performed immunoelectron microscopy using an anti-GFP antibody. The silver-enhanced gold particles were associated with isolation membranes (Figure 3D) and autophagosomes (Figure 3E). These observations confirmed that GFP-LC3 serves as a marker for autophagic membranes in the transgenic mice.
Figure 3.

Liver autophagy in response to starvation. (A–C) Liver samples were prepared from GFP-LC3 transgenic mice before (A) or after 24-h starvation (B and C) and fixed with 4% paraformaldehyde. Cryosections were analyzed by fluorescence microscopy. Panel C demonstrates the most highly induced case. Bar, 10 μm. (D and E) Localization of GFP-LC3 in hepatocytes from 24-h starved GFP-LC3 mice. Liver samples were prepared from GFP-LC3 transgenic mice after 24-h starvation and fixed with 4% paraformaldehyde. The localization of GFP-LC3 was examined by silver-enhanced immunogold electron microscopy using an anti-GFP antibody. A cup-shaped isolation membrane (arrow in D) and double-membrane autophagosome (arrow in E) were shown. Bar, 1 μm.
Starvation-induced autophagic activity as measured by GFP-LC3 staining differed among individual mice (e.g., Figure 3, B vs. C). We quantified the number of GFP-LC3-positive structures in 10 independent mice and found that this number was slightly but significantly increased in the livers of mice starved for 24 h (see Figure 7). However, the level of autophagy returned to almost the basal level after 48-h starvation.
Figure 7.

Quantitative analysis of the formation of GFP-LC3 dots during starvation. The number of GFP-LC3 dots was counted and divided by the corresponding area. For analysis of skeletal muscles, transverse sections were used. Because the glomerulus and thymic cortex consist of several kinds of cells, the actual densities of GFP-LC3 dots in podocytes and thymic epithelial cells were not calculated. The Y-axis indicates the number of GFP-LC3 dots (×104/mm2). Each value represents the mean ± SD of at least five mice. *p < 0.05 and **p < 0.01.
Muscle Tissues
We examined the gastrocnemius muscles and found a very clear starvation response: many GFP-LC3 dots were significantly induced between myofibrils and in the perinuclear regions after 24-h starvation. We also found that there was a population of muscle fibers showing much fewer dots. We hypothesize that this might reflect the difference in muscle fiber types, because differential regulation of protein degradation in slow- and fast-twitching muscles has been previously reported (Li and Goldberg, 1976; Frayn and Maycock, 1979; Dahlmann et al., 1986; Vary and Kimball, 1992). We compared fast-twitching extensor digitorum longus (EDL) muscles and slow-twitching soleus muscles. In EDL muscles, a very small number of GFP-LC3 dots were observed before starvation. However, many small GFP-LC3 dots appeared between myofibrils and in the perinuclear regions after 24-h starvation (Figure 4, A and C). Immunoelectron microscopy demonstrated many autophagosomes labeled with silver-enhanced gold particles between myofibrils (Figure 4F, arrows) and the perinuclar regions (Figure 4G, arrows). The level of autophagy after 48-h starvation was almost the same as at 24 h or slightly decreased in EDL muscles.
Figure 4.

Autophagy in muscle tissues. (A and B) GFP images of transverse sections of extensor digitorum longus (EDL) muscles (A) and soleus muscles (B) after the indicated starvation periods. Bar, 10 μm. (C and D) Longitudinal section of EDL muscles after 24-h starvation (C) and soleus muscles after 48-h starvation (D). Asterisks indicate nuclei. Bar, 10 μm. (E) GFP images of heart muscles at 0-, 24-, and 48-h starvation. Bar, 10 μm. (F and G) Immunoelectron microscopic analysis of gastrocnemius muscle after 24-h starvation. Autophagosomes associated with GFP-LC3 (arrows) accumulate between myofibrils (F) and at perinuclear regions (G). Bar, 1 μm. (H) Immunoelectron microscopic analysis of heart muscles from 24-h starved mice. A cluster of autophagosomes is generated between myofibrils (arrows). The double arrow indicates an autophagosome enclosing a mitochondrion. Bar, 1 μm.
In contrast, in slow-twitching soleus muscles, very little autophagy was induced after 24-h starvation (Figure 4B). Moderate autophagy was observed after 48-h starvation. Localization of the GFP-LC3 dots in soleus muscles was different from that in EDL muscles. Most GFP-LC3 dots appeared in the periphery of muscle fibers, particularly in the perinuclear regions (Figure 4, B and D). Very few dots were detected between myofibrils even after 48-h starvation.
The heart muscles also showed drastic changes during starvation. The heart muscle from fed mice showed very low levels of autophagy (Figure 4E). The number and size of autophagosomes were increased during 48-h starvation. At 48 h, many large ring-shaped structures were easily recognized by fluorescence microscopy. Immunoelectron microscopy revealed that many autophagosomes were generated in the cytoplasm (Figure 4H, arrows), and these autophagosomes frequently enclosed mitochondria (Figure 4H, double arrow).
Pancreas
The acinar cells in the exocrine pancreas have numerous zymogen granules containing a variety of proteases (precursors). Although a few GFP-LC3 dots were present before the starvation treatment, they were relatively small. After 24-h starvation, the size and number of zymogen granules were decreased (Figure 5A). In addition, many autophagosomes were generated in apical areas overlapping or surrounding the zymogen granule areas during starvation (Figure 5A). After 24-h starvation, the GFP-LC3 structures were large enough to be recognized as ring-shaped structures. This size difference was also evidenced by immunoelectron microscopy. In the fed state, large zymogen granules and small GFP-LC3–positive autophagosomes were observed (Figure 5B). In contrast, after 24-h starvation, larger autophagosomes and autolysosomes were detected, some of which indeed enclosed zymogen granules (Figure 5B). The amounts of proteases such as trypsin, amylase, elastase, and carboxypeptidase decreased during the 24-h starvation (Figure 5C). Therefore, massive induction of autophagy during starvation correlates with the loss of zymogen granules, suggesting that zymogen granules in pancreatic acinar cells might be degraded by autophagy. After 48-h starvation, most zymogen granules disappeared and generation of autophagosomes was also decreased (see Figure 7).
Figure 5.

Autophagy in pancreatic exocrine cells. (A) GFP images (top panels) and corresponding differential interference contrast (DIC) images (bottom panels) at the indicated starvation periods are shown. Bars, 10 μm. (B) Immunoelectron microscopic analysis of pancreatic acinar cells. Before starvation, large zymogen granules and small autophagic vacuoles (arrows) are observed (left panel). After 24-h starvation, the size of autophagosomes is enlarged, and some zymogen granules are observed in the autophagosomes (double arrows; right panel). Bar, 1 μm. (C) Amounts of pancreatic proteases decrease during starvation. Homogenates were prepared from three independent mice before and after 24-h starvation and subjected to immunoblot analysis using antitrypsin, antiamylase, antielastase, and anticarboxypeptidase antibodies. Anti-tubulin antibody was used as a control.
Kidney
Punctate GFP-LC3 dots were observed in glomeruli in the kidney (Figure 6A). The glomerulus consists of three cell types: endothelial cells, visceral epithelial cells (podocytes), and mesangial cells. The distribution pattern of cells with GFP-LC3 dots suggested that these cells were podocytes. By immunoelectron microscopy, typical autophagosomes labeled with GFP-LC3 were observed in podocytes with characteristic foot processes (Figure 6B). Interestingly, the basal level of autophagy was relatively high in podocytes, and it was further enhanced by starvation (Figure 7).
Figure 6.

Tissues showing high levels of autophagy before starvation. (A) GFP fluorescence of the glomerulus from a fed mouse. Bar, 10 μm. Inset: high-magnification images from other areas. Bar, 2 μm. (B) Immunoelectron microscopy of a podocyte using anti-GFP antibody. The arrows indicate autophagosomes. Bar, 1 μm. (C) The thymus cortex from a fed mouse. GFP fluorescence, anticytokeratin staining, and a merged image are shown. Bar, 10 μm. (D) Immunoelectron microscopy of the thymic cortex. The arrow indicates an autophagosome in a stromal epithelial cell. Bar, 1 μm. (E) A GFP image of the lens epithelial cells of a fed mouse. Bar, 10 μm. Insets: high-magnification images from other sections. Some GFP-LC3–positive structures in epithelial cells are detected as ring- and cup-shaped. Bar, 1 μm. (F) Immunoelectron microscopy of the lens. An arrow and asterisk indicate the autophagosome and a lens fiber, respectively. Bar, 1 μm.
We also observed specific GFP-LC3 dots and ring-shaped structures in the renal tubules of starved mice (our unpublished results). However, the expression level of GFP-LC3 was not high and there were autofluorescent signals in tubules. Because it was difficult to quantify the number of dots precisely, we excluded the analysis of tubules from this study.
Thymus
In the thymus, many GFP-LC3 dots were observed in stromal cells with stellate shapes in both the cortex and medulla (Figure 6C). Because they stained positively with anticytokeratin antibody, these cells were identified as epithelial reticular cells. Small GFP-LC3 dots were present in both the cell bodies and processes of the reticular cells. Immunoelectron microscopy confirmed that reticular cells surrounded by lymphocytes contained GFP-LC3–positive autophagosomes (Figure 6D). It is noteworthy that autophagy in the thymic epithelial cells occurs even without starvation treatment. We quantified the number of GFP-LC3 dots in the cortex and found that food withdrawal did not further induce autophagy in these cells (see Figure 7). Therefore, autophagy in thymic epithelial cells is constitutively active, irrespective of nutrient conditions. Such a pattern is almost the same for the medulla. However, we did not perform quantitative analysis of the medullar because there was some autofluorescence in this area (see Figure 9). In addition, thymic autophagy was more active in newborns (our unpublished observation).
Figure 9.

Distinction between GFP-LC3 and autofluorescence signals. Samples from the frontal cortex of the brain (A) and the medulla of the thymus (B) were analyzed for green (left panels) and red (middle panels) fluorescence. Merged images are shown in the right panels. GFP-specific signals (arrows) and autofluorescent signals (arrowheads) are indicated. A neuron-like cell (A) and stromal cells show autofluorescence. Similar autofluorescent signals were also observed in samples from nontransgenic mice. Bar, 10 μm.
Lens
The anterior surface of the lens is covered with a single layer of epithelial cells. At the equator region, these epithelial cells are transformed into lens fibers. Although the expression level of GFP-LC3 was relatively low in the lens epithelial cells, a number of GFP-LC3 dots were observed in these cells (Figure 6E). Some of them were large enough to be identified as cup- and ring-shaped structures (Figure 6E, insets). Autophagosomes labeled with GFP-LC3 were frequently observed in the lens epithelial cells but not in the lens fibers (Figure 6F). Autophagosomes were ubiquitously found in the outer epithelial cells, not limited to the transition sites at the equatorial region. As in the thymic epithelial cells, autophagy in the lens epithelial cells is also constitutively active and does not depend on nutrient conditions (see Figure 7)
Other Tissues
In addition to pancreatic acinar cells, other exocrine gland cells such as gastric chief cells and seminal vesicle cells showed moderate basal levels of autophagy, which were further enhanced by nutrient starvation (our unpublished results). Type II alveolar cells also showed punctate GFP-LC3 dots, and the number of dots increased after starvation (our unpublished results). Despite high-enough expression of GFP-LC3, GFP-LC3 dots were not observed in the brain, ovary, testis, and adipocytes (our unpublished results).
Quantitative Comparison of the Starvation Response between Organs
We quantified the number of GFP-LC3–positive structures in each organ. The patterns of response to starvation differ between organs (Figure 7). We have roughly divided these responses into five patterns. In the first pattern, autophagy is increasingly induced during the 48-h starvation. The heart is the best example of this group. Soleus muscles (slow twitching) show a similar pattern, but the level is much lower. The second group consists of EDL muscles (fast-twitching), gastrocnemius muscles (mostly fast-twitching; Ariano et al., 1973), and podocytes, in which autophagy is significantly induced during the first 24-h starvation and then sustained. In the third group, which includes liver and pancreas, autophagy is induced during the first 24-h starvation but returns to almost the basal level during the next 24-h starvation. The fourth group demonstrates a constitutively active pattern such as that seen in thymic epithelial cells and lens epithelial cells. In these cells, active autophagy was observed before starvation treatment and is not further enhanced by starvation. The fifth pattern is that autophagy is not induced even after the 48-h starvation treatment. The brain shows this pattern (our unpublished results). These results indicate that autophagy does not occur uniformly in mice and suggest that regulation of autophagy is organ specific.
Conversion of LC3-I to LC3-II in Tissues
The autophagic activity in each tissue was also assessed by immunoblotting of endogenous LC3 using wild-type mice (Figure 8). In the skeletal muscles, heart, and pancreas, conversion of LC3-I to LC3-II during the 24-h starvation was apparent. Slight increases in the level of LC3-II were also observed in kidney and liver. In contrast, almost no conversion was detected in the brain. These results are consistent with the morphological examination.
Figure 8.
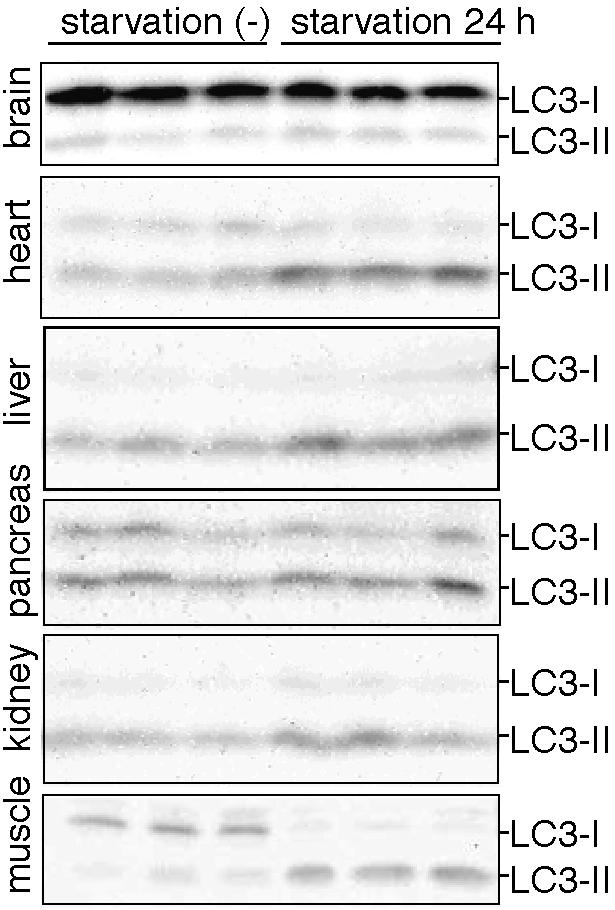
Conversion of LC3-I to LC3-II in mouse tissues during 24-h starvation. Tissue homogenates were prepared from three fed mice and three 24-h starved mice and subjected to immunoblot analysis using anti-LC3 antibody. The positions of LC3-I and LC3-II are indicated.
DISCUSSION
A Novel Monitoring Method for Autophagy
We have described a novel method to monitor autophagy in vivo. To date, assessment of autophagic activity in vivo has been achieved only by morphometric analysis by electron microscopy. In contrast, our method using the GFP-LC3 transgenic mice has several advantages. First, it is a simple and quick procedure. It enables us to examine many samples for a short time. Because autophagic activity is very dynamic, it may be important to analyze a large number of samples to obtain statistically significant results. Second, labeling of autophagosomes with GFP-LC3 is specific. Our examination by immunoelectron microscopy demonstrated that GFP-LC3 localizes only on autophagic membranes but not on other membrane structures (Kabeya et al., 2000). Therefore, this technique is very useful for the accurate identification of autophagic vacuoles that are ambiguous structures by electron microscopy. We have also confirmed that overexpression of GFP-LC3 did not affect the endogenous autophagic process. Third, we can easily observe not only the distribution of autophagosomes in cells but also the distribution of autophagy-active cells in tissues. Forth, usual fluorescence techniques, such as multicolor staining and three-dimensional reconstitution are feasible. Fifth, if we prepare primary cultures from the transgenic mice, real-time observation of living cells can be performed without any staining.
However, fluorescence images should be carefully interpreted. Some cells such as neurons (Figure 9A) and thymic stromal cells (particularly in the medulla; Figure 9B) show autofluorescent dot structures like lipofuscin. We distinguish specific GFP-LC3 signals from these autofluorescent signals by two methods. First, it is particularly important to compare samples from transgenic mice with samples from wild-type littermates. Specific GFP signals must be detected only in transgenic animals. Second, the GFP-LC3 signals must not be detected using other fluorescence filter sets such as for rhodamine and Cy5 (Figure 9). True GFP-LC3 signals should be detected specifically by the GFP or FITC filter set. All the fluorescence images demonstrated here (except for Figure 9) meet the criteria. We believe that we can eliminate nonspecific green signals by these procedures. In addition, we cannot draw any conclusions from the examination of cells that show extremely low expression of GFP-LC3. Such cells include endocrine pancreatic cells (islet cells), blood cells including erythrocytes, and type I alveolar cells. However, strong expression is not required for this assay because autophagy is clearly visible in cells with low levels of GFP-LC3 expression, such as lens epithelial cells.
Autophagy as a Starvation Response
Using the GFP-LC3 transgenic mouse, we performed systematic analyses of the autophagic response to starvation in each tissue. Although much attention has been paid to liver autophagy, we found that autophagy is induced in almost all tissues. However, the pattern of response to starvation differs among organs (Figure 7). The mechanism of autophagy regulation in vivo is not well understood. A decrease in plasma amino acid levels would induce autophagy, whereas hormonal control should also exist. Our results demonstrating various patterns of starvation response suggest that regulation of autophagy is not uniform but rather is organ dependent.
Although autophagy is regulated differently among organs, autophagy is induced in most organs in response to nutrient starvation. This indicates that the major role of autophagy is the starvation response. The liver is the principle organ responsible for recycling cytoplasmic components to provide nutrients to other tissues during the starvation. However, it is controversial whether or not autophagy is induced in the liver in vivo by starvation. It has been reported that autophagy is unchanged or slightly induced (Mortimore et al., 1983) or downregulated during starvation (de Waal et al., 1986). We found a small but significant difference between the fed and 24-h fasting states. However, the SD at 24 h was very large, indicating that autophagic activity in the liver is highly variable among individual mice (Figures 3 and 7). Some mice showed almost basal levels of autophagy even after 24-h starvation, although autophagy was clearly induced in other organs such as muscles in the same mice. Thus, regulation of liver autophagy might be more complex than regulation in other organs. Further experiments with controlled feeding regimens will be required to describe the starvation response of liver autophagy in more detail.
In contrast, a much clearer change was observed in muscles during starvation. In particular, fast-twitching muscles respond dramatically to starvation. As shown in previous studies measuring bulk degradation, slow-twitching muscles involved in tonic construction and maintenance of posture are better preserved during starvation (Li and Goldberg, 1976; Frayn and Maycock, 1979). Protein degradation in muscle fibers has been an issue of controversy, but the recent consensus is that the ubiquitin-proteasome system is responsible for degradation of myofibrils, whereas the lysosome is involved in degradation of nonmyofibril cytoplasmic proteins and organelles (Attaix and Taillandier, 1998). In the present study, we detected GFP-LC3–labeled autophagosomes only in the cytoplasm and could not find intact myofibrils inside them (Figure 4). However, this observation does not exclude the possibility that autophagy participates in myofibril degradation. For example, autophagosomes may enclose partially degraded myofibrils.
It is interesting that autophagy was not observed in the brain even after 48-h food withdrawal. The brain is probably not starved by such food withdrawal treatment; other organs could supply nutrients (glucose and ketone bodies) to the brain. Therefore, more severe treatments (e.g., ischemic injury) may be required to induce autophagy in the brain.
Autophagome Size Regulation
We were able to analyze not only the distribution but also the size of autophagosomes by fluorescence microscopic examination of the mouse tissues. If the size of the autophagosome is larger than 1 μm, it is visible as a ring-shaped structure. We have observed that the size of autophagosomes differs among culture cell lines. Embryonic stem cells generate the largest autophagosomes among the cell lines we examined (Mizushima et al., 2001, 2003), followed by F9 cells and fibroblasts (our unpublished observations). In contrast, autophagosomes in HeLa cells are small and can be detected only as dots (Kabeya et al., 2000). Such variation was also observed among mouse tissues. Cells that form large autophagosomes include hepatocytes, heart muscle cells (48-h starved), and sometimes lens epithelial cells. Autophagosomes are relatively small in pancreatic acinar cells in the fed state, but larger autophagosomes are generated after starvation. In contrast, skeletal muscles, podocytes and thymic epithelial cells generate small autophagosomes even after starvation. Therefore, the size of autophagosomes is not random and must be regulated in a cell lineage– and nutrient condition–dependent manner.
Organelle Degradation and Autophagy
It has been reported that the number of zymogen granules decreases in starved rodents (Nevalainen and Janigan, 1974; Bendayan et al., 1985; Kitagawa and Ono, 1986). So-called crinophagy, in which secretory granules are directly fused with lysosomes, is the classical degradation pathway. However, because autophagosome-like structures have been detected in acinar cells, autophagy has been thought to be a possible mechanism for the degradation. In this study, we have clearly demonstrated that autophagosomes appear in the apical portion of pancreatic acinar cells during starvation and sequester some of the zymogen granules. Although we do not know at the molecular level whether these zymogen granules are sequestered with any specificity, massive induction of autophagosomes in the area overlapping or surrounding the zymogen granules could account for the effective degradation of the granules. Moderate basal level of autophagy in other exocrine cells such as gastric chief cells and seminal vesicle cells may also participate in the degradation of secretory vesicles.
The lens fibers have few ER, mitochondria, ribosomes, and no nuclei. Thus, one possibility is that autophagy in lens cells might be involved in the elimination of these organelles (Walton and McAvoy, 1984). However, active autophagy is not limited to the regions where organelle degradation occurs. Autophagosomes were also observed in lens epithelial cells at the anterior pole, where cells are not undergoing differentiation. Furthermore, lens autophagy is not active during late embryogenesis when most lens fibers are differentiated (our unpublished observation). Therefore, autophagy in the lens epithelial cells might be important for cellular functions other than organelle degradation. Recently, 15-lipoxygenase was proposed to be responsible for such organelle degradation (van Leyen et al., 1998).
The erythrocyte is another cell that has few intracellular organelles. However, we could not examine erythrocytes because the expression of GFP-LC3 was very low in red blood cells (Okabe et al., 1997).
Constitutively Active Autophagy
Several tissues, including thymic epithelial cells, lens epithelial cells, podocytes (Figure 7), and some exocrine gland cells such as gastric chief cells and seminal vesicle cells (our unpublished results) showed high basal levels of autophagy under nutrient rich conditions. Although the volume of thymic epithelial cells is difficult to estimate by fluorescence microscopy, the number of autophagosomes in these cells might be the highest of any nonstarved tissue. One report has also demonstrated the extensive acid phosphatase activity in some vacuoles in the thymic epithelial cells (Bowen and Lewis, 1980). Younger mice and late-stage embryos show more active autophagy in the thymus (our unpublished observation). In general, cytoplasmic proteins are processed by the proteasome, delivered into the lumen of the ER and presented by the MHC class I pathway. However, there is a growing amount of data demonstrating that endogenous proteins are also presented on class II MHC (Lechler et al., 1996). How cytoplasmic proteins are loaded on class II MHC has not been well studied, but autophagy is likely responsible for this pathway (Brazil et al., 1997; Nimmerjahn et al., 2003). During positive and negative selections, thymic epithelial cells present self-antigens to lymphocytes. Because thymic epithelial cells are not thought to have phagocytic activity, it is reasonable to hypothesize that they provide self-antigens from their own cytoplasm. In this scenario, autophagy in thymic epithelial cells might be involved in T-cell development and central tolerance.
As mentioned above, it is unlikely that autophagy plays a major role in organelle degradation during lens fiber differentiation. Because lens epithelial cells are the center of metabolism and detoxification in the lens (Bhat, 2001), autophagy may be involved in such activities. The purpose of constitutively active autophagy in podocytes is not known.
CONCLUSION
We have developed a novel method to monitor autophagy in the mouse. It is simple, quick, and accurate. The systematic analysis of autophagy in response to starvation by this method provided very fundamental and novel insights. This system can be utilized to study a number of yet unsolved problems regarding autophagy such as the regulation of autophagy, significance of autophagy in disease, cell death/degeneration and development, and substrate specificity.
Acknowledgments
We thank Dr. Mohamad Zubair for providing the plasmid and helpful suggestions on generation of the transgenic mice; Yukiko Kabeya and Masami Miwa for technical assistance and Dr. Akiko Kuma for productive discussion; and Drs. Ichizo Nishino and Ikuya Nonaka for instruction on muscle preparation. This work was supported in part by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Abbreviations used: GFP, green fluorescent protein; PE, phosphatidylethanolamine; ES cells, embryonic stem cells; FCS, fetal calf serum; PFA, paraformaldehyde; PB, phosphate buffer; EDL, extensor digitorum longus; ER, endoplasmic reticulum; MHC, major histocompatibility complex; DIC, differential interference contrast.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E03–09–0704. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E03-09-0704.
References
- Ariano, M.A., Armstrong, R.B., and Edgerton, V.R. (1973). Hindlimb muscle fiber populations of five mammals. J. Histochem. Cytochem. 21, 51-55. [DOI] [PubMed] [Google Scholar]
- Attaix, D. and Taillandier, D. (1998). The critical role of the ubiquitin-proteasome pathway in muscle wasting in comparison to lysosomal and Ca2+- dependent system. JAI Press Inc., London.
- Bendayan, M., Bruneau, A., and Morisset, J. (1985). Morphometrical and immunocytochemical studies on rat pancreatic acinar cells under control and experimental conditions. Biol. Cell 54, 227-234. [DOI] [PubMed] [Google Scholar]
- Bhat, S.P. (2001). The ocular lens epithelium. Biosci. Rep. 21, 537-563. [DOI] [PubMed] [Google Scholar]
- Blommaart, E.F.C., Luiken, J.J.F.P., and Meijer, A.J. (1997). Autophagic proteolysis: control and specificity. Histochem. J. 29, 365-385. [DOI] [PubMed] [Google Scholar]
- Bowen, I.D. and Lewis, G.H. (1980) Acid phosphatase activity and cell death in mouse thymus. Histochemistry 65, 173-179. [DOI] [PubMed] [Google Scholar]
- Brazil, M.I., Weiss, S., and Stockinger, B. (1997). Excessive degradation of intracellular protein in macrophages prevents presentation in the context of major histocompatibility complex class II molecules. Eur. J. Immunol. 27, 1506-1514. [DOI] [PubMed] [Google Scholar]
- Clarke, P.G. (1990). Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.) 181, 195-213. [DOI] [PubMed] [Google Scholar]
- Dahlmann, B., Rutschmann, M., and Reinauer, H. (1986). Effect of starvation or treatment with corticosterone on the amount of easily releasable myofilaments in rat skeletal muscles. Biochem. J. 234, 659-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waal, E.J., Vreeling-Sindelarova, H., Schellens, J.P., Houtkooper, J.M., and James, J. (1986). Quantitative changes in the lysosomal vacuolar system of rat hepatocytes during short-term starvation. A morphometric analysis with special reference to macro- and microautophagy. Cell Tissue Res. 243, 641-648. [DOI] [PubMed] [Google Scholar]
- Doelling, J.H., Walker, J.M., Friedman, E.M., Thompson, A.R., and Vierstra, R.D. (2002). The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J. Biol. Chem. 277, 33105-33114. [DOI] [PubMed] [Google Scholar]
- Dunn, W.A.J. (1994). Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 4, 139-143. [DOI] [PubMed] [Google Scholar]
- Frayn, K.N., and Maycock, P.F. (1979). Regulation of protein metabolism by a physiological concentration of insulin in mouse soleus and extensor digitorum longus muscles. Effects of starvation and scald injury. Biochem. J. 184, 323-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaoka, H., Noda, T., Shirano, Y., Kato, T., Hayashi, H., Shibata, D., Tabata, S., and Ohsumi, Y. (2002). Leaf senescence and starvation-induced chlorosis are accelerated by the disruption of an Arabidopsis autophagy gene. Plant Physiol. 129, 1181-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425-479. [DOI] [PubMed] [Google Scholar]
- Hochstrasser, M. (1996). Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405-439. [DOI] [PubMed] [Google Scholar]
- Ichimura, Y. et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488-492. [DOI] [PubMed] [Google Scholar]
- Juhasz, G., Csikos, G., Sinka, R., Erdelyi, M., and Sass, M. (2003). The Drosophila homolog of Aut1 is essential for autophagy and development. FEBS Lett. 543, 154-158. [DOI] [PubMed] [Google Scholar]
- Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720-5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, T., and Ono, K. (1986). Ultrastructure of pancreatic exocrine cells of the rat during starvation. Histol. Histopathol. 1, 49-57. [PubMed] [Google Scholar]
- Klionsky, D.J. et al. (2003). A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539-545. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J., and Ohsumi, Y. (1999). Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol. 15, 1-32. [DOI] [PubMed] [Google Scholar]
- Kuma, A., Mizushima, N., Ishihara, N., and Ohsumi, Y. (2002). Formation of the ∼350 kD Apg12-Apg5·Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 277, 18619-18625. [DOI] [PubMed] [Google Scholar]
- Lechler, R., Aichinger, G., and Lightstone, L. (1996). The endogenous pathway of MHC class II antigen presentation. Immunol. Rev. 151, 51-79. [DOI] [PubMed] [Google Scholar]
- Li, J.B., and Goldberg, A.L. (1976). Effects of food deprivation on protein synthesis and degradation in rat skeletal muscles. Am. J. Physiol. Gastrointest. Liver Physiol. 231, 441-448. [DOI] [PubMed] [Google Scholar]
- Liang, X.H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., and Levine, B. (1999). Induction of autophagy and inhibition of tumorigenesis by. beclin 1. Nature 402, 672-676. [DOI] [PubMed] [Google Scholar]
- Lindsten, K., Menendez-Benito, V., Masucci, M.G., and Dantuma, N.P. (2003). A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 21, 897-902. [DOI] [PubMed] [Google Scholar]
- Melendez, A., Talloczy, Z., Seaman, M., Eskelinen, E.L., Hall, D.H., and Levine, B. (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387-1391. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Kuma, A., Kobayashi, Y., Yamamoto, A., Matsubae, M., Takao, T., Natsume, T., Ohsumi, Y., and Yoshimori, T. (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679-1688. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Noda, T., and Ohsumi, Y. (1999). Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888-3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M.D., Klionsky, D.J., Ohsumi, M., and Ohsumi, Y. (1998a). A protein conjugation system essential for autophagy. Nature 395, 395-398. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002a). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421-429. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Sugita, H., Yoshimori, T., and Ohsumi, Y. (1998b). A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889-33892. [DOI] [PubMed] [Google Scholar]
- Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., Tokuhisa, T., Ohsumi, Y., and Yoshimori, T. (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N., Yoshimori, T., and Ohsumi, Y. (2002b). Mouse Apg10 as an Apg12 conjugating enzyme: analysis by the conjugation-mediated yeast two-hybrid method. FEBS Lett. 532, 450-454. [DOI] [PubMed] [Google Scholar]
- Mortimore, G.E., Hutson, N.J., and Surmacz, C.A. (1983). Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc. Natl. Acad. Sci. USA 80, 2179-2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimore, G.E., and Pösö, A.R. (1987). Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu. Rev. Nutr. 7, 539-564. [DOI] [PubMed] [Google Scholar]
- Nevalainen, T.J., and Janigan, D.T. (1974). Degeneration of mouse pancreatic acinar cells during fasting. Virchows Arch. B Cell Pathol. 15, 107-118. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E.M., Pardoll, D.M., Bornkamm, G.W., and Mautner, J. (2003). Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 33, 1250-1259. [DOI] [PubMed] [Google Scholar]
- Nishino, I. et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906-910. [DOI] [PubMed] [Google Scholar]
- Niwa, H., Yamamura, K., and Miyazaki, J. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193-199. [DOI] [PubMed] [Google Scholar]
- Noda, T., Suzuki, K., and Ohsumi, Y. (2002). Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Biol. 12, 231-235. [DOI] [PubMed] [Google Scholar]
- Ohsumi, Y. (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell. Biol. 2, 211-216. [DOI] [PubMed] [Google Scholar]
- Okabe, M., Ikawa, M., Kominami, K., Nakanishi, T., and Nishimune, Y. (1997). `Green mice' as a source of ubiquitous green cells. FEBS Lett. 407, 313-319. [DOI] [PubMed] [Google Scholar]
- Otto, G.P., Wu, M.Y., Kazgan, N., Anderson, O.R., and Kessin, R.H. (2003). Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J. Biol. Chem. 278, 17636-17645. [DOI] [PubMed] [Google Scholar]
- Seglen, P.O., and Bohley, P. (1992). Autophagy and other vacuolar protein degradation mechanisms. Experientia 48, 158-172. [DOI] [PubMed] [Google Scholar]
- Suzuki, K., Kirisako, T., Kamada, Y., Mizushima, N., Noda, T., and Ohsumi, Y. (2001). The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971-5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida, I., Tanida-Miyake, E., Komatsu, M., Ueno, T., and Kominami, E. (2002). Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 277, 13739-13744. [DOI] [PubMed] [Google Scholar]
- Tanida, I., Tanida-Miyake, E., Ueno, T., and Kominami, E. (2001). The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J. Biol. Chem. 276, 1701-1706. [DOI] [PubMed] [Google Scholar]
- Tsukada, M., and Ohsumi, Y. (1993). Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 333, 169-174. [DOI] [PubMed] [Google Scholar]
- van Leyen, K., Duvoisin, R.M., Engelhardt, H., and Wiedmann, M. (1998). A function for lipoxygenase in programmed organelle degradation. Nature 395, 392-395. [DOI] [PubMed] [Google Scholar]
- Vary, T.C., and Kimball, S.R. (1992). Sepsis-induced changes in protein synthesis: differential effects on fast- and slow-twitch muscles. Am. J. Physiol. 262, C1513-C1519. [DOI] [PubMed] [Google Scholar]
- Walton, J., and McAvoy, J. (1984). Sequential structural response of lens epithelium to retina-conditioned medium. Exp. Eye Res. 39, 217-229. [DOI] [PubMed] [Google Scholar]
- Yoshimori, T. et al. (2000). The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol. Biol. Cell 11, 747-763. [DOI] [PMC free article] [PubMed] [Google Scholar]