

Background: StAR proteins in steroidogenic cells transport cholesterol to/into mitochondria. Under oxidative stress, StARs might deliver damaging cholesterol hydroperoxides (ChOOHs).
Results: Steroidogenic activation of MA-10 Leydig cells results in StarD1/D4 expression, ChOOH delivery to mitochondria, membrane potential loss, and reduced progesterone output.
Conclusion: Activated cells are susceptible to mitochondrial damage/dysfunction by ChOOHs.
Significance: Novel insights into how steroidogenic tissues can be damaged under oxidative stress are provided.
Keywords: Apoptosis, Cholesterol, Mitochondria, Oxidative Stress, Progesterone, Steroidogenesis, Cholesterol Hydroperoxides, Lipid Trafficking, Membrane Potential, StAR Proteins
Abstract
Steroidogenic acute regulatory (StAR) proteins in steroidogenic cells are implicated in the delivery of cholesterol (Ch) from internal or external sources to mitochondria (Mito) for initiation of steroid hormone synthesis. In this study, we tested the hypothesis that under oxidative stress, StAR-mediated trafficking of redox-active cholesterol hydroperoxides (ChOOHs) can result in site-specific Mito damage and dysfunction. Steroidogenic stimulation of mouse MA-10 Leydig cells with dibutyryl-cAMP (Bt2cAMP) resulted in strong expression of StarD1 and StarD4 proteins over insignificant levels in nonstimulated controls. During incubation with the ChOOH 3β-hydroxycholest-5-ene-7α-hydroperoxide (7α-OOH) in liposomes, stimulated cells took up substantially more hydroperoxide in Mito than controls, with a resulting loss of membrane potential (ΔΨm) and ability to drive progesterone synthesis. 7α-OOH uptake and ΔΨm loss were greatly reduced by StarD1 knockdown, thus establishing the role of this protein in 7α-OOH delivery. Moreover, 7α-OOH was substantially more toxic to stimulated than nonstimulated cells, the former dying mainly by apoptosis and the latter dying by necrosis. Importantly, tert-butyl hydroperoxide, which is not a StAR protein ligand, was equally toxic to stimulated and nonstimulated cells. These findings support the notion that like Ch itself, 7α-OOH can be transported to/into Mito of steroidogenic cells by StAR proteins and therein induce free radical damage, which compromises steroid hormone synthesis.
Introduction
Cells of the adrenal cortex, ovary, and testis use nonesterified cholesterol (Ch)3 to synthesize steroid hormones (1–3). Ch can be derived from external sources, viz. low density lipoprotein (LDL) via the LDL receptor and high density lipoprotein (HDL) via the class B type I scavenger receptor (SR-BI) scavenger receptor (3, 4). Upon delivery, cholesteryl esters are hydrolyzed by hormone-sensitive lipase, giving free Ch (3, 5). Ch can also be supplied internally, e.g. via de novo synthesis in endoplasmic reticulum, removal from plasma membrane, or hydrolysis of cholesteryl esters in lipid droplets (3). Hormone production is initiated in mitochondria (Mito) by hydroxylation and cleavage of the Ch side chain to give pregnenolone, a reaction carried out by cytochrome P450 side-chain cleavage enzyme (P450scc/Cyp11A1) on the Mito inner membrane (IM) (2, 3). Proteins of the steroidogenic acute regulatory (StAR) family play a major role in steroid hormone synthesis by selectively transporting Ch to and into Mito (3, 6–8). These proteins contain a C-terminal segment of ∼200 amino acids, the StAR-related lipid transfer (START) domain, which binds a single Ch molecule in highly selective fashion (9, 10). StarD1, the family prototype, localizes in the Mito outer membrane (OM), and in conjunction with peripheral benzodiazepine receptor and other proteins (3, 7, 11), facilitates the translocation of incoming Ch to the inner membrane (IM) for processing by the P450scc system (2, 3). Structural homologues of StarD1 have been identified (StarD1–D6), which probably function in the cytosol because they lack organelle-targeting sequences (6, 12–14). This has prompted the notion that StarD4, for example, transports Ch through cytosol to the OM, where resident StarD1 then assists in moving it to the IM (7, 8).
There is growing awareness that functionality of steroidogenic tissues may decline as a function of increasing oxidative stress associated with natural aging or vascular disorders such as atherogenesis (15–17). A common feature of these conditions is the increasing level of lipid oxidation products in the circulation, reflecting greater free radical-mediated peroxidation of unsaturated phospholipids and Ch in cell membranes and lipoproteins (18). Lipid hydroperoxides generated during this process are susceptible to reductive turnover, undergoing either iron/copper-catalyzed one-electron reduction to oxyl radicals or enzyme-catalyzed two-electron reduction to alcohols, the former intensifying peroxidative damage and the latter attenuating it (18, 19). Due to increased hydrophilicity, most lipid hydroperoxides, including Ch-derived species (ChOOHs), are capable of translocating between membranes or lipoproteins and membranes, and this can greatly expand their oxidative toxicity and signaling ranges (20–22). Our previous studies revealed that intermembrane ChOOH transfer in cell-free and cellular systems could be accelerated by sterol carrier protein-2 (SCP-2), the first reported examples of enhanced lipid hydroperoxide translocation by a lipid transfer protein (23). More recently, we showed that transfer of 7α-hydroperoxycholesterol (7α-OOH) from liposomes to isolated Mito was strongly enhanced by recombinant StarD4 and that this induced Mito peroxidative damage and loss of membrane potential (24). This was the first reported evidence for a StAR family protein acting in this manner. We now report that steroidogenic activation of mouse MA-10 Leydig cells, as evidenced by StAR protein expression and progesterone synthesis, makes these cells remarkably more sensitive to redox damage and dysfunction by Mito-targeted 7α-OOH.
EXPERIMENTAL PROCEDURES
General Materials
Sigma-Aldrich supplied the Ch, Chelex 100, desferrioxamine, dibutyryl cyclic AMP (Bt2cAMP), dithiothreitol (DTT), tert-butyl hydroperoxide (t-BuOOH), 3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine iodide (JC-1), horse serum, and Dulbecco's modified Eagle's/Ham's nutrient F12 (DME/F12) growth medium. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was obtained from Avanti Polar Lipids. Complete Mini, a mixture of protease inhibitors included in cell lysates for Western analysis, was from Roche Applied Science. Propidium iodide (PI) and fluorescein isothiocyanate (FITC)-conjugated annexin V were from Cayman Chemicals. Calbiochem supplied the fluorogenic substrate Ac-DEVD-AMC for monitoring caspase-3/7 activation, and Promega supplied the Caspase-Glo 9 kit for monitoring caspase-9 activation. Primary antibodies against mouse StarD1, mouse StarD4, and human β-actin were obtained from Santa Cruz Biotechnology. A horseradish peroxidase-conjugated IgG secondary antibody was from Cell Signaling Technology. Amersham Biosciences supplied the [4-14C]Ch (∼50 mCi/ml), referred to as [14C]Ch. 7α-OOH was prepared by dye-sensitized photoperoxidation of Ch and isolated by reverse-phase and normal-phase HPLC, as described (25–27). Stock solutions of 7α-OOH in 2-propanol were standardized for hydroperoxide content by iodometric analysis (27) and stored at −20 °C. [14C]7α-OOH was prepared similarly, using [14C]Ch that had been prepurified by HPLC. 7α-OH was generated by 7α-OOH reduction with triphenylphosphine followed by HPLC-UV isolation, as described previously (26).
Cell Culture and Steroidogenic Stimulation
Samples of MA-10 mouse Leydig tumor cells were kindly supplied by Dr. Mario Ascoli (University of Iowa) as a research gift. This cell line was initially described in 1981 (28). The cells were cultured in humidified 5% CO2, 95% air at 37 °C, using gelatin-coated plasticware and DME/F12 medium supplemented with 15% horse serum, penicillin (100 units/ml), and streptomycin (0.1 mg/ml). Prior to an experiment, cells at 60–70% confluency were washed twice with DME medium without F12 and serum and then incubated for 1.5 h at 37 °C in DME that either contained 1.0 mm Bt2cAMP or lacked it (control).
Western Blot Analyses
The effect of Bt2cAMP treatment on StAR protein expression in MA-10 cells was monitored by immunoblot analysis as described (29) with some minor modification. Cells were harvested by scraping and lysed by treating with lysing buffer (50 mm Tris-Cl, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% deoxycholate at pH 7.4) for 30 min on ice; a mixture of protease inhibitors was also present. After centrifugation (16,000 × g, 2 min), samples were analyzed for protein content by bicinchoninic assay, using reagents from Pierce Chemical, and then subjected to SDS-PAGE. Resolved proteins were transferred to a polyvinylidene difluoride membrane and incubated for 16 h at 4 °C with primary and secondary antibodies diluted according to supplier recommendations. Visualization of resolved proteins, including β-actin as a loading standard, was accomplished using the SuperSignal West Pico chemiluminescence detection kit from Thermo Scientific. Quantitation of protein bands was carried out using LabWorks image acquisition and analysis software from UVP (Upland, CA).
StarD1 Knockdown
Silencer siRNAs for mouse StarD1 were purchased from Ambion. The following two pairs of small interfering oligonucleotide constructs were used: (i) 5′-GCUGUCCUACAUCCAGCAAtt-3′ (sense), 5′-UUGCUGGAUGUAGGACAGCtc-3′ (antisense); (ii) 5′-GGCCUUGGGCAUACUCAACtt-3′ (sense), 5′-GUUGAGUAUGCCCAAGGCCtt-3′ (antisense). Two scrambled oligonucleotide pairs (Ambion) were used as a control. Single transfections of duplex mixtures were carried out using X-tremeGENE (20 μl/ml) as a cell-permeabilizing reagent (Roche Diagnostics). Cells seeded 16 h before transfection and reaching 60% confluence were switched to Opti-MEM medium (Gibco/Life Technologies) and treated for 6–8 h with 100 nm siRNA (1:1 mixture of constructs i and ii) followed by a 32–48-h recovery period in full growth medium without siRNA or X-tremeGENE. The cells were then washed, overlaid with DME medium, and stimulated with 1 mm Bt2cAMP for 3 h, after which they were scraped into cold protease inhibitor-containing PBS (Chelex-treated phosphate-buffered saline (25 mm sodium phosphate, 125 mm NaCl, pH 7.4)) and analyzed for StarD1 expression by Western blotting.
Preparation of Liposomes
Small unilamellar liposomes (50-nm SUVs) consisting of POPC/Ch/7α-OOH (1.0:0.8:0.2 by mol), POPC/Ch/7α-OH (1.0:0.8:0.2 by mol), or POPC alone were prepared by an extrusion process (30). A chloroform solution of chosen lipids was dried under argon and then held under a vacuum for several hours at room temperature. After hydration in Chelex-treated PBS followed by five cycles of freezing in liquid nitrogen and thawing, the vesicle suspension was passed 10 times through two stacked 0.05-μm-pore polycarbonate filters in an Extruder apparatus (Lipex Biomembranes, Vancouver, British Columbia, Canada). The resulting SUVs (2.0 mm total lipid in bulk suspension in PBS) were stored under argon at 4 °C until used experimentally, typically within 3 days.
Determination of Total Cellular Uptake and Mitochondrial Uptake of 7α-OOH
For accomplishing this, Bt2cAMP-treated MA-10 cells, along with untreated controls, were exposed to liposomal [14C]7α-OOH (0.2 μCi/ml) at an initial concentration of 50 or 100 μm, using a stock suspension of POPC/Ch/[14C]7α-OOH (1.0:0.8:0.2 by mol) SUVs. After incubation at 37 °C for increasing periods up to 5 h, the cells were washed twice, detached, and collected by centrifugation. In some preparations, Mito were isolated by differential centrifugation (23). Total cell samples and Mito fractions were analyzed for incorporated radioactivity by scintillation counting. 7α-OOH uptake by isolated Mito from stimulated and nonstimulated cells was determined similarly, using 50 μm SUV [14C]7α-OOH in this case. The effect of StarD1 knockdown on 7α-OOH uptake by Mito in stimulated versus nonstimulated was also assessed, the general approach being similar to that described above for wild type cells.
Measurement of Mitochondrial Membrane Potential (ΔΨm)
In initial experiments, control and Bt2cAMP-stimulated MA-10 cells were incubated with 50 μm SUV 7α-OOH for a single time (3 h) and then separated from the SUVs, washed, and treated with the ratiometric probe JC-1 (2.5 μg/ml) for 30 min. After washing, the cells were recovered into PBS (4 × 106 cells/ml), and fluorescence was recorded, using a PTI QM-7SE spectrofluorometer. Instrument settings were as follows: red fluorescence (λex 560 nm; λem 590 nm); green fluorescence (λex 488 nm; λex 525 nm). The ΔΨm strength is reflected by the magnitude of 590 nm (red) emission relative to 525 nm (green) emission, referred to as the fluorescence intensity ratio (RFI) (31). Other details were as described previously (31, 32).
The consequence of StarD1 knockdown on 7α-OOH-induced Mito depolarization was examined as follows. Wild type and knockdown cells (after 36 h of recovery from transfection) were either not stimulated or stimulated with 0.15 mm Bt2cAMP in DME medium for 1.5 h, after which 100 μm liposomal 7α-OOH was introduced and incubation continued at 37 °C. At various time points up to 7 h, the cells were washed once, treated with JC-1 (see above), washed again, and analyzed immediately using a BioTek Synergy MX fluorescence plate reader (Winooski, VT) set as follows: red (λex 560 nm; λem 595 nm); green (λex 485 nm; λem 535 nm). Experiments were carried out at least in triplicate.
Measurement of Caspase-3/7 and Caspase-9 Activation
At selected times after exposure to 7α-OOH, cells were recovered by scraping into ice-cold PBS, pelleted, and lysed as described previously (33). After protein determination, lysates for determining caspase-3/7 activity were incubated with 25 μm Ac-DEVD-AMC for 30 min in a 96-well plate at 25 °C. Fluorescence of liberated AMC was recorded with the Synergy MX plate reader, using 360 nm excitation and 460 nm emission. Lysates for determining caspase-9 activity were treated with Caspase-Glo 9 reagents in a 96-well plate according to supplier instructions, and chemiluminescence was measured with a Victor 3V Multilabel Counter (PerkinElmer Life Sciences).
Determination of Progesterone
The effect of 7α-OOH on the ability of stimulated MA-10 cells to synthesize progesterone from Ch was examined using an enzyme immunoassay approach. The assay is based on competition between progesterone and a progesterone-acetylcholinesterase conjugate for binding to an immobilized progesterone-specific antibody in a 96-well format. The signal derives from hydrolysis of acetylthiocholine to thiocholine, which reacts with Ellman's reagent to give 5,5′-dithio-bis-(2-nitrobenzoic acid), the absorbance of which at 412 nm is monitored (34, 35). A progesterone-enzyme immunoassay kit for carrying out this assay was obtained from Cayman Chemicals. Detailed instructions supplied with the kit were followed throughout.
Assessment of Hydroperoxide-induced Cell Death
Cells preincubated in the absence or presence of Bt2cAMP for 1.5 h were washed and either not treated (control) or treated with liposomal 7α-OOH or 7α-OH in increasing concentrations up to 100 μm for 16 h at 37 °C, after which viability was assessed by MTT (thiazolyl blue) assay (32). Treated cells were also examined for death mechanism (apoptosis versus necrosis). Overall procedures were similar to those described for the MTT assay except that cell status was evaluated every hour up to 6 h of treatment, using fluorescence microscopy with annexin V-FITC for detecting early apoptosis and PI for detecting necrosis (36). Fluorescence intensities of annexin V-FITC-stained versus PI-stained cells were quantified using the MetaMorph software program.
Statistical Analyses
The two-tailed Student's t test was used for determining the significance of perceived differences between experimental values; a p value of >0.05 was considered statistically insignificant.
RESULTS
StAR Protein Expression in Steroidogenically Activated MA-10 Cells
Unstimulated Leydig MA-10 cells in vitro lack the ability to synthesize steroid hormones, but gain it after treatment with chorionic gonadotropin or its downstream effector cyclic AMP in cell-permeating dibutyryl form (Bt2cAMP) (37, 38). Steroidogenic competency has been linked to expression of StAR family proteins and the P450scc system (6–8, 37). Consistent with this, we observed a strong induction of immunodetectable StarD1 (∼27 kDa) in MA-10 cells after treating with 1 mm Bt2cAMP in serum-free DME/F12 medium (Fig. 1A). StarD1 was detected after 0.5 h and increased to an apparent maximal level of ∼17-fold above basal after 1.5 h with Bt2cAMP. StarD4 (∼21 kDa) showed a comparable time course of protein expression, its level at 0.5 h being ∼20-fold above basal and remaining there up to at least 6 h (Fig. 1B). This appears to be the first direct evidence for both StarD1 and StarD4 induction in Bt2cAMP-treated MA-10 cells, suggesting that these proteins might be coordinately expressed during steroidogenic activation.
FIGURE 1.

StAR protein expression in steroidogenically activated MA-10 cells. Cells at 60–70% confluency in serum-free DME medium were incubated with 1 mm Bt2cAMP for up to 6 h, after which cells were collected, lysed, and subjected to Western analysis for StarD1 (A) and StarD4 (B), using β-actin as a loading standard. Control cells (Con) incubated in the absence of Bt2cAMP (db-cAMP) for 6 h are also represented. Total protein load: 60 μg per lane. The number below each lane in A and B is StAR protein band intensity relative to β-actin and normalized to the control.
7α-OOH Uptake by Activated MA-10 Cells and MA-10 Mitochondria
We predicted that steroidogenic activation with accompanying StarD1 and StarD4 expression would enhance 7α-OOH uptake by whole MA-10 cells, and specifically, by their mitochondria, as has been demonstrated for Ch uptake (1–3). In an initial test for this, we incubated Bt2cAMP-stimulated cells and nonstimulated controls with liposomal [14C]7α-OOH at two different concentrations, 50 and 100 μm. After 5 h, the cells were washed and subjected to scintillation counting. As shown in Fig. 2A, stimulated cells took up substantially more 7α-OOH than nonstimulated controls, the level at 50 μm hydroperoxide being ∼35% greater and the level at 100 μm hydroperoxide being ∼70% greater. Mito isolated from stimulated cells after exposure to 50 μm liposomal [14C]7α-OOH for 2 h contained ∼90% more radioactivity than those from nonstimulated counterparts (Fig. 2B). The Mito lipid pool is clearly much smaller than that of the whole cell. Therefore, a direct comparison of the effects of stimulation on uptake by Mito and whole cells supports the notion that [14C]7α-OOH was preferentially delivered to Mito and that plasma membrane and other compartments were relatively unimportant acceptors.
FIGURE 2.
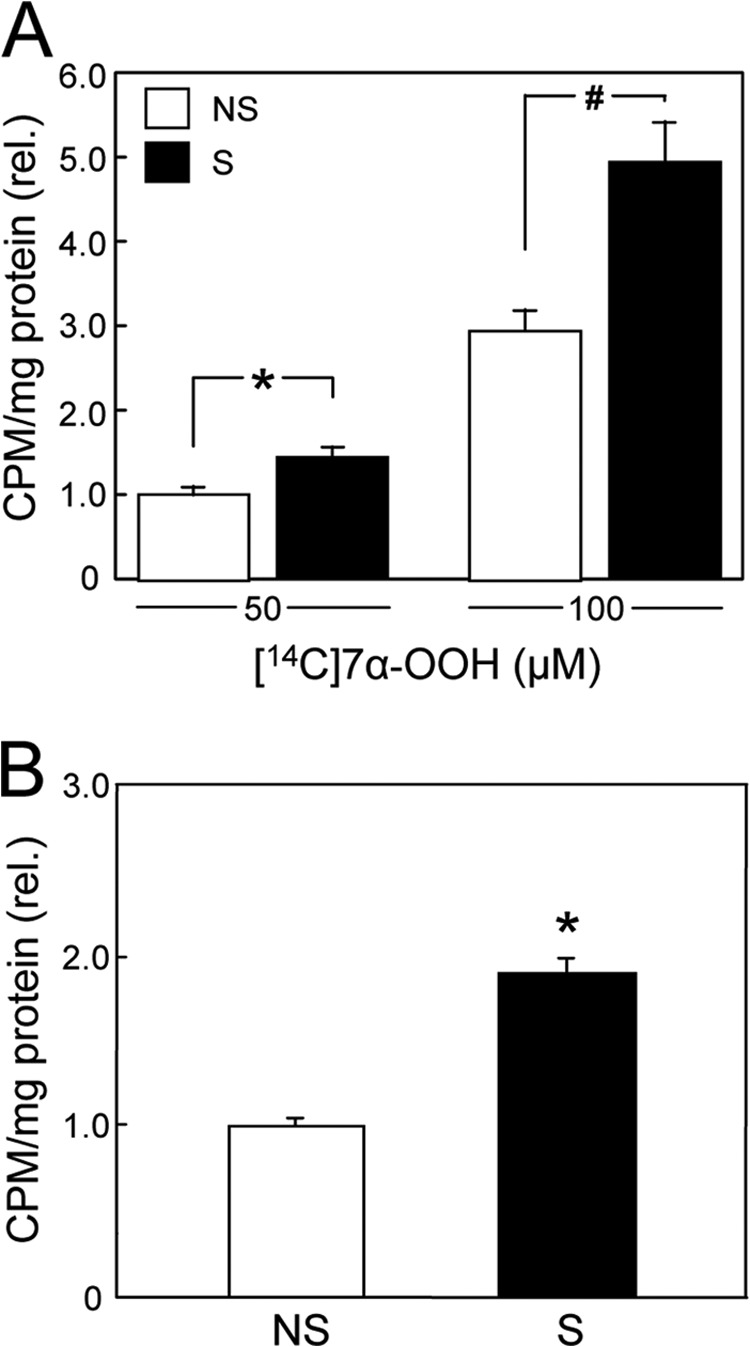
Uptake of radioactive 7α-OOH by whole MA-10 cells and by their mitochondria. A, cells stimulated (S) with 1 mm Bt2cAMP for 3 h (cf. Fig. 1), along with nonstimulated (NS) controls, were incubated with 50 or 100 μm [14C]7α-OOH in POPC/Ch/[14C]7α-OOH (1.0:0.8:0.2 by mol) SUVs for 5 h and then washed thoroughly, recovered, and analyzed by scintillation counting. rel., relative. B, cells from the same stimulated (S) and nonstimulated (NS) populations in A were recovered after a 2-h exposure to 50 μm SUV-[14C]7α-OOH. Cells were homogenized, and Mito were isolated by differential centrifugation and analyzed by scintillation counting. A and B: means ± S.E. of values from three separate experiments are plotted. A, 50 μm: *, p < 0.05 (stimulated versus nonstimulated); 100 μm: #, p < 0.01 (stimulated versus nonstimulated). B, *, p < 0.01 (stimulated versus nonstimulated).
Effect of 7α-OOH on ΔΨm of Stimulated versus Nonstimulated Cells
One-electron redox activity of 7α-OOH upon delivery to Mito could result in free radical-mediated peroxidative damage and dysfunction in this compartment. To begin assessing this, we determined how 7α-OOH uptake would affect Mito transmembrane potential (ΔΨm). Cells stimulated with Bt2cAMP for 3 h, along with nonstimulated controls, were incubated with 50 μm liposomal 7α-OOH for 3 h and then separated from the liposomes and analyzed for ΔΨm, using the fluorescent probe JC-1 (31). As shown in Fig. 3, the ΔΨm signal of stimulated cells (presented as 590 nm/525 nm emission intensity ratio) was only 45–50% that of nonstimulated controls. This supports our hypothesis that steroidogenic activation increases the risk of ChOOH-induced damage/dysfunction in MA-10 cells.
FIGURE 3.
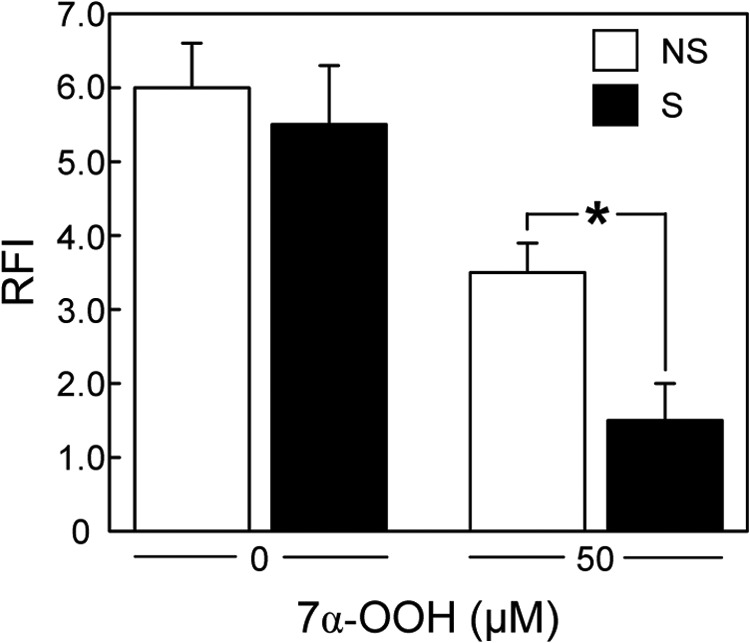
Effect of 7α-OOH on ΔΨm of Bt2cAMP-stimulated versus nonstimulated MA-10 cells. Nonstimulated (NS) and 3-h Bt2cAMP-stimulated (S) cells were incubated with 50 μm 7α-OOH in POPC/Ch/7α-OOH (1.0:0.8:0.2 by mol) SUVs for 3 h and then washed, treated with 2 μm JC-1 for 30 min, washed again, and examined for fluorescence intensity at 590 and 525 nm, using 488 nm excitation. The 590 nm/525 nm fluorescence intensity ratio (RFI), which reflects membrane potential (ΔΨm), is plotted. Control cells (nonstimulated and stimulated) were incubated for 3 h with liposomes lacking 7α-OOH, i.e. POPC/Ch (1:1 by mol) SUVs at the same total lipid concentration, after which RFI was determined. Values are means ± S.E. (n = 3). *, p < 0.05 (stimulated versus nonstimulated).
StarD1 Knockdown in MA-10 Cells with Effects on 7α-OOH Uptake and ΔΨm Loss after Stimulation
Direct evidence for StAR protein involvement in 7α-OOH trafficking and damage to Mito was sought by using an siRNA approach to minimize StarD1 expression during cell stimulation. As shown in Fig. 4A, Western blotting of an extract from cells treated with control siRNA for StarD1 and stimulated with Bt2cAMP revealed robust StarD1 expression similar to that observed for stimulated wild type cells (Fig. 1A). In contrast, cells treated with an active siRNA construct expressed ∼50% less StarD1 after stimulation (Fig. 4A). Increasing the incubation time with siRNA and X-tremeGENE transfection reagent from 6 to 8 h did not increase knockdown further, so we used 6 h for all experiments to limit any irreversible deleterious effects of X-tremeGENE on the cells. Cells were switched to fresh medium after siRNA treatment, and 36 h later, they were stimulated with Bt2cAMP and then incubated with 100 μm radiolabeled or unlabeled liposomal 7α-OOH for determination of peroxide uptake or effects on ΔΨm, respectively. Fig. 4B shows time courses of radioactivity accumulating in Mito during exposure of cells to [14C]7α-OOH. Uptake by stimulated control knockdown cells increased rapidly over the first 2 h and leveled off thereafter, as also observed with stimulated wild type cells (not shown). Uptake by nonstimulated cells was much slower, reaching ∼50% that of stimulated counterparts at 2 h (Fig. 4B), in good agreement with the 2-h data in the Fig. 2B experiment, where 50 μm 7α-OOH was used. Stimulated StarD1 knockdown cells showed a substantial reduction in peroxide uptake relative to stimulated control knockdown cells, reaching only ∼40% that of the latter at 2 h, corrected for nonstimulated background (Fig. 4B). As shown in Fig. 4C, StarD1 knockdown also reduced the extent of 7α-OOH-induced ΔΨm loss, which went from ∼70% in stimulated control knockdown to only ∼25% in stimulated knockdown at 3 h, both values relative to nonstimulated cells. (That depolarization of stimulated versus nonstimulated cells is greater in this experiment than in the Fig. 3 experiment reflects the lower peroxide concentration used in the latter.) Taken together, the data in Fig. 4 provide strong evidence for StarD1 involvement in deleterious 7α-OOH trafficking to MA-10 Mito.
FIGURE 4.
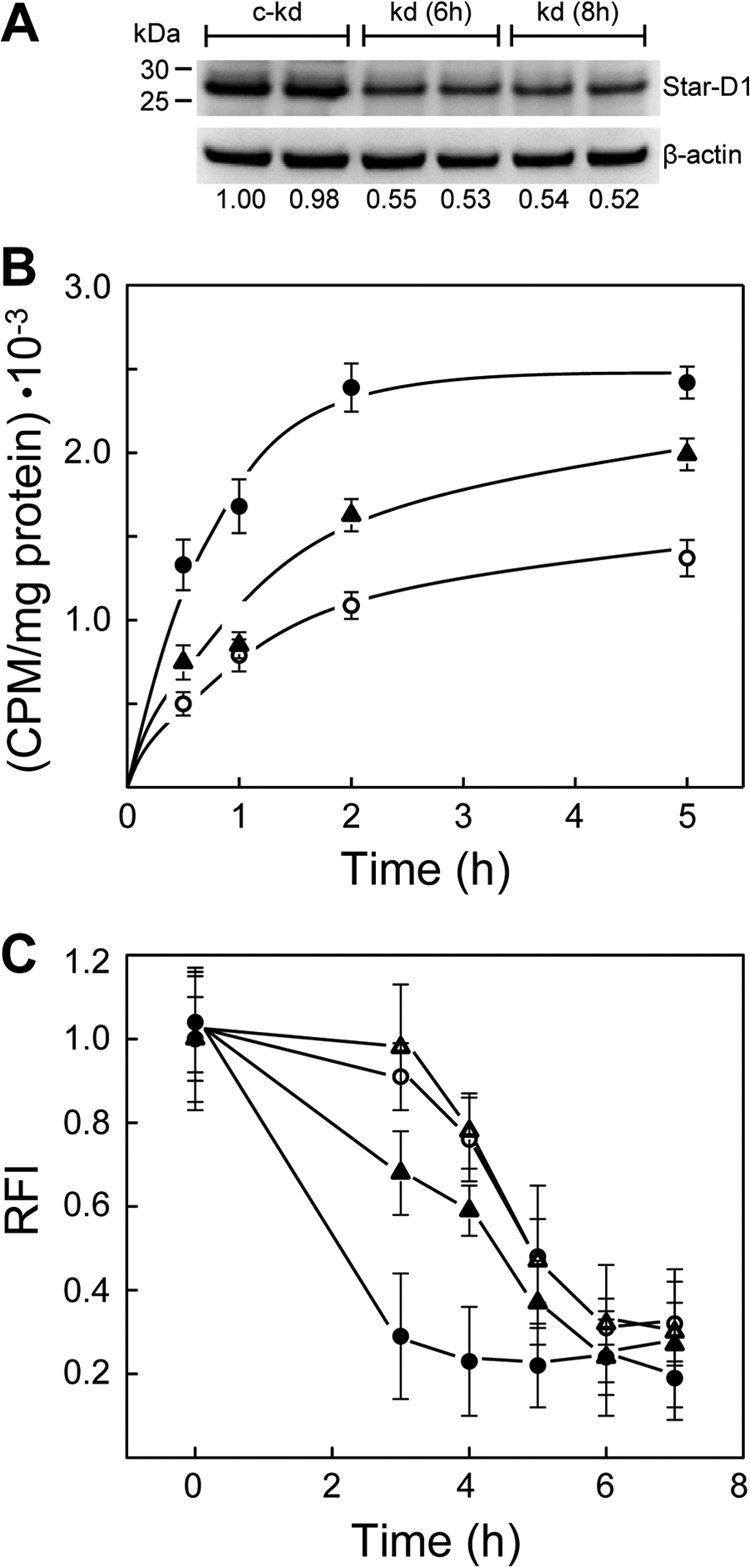
Effect of StarD1 knockdown on mitochondrial uptake of 7α-OOH and on peroxide-induced ΔΨm decay. A, StarD1 protein levels. MA-10 cells were treated with StarD1 siRNA for 6 and 8 h, or a control construct for 8 h, allowed to recover for 36 h, and then stimulated with 1 mm Bt2cAMP for 3 h. Immediately thereafter, cells were lysed and analyzed for StarD1 by immunoblotting. Duplicate samples of 40 μg of total protein per lane are represented: kd, knockdown; c-kd, control knockdown. Numbers below lanes indicate normalized StarD1 band intensities relative to β-actin. B, 7α-OOH uptake. A 6-h exposure to control or active siRNA was used followed by 36 h of recovery. Bt2cAMP (1 mm, 3 h)-stimulated knockdown and control knockdown cells, along with nonstimulated control knockdown cells, were incubated in the presence of SUV-borne [14C]7α-OOH (50 μm, ∼150 nCi/μmol) in DME medium at 37 °C. At the indicated times, cells were washed and homogenized, and the Mito fraction was isolated by differential centrifugation. Lipid extracts were prepared and analyzed by scintillation counting. Protein-based specific radioactivity of the lipid extracts is plotted as a function of incubation time. ●, stimulated control knockdown cells; ○, nonstimulated control knockdown cells; ▴, stimulated knockdown cells. Calculated 7α-OOH content at 2 h (nmol/mg of Mito protein) was 0.72 ± 0.05, 0.33 ± 0.02, and 0.48 ± 0.03, respectively. C, effects on ΔΨm. Control and active siRNA treatment conditions were as described in B. Cell stimulation in this experiment was carried out using 150 μm Bt2cAMP, which remained in the system after peroxide was added. Cells were incubated for the indicated times with 100 μm SUV 7α-OOH and then washed and incubated with JC-1 (2.5 μg/ml) for 30 min. After washing, the cells were examined by fluorescence plate reader using 560 nm excitation/595 nm emission and 485 nm excitation/535 nm emission. Time-dependent changes in 595 nm/535 nm fluorescence intensity ratio (RFI) are plotted. Cells were as follows: ●,▴, stimulated; ○,▵, nonstimulated; ▴,▵, knockdown; ●,○, control knockdown. Means ± S.E. of values from 3–4 replicate experiments are plotted in B and C.
Effect of 7α-OOH Uptake on Progesterone Biosynthesis
To further investigate the functional ramifications of 7α-OOH delivery to Mito of steroidogenically activated MA-10 cells, we used an ELISA-type assay to monitor de novo formation of progesterone, which lies immediately downstream of pregnenolone, the initial product of Ch side-chain cleavage by Mito Cyp11A1 (1–3). As shown in Fig. 5A, 50 μm SUV 7α-OOH had a significant inhibitory effect on progesterone formation in these cells. After 3 h of Bt2cAMP stimulation with 7α-OOH present, progesterone yield was reduced to ∼50% of that in the Ch-only control. At that point, peroxide-treated cells were all still attached, had normal morphology, and exhibited no overall signs of toxicity (Fig. 5B). Therefore, the drop in progesterone under the conditions described is attributed to damage-impaired synthesis rather than a large number of cells being already dead.
FIGURE 5.
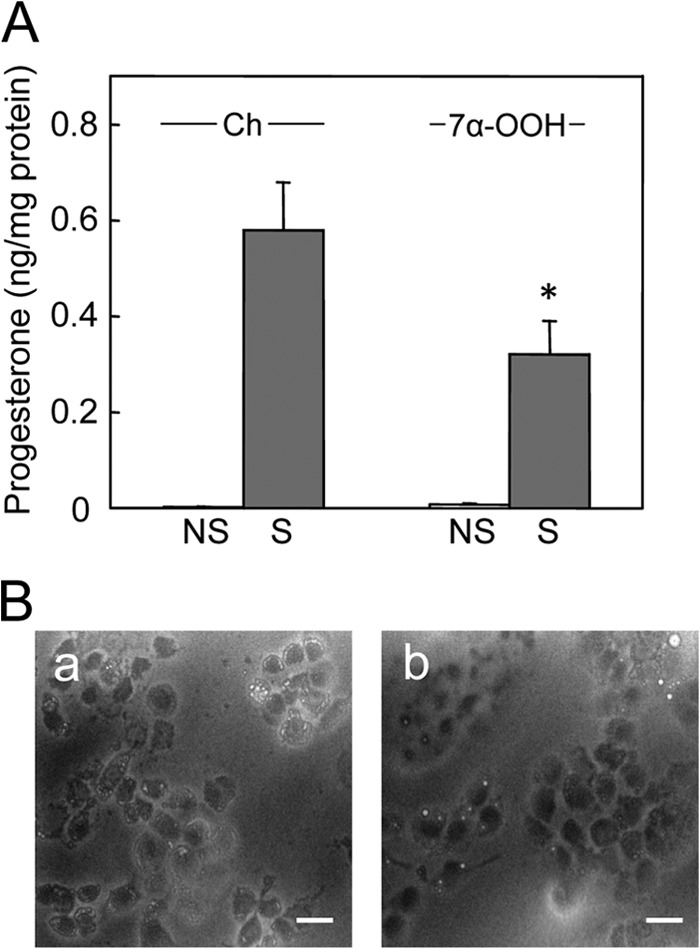
Progesterone formation in 7α-OOH-challenged MA-10 cells. A, cells in a 96-well plate were preincubated overnight in serum-free DMEM/F12 medium. They were then either not treated (NS) or treated (S) with 1 mm Bt2cAMP in the presence of POPC/Ch/7α-OOH (0.8:1.0:0.2 by mol) SUVs (7α-OOH) or POPC/Ch (1:1 by mol) SUVs (Ch); total SUV lipid was 0.8 mm. After 3 h of incubation, the medium was removed and analyzed for progesterone by enzyme immunoassay, whereas cells were scraped into cold lysis buffer and analyzed for total protein. Plotted values are means ± S.E. (n = 3); *, p < 0.01 as compared with stimulated, Ch-treated. B, bright field microscopic images from experiment described in A. Stimulated cells were examined after being exposed to POPC/Ch (1:1 by mol) (panel a) or POPC/Ch/7α-OOH (0.8/1.0/0.2 by mol) (panel b) SUVs for 3 h. Bar: 75 μm.
Effect of Steroidogenic Activation on Mechanism of 7α-OOH-provoked Cell Death
We predicted that preferential trafficking of 7α-OOH to Mito of activated cells expressing StarD1/D4 would not only impair steroid formation, but also decrease cell viability. To assess this, we compared the lethal effects of liposomal 7α-OOH on 1.5-h Bt2cAMP-stimulated MA-10 cells versus nonstimulated controls. As shown in Fig. 6A, the MTT-detected viable fraction for control cells decreased progressively with increasing 7α-OOH concentration, the lethal dose for 50% loss (LD50) after 16 h with 50 μm hydroperoxide being ∼165 μm. Stimulated cells proved to be substantially more sensitive, their LD50 under the same conditions of 7α-OOH treatment being ∼40 μm (Fig. 6A). When 7α-OH, a redox-inactive alcohol analog, was substituted for 7α-OOH, little or no cytotoxicity was observed for stimulated or nonstimulated cells over the concentration range used (Fig. 6A). Thus, the cytotoxic effects observed with 7α-OOH were most likely due to its damaging redox activity. We also asked how cell killing by a non-steroid hydroperoxide, t-BuOOH, would compare with that by 7α-OOH. As shown in Fig. 6B, the LD50 of t-BuOOH was ∼220 μm, and there was no significant difference between nonstimulated and stimulated cells in this case, nor was there in the extent of ΔΨm loss (not shown). Because t-BuOOH is not a ligand for StAR proteins, these results further support our argument that 7α-OOH toxicity was not due to random attack, but rather specific StAR-mediated targeting.
FIGURE 6.
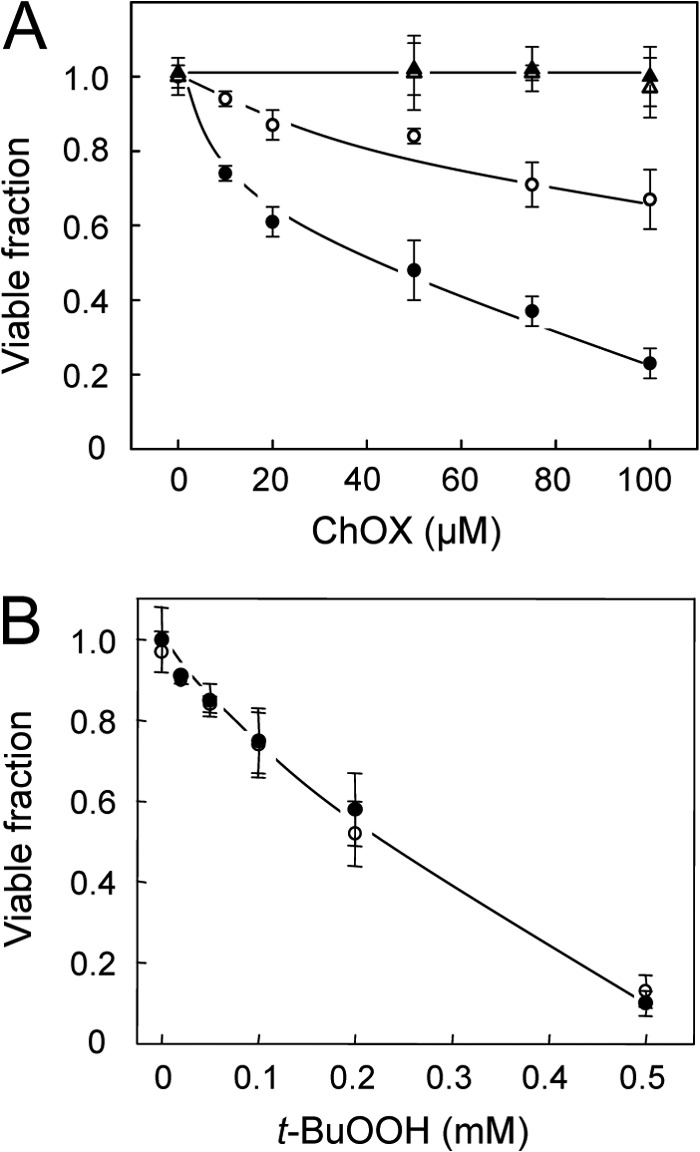
Effect of steroidogenic activation on MA-10 cell sensitivity to 7α-OOH versus t-BuOOH toxicity. A, cells at ∼70% confluency were incubated in the absence (nonstimulated) or presence (stimulated) of 1 mm Bt2cAMP for 1.5 h in serum-free medium, after which POPC/Ch/7α-OOH (1.0:0.5;0.5 by mol) or POPC/Ch/7α-OH (1.0:0.5:0.5 by mol) SUVs were introduced at the indicated cholesterol oxide (ChOX:7α-OOH or 7α-OH) concentration in bulk phase. B, similarly prepared cells were also challenged with t-BuOOH. After 16 h of incubation, cell viability was assessed by thiazolyl blue (MTT) assay. Data points are as follows: A, 7α-OOH, nonstimulated (○); 7α-OOH, stimulated (●); 7α-OH, nonstimulated (▵); 7α-OH, stimulated (▴). B, nonstimulated (○); stimulated (●). Means ± S.E. of values from 3–4 separate experiments are plotted in A and B.
We predicted that if StarD1 and D4 were able to transport 7α-OOH specifically to Mito of steroidogenically stimulated cells, then these cells would likely die via activation of the intrinsic apoptotic pathway (39, 40), whereas nonstimulated counterparts might die via indiscriminate membrane damage leading to necrosis, although extrinsic pathway apoptosis (41) might also make a contribution in this case. To investigate these possibilities, we exposed 1.5-h Bt2cAMP-stimulated cells to 50 μm liposomal 7α-OOH and at various times examined them for caspase activation along with extent of apoptosis versus necrosis. As shown in Fig. 7A, caspase-9 activity in stimulated cells increased steadily during incubation with 7α-OOH, reaching ∼12 times the basal signal level after 8 h. An increase in caspase-9 activity also occurred in nonstimulated cells, but after a long delay (∼4 h), and the level attained at 8 h was only ∼4-times basal (Fig. 7A). Thus, activation of this proapoptotic enzyme was substantially more advanced in stimulated cells (at least 5-fold at 4 h), in general agreement with the overall viability loss described in Fig. 6A. A similar trend was observed for caspase-3/7 activation in response to 7α-OOH, stimulated cells in this case exhibiting twice the activity of nonstimulated controls after 16 h (data not shown). However, activation of caspase-3/7 began significantly later than that of caspase-9 (Fig. 7A), which is consistent with the known dependence of the former on the latter (40). Control and Bt2cAMP-stimulated cells were also analyzed for the extent of apoptosis versus necrosis after several hours of incubation with 50 μm liposomal 7α-OOH. As shown by the microscopic images in Fig. 7B, which were taken after 7 h with 7α-OOH, stimulated cells exhibited strong signs of early apoptosis upon annexin V-FITC staining, but little, if any, signs of necrosis upon PI staining. In striking contrast, nonstimulated cells showed relatively weak annexin V-FITC signals, but strong PI signals, suggesting increased membrane permeability and loss of metabolic homeostasis, leading to necrosis as opposed to apoptosis. Bright field images in Fig. 7B indicate that control cells were elongated like nonchallenged counterparts and still attached at this point, whereas stimulated cells undergoing apoptosis were rounded up, but also still attached. Quantified image intensities from a replicate experiment taken after 3 and 7 h of continuous cell exposure to 50 μm 7α-OOH are shown in Fig. 7C. Note that for stimulated cells after 3 h, >80% of all cells shown to be dead/dying were apoptotic and only ∼15% were necrotic, whereas for the 3-h controls, ∼65% were necrotic and ∼35% were apoptotic. This sharp contrast in death mechanism was even more striking after 7 h. For stimulated cells at this point, the proportion of dead/dying cells classified as apoptotic was ∼85% and the proportion of cells classified as necrotic was ∼10%, whereas for nonstimulated controls, the proportion classified as apoptotic was ∼5% and that classified as necrotic was >90% (Fig. 7C). These findings further support our hypothesis that steroidogenic activation of MA-10 cells, including the expression of StAR family proteins, promotes not only the delivery of nonoxidized Ch to Mito, but peroxidized Ch as well. The latter puts these cells at risk of free radical damage to Mito, resulting in metabolic dysfunction and intrinsic (Mito-centered) apoptotic cell death.
FIGURE 7.

Effect of steroidogenic activation on death mechanism of 7α-OOH-challenged cells. A, caspase-9 activation in 7α-OOH-challenged cells. MA-10 cells that had been stimulated for 3 h with 1 mm Bt2cAMP, along with nonstimulated controls, were incubated with 50 μm SUV-7α-OOH for the indicated times and then recovered by scraping and analyzed for caspase-9 activity using a chemiluminescence assay. ○, nonstimulated; ●, stimulated. Plotted values are means ± S.E. (n = 3). B, fluorescence images (annexin V-FITC (Anx V) and PI) and bright field (BF) images of cells after exposure to 7α-OOH. Nonstimulated (NS) and 1.5-h Bt2cAMP-stimulated (S) cells were incubated with 50 μm SUV-7α-OOH for 7 h. Immediately thereafter, the cells were washed free of SUVs and analyzed for death mode by fluorescence microscopy with annexin V-FITC staining to detect early apoptosis and PI staining to detect necrosis. Bar: 50 μm. C, integrated fluorescence image intensities after 3 and 7 h of cell exposure to 7α-OOH. Plots show percentage of apoptotic (Apo) or necrotic (Nec) cells relative to total dead/dying cells in the nonstimulated (NS) and stimulated (S) populations. Means ± S.E. of values from 3 separate experiments are plotted. *, p < 0.01 versus necrotic; #, p < 0.001 versus apoptotic; †, p < 0.0001 versus necrotic ; §, p < 0.0001 versus apoptotic.
DISCUSSION
There is increasing evidence that functionality of steroidogenic tissues might be compromised in physiological states encumbered by persistent oxidative stress with generation of reactive oxygen species. Steroidogenic dysfunction might arise in oxidant-associated disease states such as atherosclerosis, type-2 diabetes, and chronic inflammation, although the pro-oxidant effects of natural aging have received the most attention in this regard. An early study (42) showed that rat adrenocortical cells were highly susceptible to oxidative injury and dysfunction, which was attributed to the vigorous Mito electron transport activity required by these cells. These negative effects increased dramatically with animal age in parallel with diminished levels of nonenzymatic and enzymatic antioxidants, including vitamin C, glutathione, glutathione peroxidase-1, and two superoxide dismutases (43). More recent studies with rat primary testicular Leydig cells showed that levels of steroidogenic enzymes declined with advancing animal age along with testosterone output (15, 43). Leydig cells, which reside in the testicular interstitium, are reported to be particularly susceptible to oxidative damage in vivo due to their close proximity to reactive oxygen species-producing testicular interstitial macrophages (44). Decreased antioxidant enzyme activities, gene expression, and protein levels, along with lower glutathione, have been reported to play a key role in the diminished ability of Leydig cells from aging rat populations to synthesize testosterone (43, 45–47). Other work showed that steroidogenic luteal cells in vitro became damaged and dysfunctional when exposed to cumene hydroperoxide or a fatty acid hydroperoxide (48). Similarly, the progesterone-synthesizing ability of cAMP-stimulated mouse Leydig cells was found to be significantly reduced after cell exposure to H2O2 (49), an effect attributed to diminished Ch trafficking due to impaired StAR expression. More recent work showed that superoxide (O2˙̄)- or H2O2-provoked inhibition of adrenal cell steroidogenesis was mediated by p38 mitogen-activated protein kinase stress signaling (50). Although these previous studies (48–50) are clearly relevant to this one, it is important to realize that although the cells used expressed a network of steroidogenic proteins, including StAR family proteins, none of the primary oxidants used (H2O2, cumene hydroperoxide, fatty acid hydroperoxide) were actual StAR ligands. It is likely, therefore, that the subcellular distribution of those oxidants upon entering cells was random rather than site-directed, given that StARs are known to be highly selective in binding and trafficking only molecules with a steroid ring (9, 10, 51). The present study was based on a more realistic and physiologically relevant approach for testing the effects of oxidative stress on steroidogenic cells, viz. employing a naturally occurring hydroperoxide of Ch, 7α-OOH, which is not only redox-active, but a transportable StAR ligand as well (22).
Free radical-mediated peroxidation of Ch in cell membranes and lipoproteins gives rise to 7α-OOH and 7β-OOH as prominent reactive intermediates (19). These species have been identified and quantified in tissues from oxidatively stressed experimental animals, e.g. ethanol-fed (52) or diabetic rats (53). For streptozotocin-induced diabetic rats, total 7-OOH (nmol/mg of protein) amounted to 0.90 ± 0.14, 0.16 ± 0.05, and 0.67 ± 0.03 in kidney, heart, and liver, respectively (53). Control animals exhibited lower yet significant 7-OOH levels, e.g. 0.41 ± 0.12 nmol/mg of protein in liver. Our values for 7α-OOH uptake by MA-10 cell Mito in 2 h ranged from ∼0.3 (nonstimulated) to ∼0.7 nmol/mg of protein (stimulated) (Fig. 4B). These levels compare well with the described values for oxidatively stressed animals (53), suggesting that our experimental conditions were physiologically relevant.
Like other lipid hydroperoxides in membranes and lipoproteins, 7α- and 7β-OOH can accumulate with increasing oxidative pressure or may undergo iron-catalyzed one-electron reduction to oxyl radicals, which trigger free radical-mediated peroxidative damage (19, 54). These reactions can be attenuated by nonenzymatic antioxidants such as α-tocopherol and β-carotene, which scavenge lipid-derived radicals, or by the enzymatic antioxidant glutathione peroxidase type-4 (GPx4), which catalyzes the two-electron reduction of ChOOHs to redox-silent diols (cholesterol hydroxides) (55, 56). About 12 years ago, we discovered that 7α-OOH and other ChOOHs can translocate between membranes or membranes and lipoproteins, and much more rapidly than Ch itself (20–22). Translocation greatly expanded the damaging ranges of these hydroperoxides if antioxidant capacity in acceptor compartments was overwhelmed or if an acceptor was relatively rich in redox iron, e.g. Mito (22). We later showed that ChOOH transfer could be further enhanced by the nonspecific lipid transfer protein, SCP-2 (23) and that SCP-2 overexpression rendered hepatoma cells more sensitive to 7α-OOH toxicity, primarily via Mito targeting (32). In recent work with more direct bearing on this study, we showed that recombinant StarD4 concentration-dependently accelerated 7α-OOH transfer from liposomal donors to isolated Mito and to a greater extent than Ch transfer (24). StarD4 had no effect on transfer of unoxidized and peroxidized phosphatidylcholine, whereas SCP-2 accelerated both. Thus, the known specificity of StarD4 and other StARs for the steroid ring (9) was shown to apply to ChOOHs. StarD4-enhanced 7α-OOH transfer to Mito caused a large loss of ΔΨm, which was attributed to an observed burst of free radical lipid peroxidation in Mito membranes (24).
The present study represents an important advance from our previous work (24) and provides the first direct evidence for deleterious Mito targeting of ChOOH in a steroidogenic cell line. It is well established that Leydig MA-10 cells express a network of proteins dedicated to steroid synthesis upon stimulation by chorionic gonadotropin or its downstream effector, cAMP, in cell-permeating dibutyryl form (3, 7, 37). These proteins include the P450scc system in the Mito IM, StarD1 in the OM, and at least one cytosolic homologue such as StarD4. It has been proposed that these StAR transporters act cooperatively in delivering Ch to the IM for pregnenolone formation by P450scc (7, 8). Our evidence revealed that Bt2cAMP-stimulated MA-10 cells (i) strongly expressed StarD1 and StarD4; (ii) channeled more 7α-OOH to Mito than nonstimulated controls; (iii) underwent a greater loss of ΔΨm during 7α-OOH exposure than controls; (iv) sustained greater inhibition of progesterone biosynthesis by 7α-OOH than controls; and (v) underwent greater Mito-centered apoptosis during a 7α-OOH challenge than controls. These findings support the idea that under oxidative stress conditions, Leydig and other steroid-synthesizing cells can deliver not only Ch to Mito, but also ChOOHs such as 7α- and 7β-OOH, leading to free radical damage/dysfunction and/or induction of intrinsic apoptosis via redox signaling. According to this model, ChOOHs would be Mito-targeted in a stealth- or Trojan Horse-like fashion. Like 7α-OOH, 7α-OH was taken up more extensively by Bt2cAMP-stimulated cells than control cells (data not shown), yet this redox-inert diol analogue was nontoxic to both populations. This confirmed that the observed cytotoxic effects of 7α-OOH were due to its damaging one-electron turnover, stimulated cells being more vulnerable to this because of their greater 7α-OOH import. Also important was our observation that t-BuOOH was no more toxic to stimulated cells than to controls. A similar result was obtained with H2O2 (data not shown). Lacking a sterol backbone, t-BuOOH and H2O2 cannot be recognized and trafficked by StAR proteins. This implies that Bt2cAMP stimulation of MA-10 cells on its own did not enhance their overall susceptibility to any type of peroxide challenge. The responses of stimulated cells to t-BuOOH and H2O2 contrasted sharply with those to SUV-borne 7α-OOH, supporting our argument that the latter was trafficked into Mito by up-regulated StarD4 and StarD1. For StarD1 at least, this idea was solidified by showing that its knockdown prior to MA-10 stimulation significantly reduced Mito uptake of 7α-OOH (Fig. 4B). Not surprisingly, Mito uptake of Ch itself was also elevated after cell stimulation, and this too was significantly reversed by StarD1 knockdown (data not shown). This appears to be the first reported evidence for StarD1-mediated Ch delivery based on a knockdown approach. Equally important to the uptake was our finding that limiting StarD1 expression reduced the extent of Mito depolarization by 7α-OOH, thereby demonstrating that this protein not only plays a role in Ch delivery, but also Mito-damaging ChOOH delivery.
Like all ChOOHs, 7α-OOH is resistant to reductive detoxification by glutathione peroxidase type-1 (GPx1), the most abundant selenoperoxidase in eukaryotic cells (57). The only enzyme known to catalyze ChOOH detoxification (albeit more slowly than phospholipid hydroperoxide detoxification) is GPx4, which can exist in multiple compartments of mammalian cells, including cytosol and Mito (58). Like rat Leydig cells (46), murine MA-10 cells probably express GPx1 and presumably also GPx4. How, then, would a highly reactive ChOOH like 7α-OOH survive trafficking through cytosol to Mito in a steroid-producing cell? The START domain of StarD4 (and by implication StarD1) consists of a hydrophobic Ch binding pocket and a lid-like fold (10). We postulate that StarD4-borne 7α-OOH would be protected against reductive turnover during transit. In the case of one-electron reduction, this is reasonable because levels of redox-active iron in cytosol of prestressed cells would be vanishingly low (59). Moreover, the hydroperoxyl group of 7α-OOH is near the C-3 hydroxyl, but distant from the hydrophobic isoprenoid tail, suggesting that like Ch, it should be tightly sequestered in the START binding pocket (9, 10). This should hinder interaction with cytosolic antioxidants like GSH and GPx4, thus limiting the possibility of two-electron reductive loss in transit. These considerations further support our model of potentially deleterious ChOOH delivery via the StarD4/StarD1 system.
For acute needs, steroidogenic cells can acquire Ch from external sources, one of the most important being high density lipoprotein (HDL) in the circulation, which delivers esterified and free Ch via the SR-BI (3, 4). Upon arrival at the inner face of the plasma membrane, cholesteryl esters are hydrolyzed by hormone-sensitive lipase, after which the Ch molecule begins its StAR-mediated delivery to/into Mito. SR-BI is also expressed in liver and is crucial for removal of excess Ch via reverse Ch transport (4). Previous studies have shown that stress-generated ChOOHs in HDL can also be disposed of in this fashion (60). Under such conditions, it is not difficult to imagine that SR-BI-expressing steroidogenic tissues might also take up some ChOOH-bearing HDL and that this might provoke damaging redox reactions in these tissues. We are in the process of testing this idea on cAMP-activated MA-10 cells, using HDL that has been transfer-loaded with various levels of 7α-OOH.
In summary, our findings are both significant and novel because they describe a previously unrecognized mechanism by which steroid hormone production in the adrenal gland, ovary, and testis may be compromised in physiological states associated with increasing oxidative stress and/or declining antioxidant capacity, important examples being diabetes (61), atherosclerosis (17), and natural aging (16, 43). Of added interest, our study provides valuable insights into how the antisteroidogenic effects of ChOOH trafficking might be attenuated by site-selective antioxidants such mitochondrial GPx4 or MitoQ (62).
Acknowledgments
We are grateful to Dr. Mario Ascoli of The University of Iowa for generously providing us with the MA-10 mouse Leydig cells as a gift and for informing us about suitable methods for growing and maintaining these cells in culture. We also thank Drs. Douglas Stocco and Pulak Manna of Texas Tech University for advice regarding optimization of conditions for StarD1 knockdown in MA-10 cells.
This work was supported, in whole or in part, by National Institutes of Health Grant HL85677 (to A. W. G.) and by Polish National Science Center Grant 2011/01/B/NZ3/02167 and Medical College of Wisconsin Research Affairs Committee Grant 3726 (to W. K.).
- Ch
- cholesterol
- ChOOH
- cholesterol hydroperoxide
- Bt2cAMP
- dibutyryl cyclic AMP
- DME
- Dulbecco's modified Eagle's
- GPx1
- glutathione peroxidase type-1
- GPx4
- glutathione peroxidase type-4
- IM
- mitochondrial inner membrane
- OM
- mitochondrial outer membrane
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolyl-carbocyanine iodide
- ΔΨm
- mitochondrial membrane potential
- MTT
- 3-(4,5-dimethylthiazolyl-2-yl)-2,5-diphenyltetrazolium bromide
- PI
- propidium iodide
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SCP-2
- sterol carrier protein-2
- StarD1 and StarD4
- type-1 and type-4 steroidogenic acute regulatory domain protein
- StAR
- steroidogenic acute regulatory
- START
- StAR-related lipid transfer
- SUV
- small unilamellar vesicle
- t-BuOOH
- tert-butyl hydroperoxide
- 7α-OOH
- 3β-hydroxycholest-5-ene-7α-hydroperoxide
- 7α-OH
- cholest-5-ene-3β,7α-diol
- AMC
- 7-amido-4-methylcoumarin
- P450scc
- P450 side-chain cleavage enzyme
- SR-BI
- class B type I scavenger receptor.
REFERENCES
- 1. Miller W. L. (1988) Molecular biology of steroid hormone synthesis. Endocr. Rev. 9, 295–318 [DOI] [PubMed] [Google Scholar]
- 2. Payne A. H., Hales D. B. (2004) Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 25, 947–970 [DOI] [PubMed] [Google Scholar]
- 3. Rone M. B., Fan J., Papadopoulos V. (2009) Cholesterol transport in steroid biosynthesis: role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta 1791, 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Connelly M. A., Williams D. L. (2003) SR-BI and cholesterol uptake into steroidogenic cells. Trends Endocrinol. Metab. 14, 467–472 [DOI] [PubMed] [Google Scholar]
- 5. Kraemer F. B., Shen W. J. (2002) Hormone-sensitive lipase: control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid Res. 43, 1585–1594 [DOI] [PubMed] [Google Scholar]
- 6. Soccio R. E., Breslow J. L. (2003) StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J. Biol. Chem. 278, 22183–22186 [DOI] [PubMed] [Google Scholar]
- 7. Miller W. L. (2007) StAR search–what we know about how the steroidogenic acute regulatory protein mediates mitochondrial cholesterol import. Mol. Endocrinol. 21, 589–601 [DOI] [PubMed] [Google Scholar]
- 8. Miller W. L. (2007) Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim. Biophys. Acta 1771, 663–676 [DOI] [PubMed] [Google Scholar]
- 9. Tsujishita Y., Hurley J. H. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol. 7, 408–414 [DOI] [PubMed] [Google Scholar]
- 10. Romanowski M. J., Soccio R. E., Breslow J. L., Burley S. K. (2002) Crystal structure of the Mus musculus cholesterol-regulated START protein 4 (StarD4) containing a StAR-related lipid transfer domain. Proc. Natl. Acad. Sci. U.S.A. 99, 6949–6954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hauet T., Yao Z-X., Bose H. S., Wall C. T., Han Z., Li W., Hales D. B., Miller W. L., Culty M., Papadopoulos V. (2005) Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into Leydig cell mitochondria. Mol. Endocrinol. 19, 540–554 [DOI] [PubMed] [Google Scholar]
- 12. Soccio R. E., Breslow J. L. (2004) Intracellular cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 24, 1150–1160 [DOI] [PubMed] [Google Scholar]
- 13. Rodriguez-Agudo D., Ren S., Hylemon P. B., Redford K., Natarajan R., Del Castillo A., Gil G., Pandak W. M. (2005) Human StarD5, a cytosolic StAR-related lipid binding protein. J. Lipid Res. 46, 1615–1623 [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez-Agudo D., Ren S., Wong E., Marques D., Redford K., Gil G., Hylemon P., Pandak W. M. (2008) Intracellular cholesterol transporter StarD4 binds free cholesterol and increases cholesteryl ester formation. J. Lipid Res. 49, 1409–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zirkin B. R., Chen H. (2000) Regulation of Leydig cell steroidogenic function during aging. Biol. Reprod. 63, 977–981 [DOI] [PubMed] [Google Scholar]
- 16. Chen H., Ge R. S., Zirkin B. R. (2009) Mol. Cell. Endocrinol. Leydig cells: From stem cells to aging. 306, 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steinberg D., Witztum J. L. (2002) Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation 105, 2107–2111 [DOI] [PubMed] [Google Scholar]
- 18. Porter N. A., Caldwell S. E., Mills K. A. (1995) Mechanisms of free radical oxidation of unsaturated lipids. Lipids 30, 277–290 [DOI] [PubMed] [Google Scholar]
- 19. Girotti A. W. (1998) Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 39, 1529–1542 [PubMed] [Google Scholar]
- 20. Vila A., Korytowski W., Girotti A. W. (2001) Spontaneous intermembrane transfer of various cholesterol-derived hydroperoxide species: kinetic studies with model membranes and cells. Biochemistry 40, 14715–14726 [DOI] [PubMed] [Google Scholar]
- 21. Vila A., Korytowski W., Girotti A. W. (2002) Spontaneous transfer of phospholipid and cholesterol hydroperoxides between cell membranes and low-density lipoprotein: assessment of reaction kinetics and prooxidant effects. Biochemistry 41, 13705–13716 [DOI] [PubMed] [Google Scholar]
- 22. Girotti A. W. (2008) Translocation as a means of disseminating lipid hydroperoxide-induced oxidative damage and effector action. Free Radic. Biol. Med. 44, 956–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vila A., Levchenko V. V., Korytowski W., Girotti A. W. (2004) Sterol carrier protein-2-facilitated intermembrane transfer of cholesterol- and phospholipid-derived hydroperoxides. Biochemistry 43, 12592–12605 [DOI] [PubMed] [Google Scholar]
- 24. Korytowski W., Rodriguez-Agudo D., Pilat A., Girotti A. W. (2010) StarD4-mediated translocation of 7-hydroperoxycholesterol to isolated mitochondria: deleterious effects and implications for steroidogenesis under oxidative stress conditions. Biochem. Biophys. Res. Commun. 392, 58–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Korytowski W., Bachowski G. J., Girotti A. W. (1991) Chromatographic separation and electrochemical determination of cholesterol hydroperoxides generated by photodynamic action. Anal. Biochem. 197, 149–156 [DOI] [PubMed] [Google Scholar]
- 26. Korytowski W., Geiger P. G., Girotti A. W. (1999) Lipid hydroperoxide analysis by high-performance liquid chromatography with mercury cathode electrochemical detection. Methods Enzymol. 300, 23–33 [DOI] [PubMed] [Google Scholar]
- 27. Girotti A. W., Korytowski W. (2000) Cholesterol as a singlet oxygen detector in biological systems. Methods Enzymol. 319, 85–100 [DOI] [PubMed] [Google Scholar]
- 28. Ascoli M. (1981) Characterization of several clonal lines of cultured Leydig tumor cells: gonadotropin receptors and steroidogenic responses. Endocrinology 108, 88–95 [DOI] [PubMed] [Google Scholar]
- 29. Wang X. J., Dyson M. T., Mondillo C., Patrignani Z., Pignataro O., Stocco D. M. (2002) Interaction between arachidonic acid and cAMP signaling pathways enhances steroidogenesis and StAR gene expression in MA-10 Leydig tumor cells. Mol. Cell Endocrinol. 188, 55–63 [DOI] [PubMed] [Google Scholar]
- 30. Mayer L. D., Hope M. J., Cullis P. R. (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta 858, 161–168 [DOI] [PubMed] [Google Scholar]
- 31. Reers M., Smiley S. T., Mottola-Hartshorn C., Chen A., Lin M., Chen L. B. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260, 406–417 [DOI] [PubMed] [Google Scholar]
- 32. Kriska T., Levchenko V. V., Korytowski W., Atshaves B. P., Schroeder F., Girotti A. W. (2006) Intracellular dissemination of peroxidative stress. Internalization, transport, and lethal targeting of a cholesterol hydroperoxide species by sterol carrier protein-2-overexpressing hepatoma cells. J. Biol. Chem. 281, 23643–23651 [DOI] [PubMed] [Google Scholar]
- 33. Bhowmick R., Girotti A. W. (2010) Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy. Free Radic. Biol. Med. 48, 1296–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 [DOI] [PubMed] [Google Scholar]
- 35. Pradelles P., Grassi J., Maclouf J. (1985) Enzyme immunoassays of eicosanoids using acetylcholine esterase as label: an alternative to radioimmunoassay. Anal. Chem. 57, 1170–1173 [DOI] [PubMed] [Google Scholar]
- 36. Spector D. L., Goldman R. D., Leinwald L. A. (1997) in Cells: A Laboratory Manual, Vol. 1, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 37. Stocco D. M., Kilgore M. W. (1988) Induction of mitochondrial proteins in MA-10 Leydig tumour cells with human choriogonadotropin. Biochem. J. 249, 95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clark B. J., Soo S-C., Caron K. M., Ikeda Y., Parker K. L., Stocco D. M. (1995) Hormonal and developmental regulation of the steroidogenic acute regulatory protein. Mol. Endocrinol. 9, 1346–1355 [DOI] [PubMed] [Google Scholar]
- 39. Danial N. N., Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 40. Kroemer G., Galluzzi L., Brenner C. (2007) Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99–163 [DOI] [PubMed] [Google Scholar]
- 41. Schulze-Osthoff K., Ferrari D., Los M., Wesselborg S., Peter M. E. (1998) Apoptosis signaling by death receptors. Eur. J. Biochem. 254, 439–459 [DOI] [PubMed] [Google Scholar]
- 42. Azhar S., Cao L., Reaven E. (1995) Alteration of the adrenal antioxidant defense system during aging in rats. J. Clin. Invest. 96, 1414–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Midzak A. S., Chen H., Papadopoulos V., Zirkin B. R. (2009) Leydig cell aging and the mechanisms of reduced testosterone synthesis. Mol. Cell Endocrinol. 299, 23–31 [DOI] [PubMed] [Google Scholar]
- 44. Hales D. B., Diemer T., Hales K. H. (1999) Role of cytokines in testicular function. Endocrine 10, 201–217 [DOI] [PubMed] [Google Scholar]
- 45. Culty M., Luo L., Yao Z-X., Chen H., Papadopoulos V., Zirkin B. R. (2002) Cholesterol transport, peripheral benzodiazepine receptor, and steroidogenesis in aging Leydig cells. J. Androl. 23, 439–447 [PubMed] [Google Scholar]
- 46. Luo L., Chen H., Trush M. A., Show M. D., Anway M. D., Zirkin B. R. (2006) Aging and the brown Norway rat leydig cell antioxidant defense system. J. Androl. 27, 240–247 [DOI] [PubMed] [Google Scholar]
- 47. Chen H., Pechenino A. S., Liu J., Beattie M. C., Brown T. R., Zirkin B. R. (2008) Effect of glutathione depletion on Leydig cell steroidogenesis in young and old brown Norway rats. Endocrinology 149, 2612–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kodaman P. H., Aten R. F., Behrman H. R. (1994) Lipid hydroperoxides evoke antigonadotropic and antisteroidogenic activity in rat luteal cells. Endocrinology 135, 2723–2730 [DOI] [PubMed] [Google Scholar]
- 49. Diemer T., Allen J. A., Hales K. H., Hales D. B. (2003) Reactive oxygen disrupts mitochondria in MA-10 tumor Leydig cells and inhibits steroidogenic acute regulatory (StAR) protein and steroidogenesis. Endocrinology 144, 2882–2891 [DOI] [PubMed] [Google Scholar]
- 50. Abidi P., Zhang H., Zaidi S. M., Shen W-J., Leers-Sucheta S., Cortez Y., Han J., Azhar S. (2008) Oxidative stress-induced inhibition of adrenal steroidogenesis requires participation of p38 mitogen-activated protein kinase signaling pathway. J. Endocrinol. 198, 193–207 [DOI] [PubMed] [Google Scholar]
- 51. Kallen C. B., Billheimer J. T., Summers S. A., Stayrook S. E., Lewis M., Strauss J. F. (1998) Steroidogenic acute regulatory protein (StAR) is a sterol transfer protein. J. Biol. Chem. 273, 26285–26288 [DOI] [PubMed] [Google Scholar]
- 52. Ariyoshi K., Adachi J., Asano M., Ueno Y., Rajendram R., Preedy V. R. (2002) Effect of chronic ethanol feeding on oxysterols in rat liver. Free Radic. Res. 36, 661–666 [DOI] [PubMed] [Google Scholar]
- 53. Yoshioka N., Adachi J., Ueno Y., Yoshida K-I. (2005) Oxysterols increase in diabetic rats. Free Radic. Res. 39, 299–304 [DOI] [PubMed] [Google Scholar]
- 54. Girotti A. W. (2002) in Sterols and Oxysterols: Chemistry, Biology, and Pathobiology (Fliesler S. J., ed) pp. 121–139, Research Signpost, Trivandrum, India [Google Scholar]
- 55. Thomas J. P., Maiorino M., Ursini F., Girotti A. W. (1990) Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation: in situ reduction of phospholipid and cholesterol hydroperoxides. J. Biol. Chem. 265, 454–461 [PubMed] [Google Scholar]
- 56. Korytowski W., Geiger P. G., Girotti A. W. (1996) Enzymatic reducibility in relation to cytotoxicity for various cholesterol hydroperoxides. Biochemistry 35, 8670–8679 [DOI] [PubMed] [Google Scholar]
- 57. Papp L. V., Lu J., Holmgren A., Khanna K. K. (2007) From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid. Redox Signal. 9, 775–806 [DOI] [PubMed] [Google Scholar]
- 58. Imai H., Nakagawa Y. (2003) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic. Biol. Med. 34, 145–169 [DOI] [PubMed] [Google Scholar]
- 59. Hallwell B., Gutteridge J. M. C. (1990) Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 186, 1–85 [DOI] [PubMed] [Google Scholar]
- 60. Fluiter K., Sattler W., De Beer M. C., Connell P. M., van der Westhuyzen D. R., van Berkel T. J. C. (1999) Scavenger receptor BI mediates the selective uptake of oxidized cholesterol esters by rat liver. J. Biol. Chem. 274, 8893–8899 [DOI] [PubMed] [Google Scholar]
- 61. Astort F., Repetto E. M., Martínez Calejman C., Cipelli J. M., Sánchez R., Di Gruccio J. M., Mercau M., Pignataro O. P., Arias P., Cymeryng C. B. (2009) High glucose-induced changes in steroid production in adrenal cells. Diabetes Metab. Res. Rev 25, 477–486 [DOI] [PubMed] [Google Scholar]
- 62. Smith R. A., Murphy M. P. (2010) Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N.Y. Acad. Sci. 1201, 96–103 [DOI] [PubMed] [Google Scholar]