

Background: Malignant peripheral nerve sheath tumor (MPNST) cells overexpress PDGF receptor-β, which increases intracellular calcium when activated.
Results: Calcium/calmodulin (CaM) is involved in sustained phosphorylation of Akt and promotion of cell survival in an MPNST cell line.
Conclusion: MPNST cells evade normal cell death through CaM-dependent sustained activation of Akt.
Significance: Activation of CaM by abnormally expressed growth factor receptors may contribute to NF1 tumor formation.
Keywords: Akt/PKB, Calmodulin, Cancer Biology, Cell Death, Growth Factors, PI 3-Kinase (PI3K), Schwann Cells, Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
Abstract
Neurofibromatosis type 1-derived Schwann cells isolated from malignant peripheral nerve sheath tumors (MPNSTs) overexpress PDGF receptor-β and generate an aberrant intracellular calcium increase in response to PDGF-BB. Using the human MPNST Schwann cell line ST88-14, we demonstrate that, in addition to a transient phosphorylation of Akt, PDGF-BB stimulation produces an atypical sustained phosphorylation of Akt that is dependent on calcium and calmodulin (CaM). The sustained Akt phosphorylation did not occur in PDGF-BB-stimulated normal human Schwann cells or ST88-14 cells stimulated with stem cell factor, whose receptor is also overexpressed in ST88-14 cells. The sustained Akt phosphorylation induced by PDGF-BB was inhibited by pretreatment of the cells with either the intracellular calcium chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl) ester (BAPTA-AM) or the CaM antagonist W7, whereas the transient portion was not inhibited. Akt also co-immunoprecipitated with CaM in a PDGF-BB-dependent manner, suggesting that direct interaction between Akt and CaM is involved in the sustained phosphorylation of Akt. Furthermore, we provide evidence that anti-apoptotic effects of PDGF-BB on serum-deprived ST88-14 cells can be inhibited by W7, implicating the PDGF-BB-induced activation of calcium/CaM in promoting cell survival, presumably through sustained Akt activation. We conclude that the activation of the calcium/CaM/Akt pathway resulting from stimulation of overexpressed PDGF receptor-β may contribute to the survival and tumorigenicity of MPNST cells.
Introduction
Elevated expression of growth factors and their receptors is a common characteristic of tumor cells, including many of the Schwann cell lines derived from neurofibromin-deficient malignant peripheral nerve sheath tumors (MPNSTs).3 These tumors are associated with neurofibromatosis type 1 (NF1) (1, 2), which is a human genetic disorder caused by mutations in the tumor suppressor gene that encodes the GTPase-activating protein neurofibromin. NF1-derived cell lines have elevated levels of receptors, such as the EGF receptor (3) and c-Kit (4), that typically are not expressed by Schwann cells. These abnormally expressed receptors likely play a role in the development of MPNSTs. Alterations in growth factor receptor-mediated signaling may also be caused by the activation of receptors characteristically present on normal Schwann cells but overexpressed in MPNST cells, which is the case for PDGF receptor-β (PDGFR-β) (5). Activation of PDGFR-β in MPNST cell lines induces an increase in intracellular calcium and phosphorylation of calmodulin (CaM) kinase II, signaling events that do not happen when PDGFR-β is stimulated in normal Schwann cells (6).
The serine/threonine kinase Akt/PKB is a central downstream component of growth factor receptor-mediated signaling and plays an essential role in controlling normal cell survival, proliferation, and migration (7). The question of how Akt signaling can mediate such diverse cellular functions may be explained in part by the three different Akt isoforms (Akt1 (α), Akt2 (β), and Akt3 (γ)) that make up the Akt family. These isoforms appear to play unique as well as overlapping roles in the cell (8).
Dysregulation of the Akt pathway can lead to tumor formation by altering critical biological outcomes within the cell. Akt activation occurs through both PI3K-dependent and -independent pathways. During PI3K-dependent activation, binding of a growth factor to its receptor tyrosine kinase recruits and activates PI3K, which then catalyzes the conversion of membrane phosphatidylinositol 3,4-bisphosphate to phosphatidylinositol 3,4,5-trisphosphate. Akt is translocated to the plasma membrane, where it is anchored through its pleckstrin homology domain to phosphatidylinositol 3,4,5-trisphosphate and becomes activated through phosphorylation in the kinase domain at Thr-308 by PDK1 (protein-dependent kinase-1) and in the regulatory domain at Ser-473 by mTORC2 (mammalian target of rapamycin complex 2) (9). The PI3K-independent pathways that activate Akt are less well characterized and include direct interactions between Ras GTPase-activating protein and Akt (10), as well as CaM-mediated activation, which can occur through direct association of Akt with either CaM (11–13) or CaM-activated CaM kinase kinase (14).
In this study, we examined the role of calcium/CaM in PDGF-BB-stimulated Akt phosphorylation in the NF1-derived MPNST cell line ST88-14, which overexpresses PDGFR-β and produces an abnormal increase in intracellular calcium as a result of PDGF-BB stimulation (6). We found that calcium/CaM-dependent and -independent mechanisms act concurrently to influence Akt phosphorylation, but with different kinetics. Furthermore, we show that the calcium/CaM-dependent Akt phosphorylation induced by PDGF-BB supports survival under conditions that promote apoptosis, suggesting a potential mechanism by which these cells evade normal programmed cell death and promote tumorigenesis.
EXPERIMENTAL PROCEDURES
Cell Culture and Growth Factor Treatment
The NF1-derived MPNST cell line ST88-14 (obtained from Jonathan Fletcher, Brigham and Women's Hospital, Boston, MA) was grown in DMEM (Invitrogen) supplemented with 5% FBS. The day before an experiment, cells were plated at a density of 150,000 cells/35-mm dish (immunoblotting experiments) or 1.5–2 × 106 cells/100-mm dish (immunoprecipitation experiments) and incubated overnight in DMEM and 5% FBS. Four hours prior to growth factor treatment, the serum-containing medium was replaced with DMEM without FBS to reduce the stimulatory effects of the serum.
Human Schwann cells were prepared from human peripheral nerve (obtained from Dr. Patrick Wood, Miami Project to Cure Paralysis, University of Miami Miller School of Medicine, Miami, FL) by the method of Casella et al. (15). The epineurium was removed from the nerves, and the nerves were cut into 2-mm segments and placed on 35-mm tissue culture dishes containing DMEM and 10% FBS. The nerve segments were allowed to incubate for 2 weeks in the presence of 2 μm forskolin (Calbiochem) and 10 ng/ml neuregulin β1 (a gift from Amgen, Thousand Oaks, CA). The fascicles were dissociated with collagenase, and the dissociated cells were placed on collagen-coated tissue culture dishes. The cells were grown in DMEM and 5% FBS supplemented with 10 ng/ml neuregulin β1. Differential adhesion was used to remove contaminating fibroblasts each time the cells were passaged and before the cells were plated for an experiment. Four hours before growth factor addition, the growth medium was removed from the cells and replaced with DMEM without serum, neuregulin β1, or forskolin to reduce the stimulatory effects of these agents.
Western Blot Analysis
The cells on 35-mm culture dishes were treated with PDGF-BB (PeproTech, Rocky Hill, NJ) or stem cell factor (SCF; R&D Systems, Minneapolis, MN) at a concentration of 20 ng/ml. For some experiments, the inhibitors LY294002 (50 μm), wortmannin (0.2 μm), BAPTA-AM (25 μm) (Calbiochem), W7 (10 μm), and W5 (10 μm) (BIOMOL, Plymouth Meeting, PA) were added 30 min prior to growth factor treatment. At the designated time points, cells were placed on ice, washed twice with cold PBS containing 1 mm NaF and 1 mm Na3VO4, and immediately solubilized in 50–100 μl of sample buffer. Cellular proteins were separated on 4–12% NuPAGE bis-tris gels (Invitrogen) and then transferred to a PVDF membrane (PerkinElmer Life Sciences). After blocking in TBS and 0.05% Tween 20 containing 5% nonfat dry milk, the membrane was incubated overnight at 4 °C with primary antibody: anti-NF1 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-phospho-ERK (pERK)1/2, anti-phospho-Akt (pAkt), anti-Akt, anti-poly(ADP-ribose) polymerase (PARP), anti-cleaved PARP (New England Biolabs, Beverly, MA); or anti-GAPDH (Trevigen, Gaithersburg, MD). The membrane was washed four times with TBS and 0.05% Tween 20 and then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (Pierce or Jackson ImmunoResearch Laboratories). Immunoreactivity was detected by enhanced chemiluminescence (Pierce and PerkinElmer Life Sciences). Exposed films were semiquantitatively analyzed by densitometry using NIH ImageJ software.
Co-immunoprecipitation
At designated time points after growth factor treatment, ST88-14 cells (1.5–2 × 106 cells) were solubilized in 1 ml of lysis/immunoprecipitation buffer (150 mm NaCl, 10 mm Tris, 1 mm EDTA, 1% Triton X-100, 0.5% Nonidet P-40, protease inhibitor mixture (Calbiochem), 50 mm NaF, and 1 mm Na3VO4), centrifuged to remove insoluble material, and incubated with 2 μg of rabbit anti-CaM monoclonal antibody (Epitomics, Burlingame, CA) for 1 h at 4 °C. Protein A/G-agarose (15 μl; Pierce) was added, and the mixture was incubated overnight at 4 °C with mixing. The agarose pellets were collected by centrifugation and washed three times with lysis/immunoprecipitation buffer. After the last wash, 15 μl of 2.5× reducing sample buffer was added to the agarose pellets. The eluted proteins were separated on 4–12% NuPAGE gels and transferred to PVDF membranes for Western blot analysis. The blots were immunostained for Akt and CaM. The protein bands on films were semiquantitatively analyzed by densitometry using ImageJ software.
Cell Survival Studies
ST88-14 cells were plated at a density of 150,000 cells/35-mm tissue culture dish. The following day, the medium was removed, and the cells were washed once to remove traces of serum before adding low glucose minimum essential medium (MEM; Invitrogen) without serum supplementation. The CaM inhibitor W7 or W5 or vehicle (Me2SO) was added 30 min prior to PDGF-BB stimulation. The cells were incubated for 24 h and then harvested in sample buffer. Cleavage of nuclear PARP was analyzed by Western blotting.
Statistics
All experiments were performed at least three times. Statistical analyses were done using GraphPad Prism software. Student's t test was used to compare between two groups with significance at p < 0.05.
RESULTS
ERK, but Not Akt, Shows Altered Basal Levels of Phosphorylation and Expression
Previous work from our laboratory (6) suggested that the atypical increase in intracellular calcium resulting from PDGFR-β activation in NF1-derived Schwann cells may influence receptor-mediated signaling cascades, particularly the PI3K/Akt and MAPK pathways. Prior to examining PDGF-BB-induced signaling in NF1-derived ST88-14 cells, we assessed the basal levels of phosphorylation and expression of Akt and ERK1/2 to determine whether constitutively elevated Ras-GTP, inherent to NF1-derived cells, affects these pathways in unstimulated cells. ST88-14 cells were compared with normal human Schwann cells (nhSc) under serum-free conditions to remove contributions from serum-induced signaling. The ST88-14 cells expressed an undetectable level of neurofibromin (Fig. 1) and had 4-fold more Ras-GTP than nhSc (data not shown), consistent with the original characterization of the ST88-14 cell line as well as other NF1-derived MPNST Schwann cells (16).
FIGURE 1.
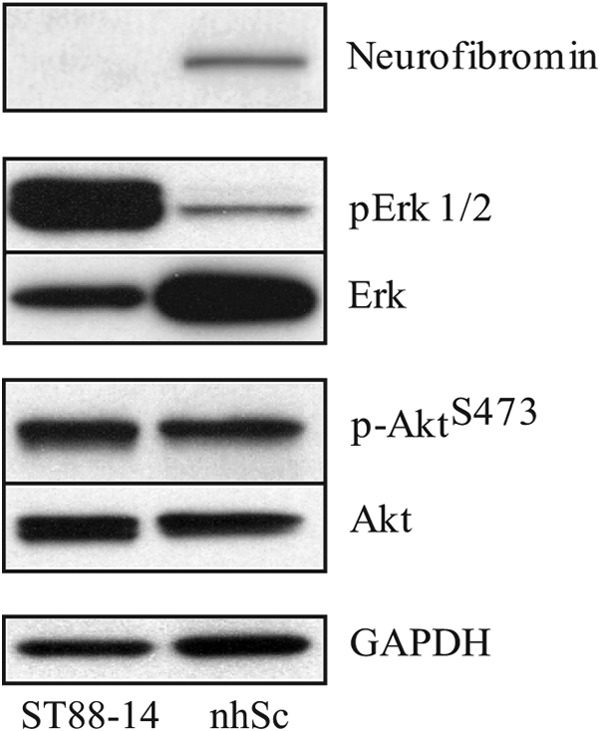
Comparison of basal levels of Akt and ERK1/2 phosphorylation and expression in ST88-14 cells and nhSc. Total cell lysates were prepared from cells that were serum-starved for 4 h prior to harvesting and analyzed by Western blotting. The difference in neurofibromin expression between the two cell types was demonstrated by immunostaining with a polyclonal antibody whose epitope maps within the C terminus of human neurofibromin. Immunostaining for pERK1/2 and pAktSer-473 was done using appropriate phosphospecific antibodies. Peroxidase-conjugated secondary antibodies were used for visualization, after which the blots were stripped and reprobed with anti-Akt and anti-ERK antibodies. GAPDH was used as a loading control. Notice that there are similar levels of total Akt and pAktSer-473 for the two cell types but markedly different amounts of total ERK and pERK. The results shown are representative of three or more independent experiments.
The basal levels of both Akt phosphorylated at Ser-473 (pAktSer-473) and total Akt were comparable between nhSc and ST88-14 cells (Fig. 1). In contrast, ST88-14 cells had increased amounts of pERK1/2 (Fig. 1). Interestingly, the amount of total ERK protein was markedly higher in nhSc, even though pERK was greater in ST88-14 cells (Fig. 1). These data imply that, at least in unstimulated ST88-14 cells, the NF1 phenotype has no appreciable effect on the PI3K/Akt pathway, whereas the MAPK pathway shows a substantial increase in basal activity relative to nhSc.
Growth Factor Activation of Akt and MAPK Pathways
We analyzed the time course of Akt and ERK1/2 phosphorylation after activation by either SCF or PDGF-BB. Each of these growth factors has its receptor overexpressed in the ST88-14 cell line (4, 5). Over a 2-h time period, maximum phosphorylation of Akt at Ser-473 occurred within 5 min following stimulation of ST88-14 cells with either SCF (Fig. 2A) or PDGF-BB (Fig. 2B). However, pAktSer-473 returned to nearly unstimulated levels (0 min) within 1 h after SCF treatment (Fig. 2A), whereas a high level of pAktSer-473 in PDGF-BB-treated cells was sustained for >2 h (Fig. 2B). A similar comparison between nhSc and ST88-14 cells after stimulation with PDGF-BB revealed a transient phosphorylation of Akt at Ser-473 in nhSc (Fig. 2C) relative to the sustained phosphorylation of Akt at Ser-473 in ST88-14 cells (Fig. 2D).
FIGURE 2.

Time course of Akt Ser-473 and ERK1/2 phosphorylation after growth factor stimulation. Cells were plated at a density of 1.5 × 105 cells/35-mm tissue culture dish for ST88-14 cells or type I collagen-coated dish for nhSc, and the following day, they were serum-starved for 4 h prior to being treated with growth factors (20 ng/ml for both SCF and PDGF-BB) for the indicated times. Total cell lysates were prepared and subjected to Western blot analysis for pAkt, pERK1/2, and GAPDH (loading control). Phosphorylation of Akt and ERK1/2 over a 2-h time period is compared in SCF-stimulated (A) and PDGF-BB-stimulated (B) ST88-14 cells and in PDGF-BB-stimulated nhSc (C) and PDGF-BB-stimulated ST88-14 cells (D). The results shown are representative of three independent experiments.
To determine whether sustained activation of the MAPK pathway also occurs in ST88-14 cells after PDGF-BB stimulation, the blots were immunostained for pERK1/2. Similar to the initial kinetics of SCF- or PDGF-BB-induced pAktSer-473 formation described above, maximum phosphorylation of ERK1/2 in ST88-14 cells occurred within 5 min. The initial phosphorylation of ERK1/2 was not as rapid in nhSc, which appeared to peak between 5 and 30 min (Fig. 2C). The PDGF-BB-induced increase in pERK1/2 levels in ST88-14 cells was sustained at least 30 min longer than that in ST88-14 cells stimulated with SCF (Fig. 2, A and B) or in nhSc stimulated with PDGF-BB (Fig. 2, C and D). However, by 120 min, the amount of pERK1/2 in PDGF-BB-treated ST88-14 cells had returned to unstimulated levels (0 min) (Fig. 2, B and D). These observations indicate that, relative to Akt, the MAPK pathway in ST88-14 cells is noticeably less susceptible to sustained activation by PDGF-BB.
Intracellular Calcium Chelator BAPTA-AM and CaM Antagonist W7 Inhibit Sustained but Not Transient Akt Ser-473 Phosphorylation
On the basis of our previous findings that PDGF-BB stimulation elicits an increase in intracellular calcium in NF1-derived Schwann cell lines, including ST88-14 cells, and that this intracellular calcium increase does not occur in PDGF-BB-stimulated nhSc (6), we designed experiments to determine whether the PDGF-BB-stimulated intracellular calcium increase was responsible for the sustained Akt Ser-473 phosphorylation. ST88-14 cells were preincubated with the intracellular calcium chelator BAPTA-AM for 30 min prior to PDGF-BB stimulation. Fig. 3A shows that BAPTA-AM treatment significantly inhibited the sustained (120 min) phosphorylation of Akt at Ser-473 without affecting the transient (30 min) phosphorylation. This result indicates that PDGF-BB induces both a transient calcium-independent and a sustained calcium-dependent phosphorylation of Akt at Ser-473.
FIGURE 3.

Effect of intracellular calcium chelator BAPTA-AM and CaM antagonist W7 on PDGF-BB-induced transient and sustained phosphorylation of Akt at Ser-473. ST88-14 cells were serum-starved for 4 h and then pretreated with vehicle, 25 μm BAPTA-AM (A), 10 μm W7 (B), or 10 μm W5 (B) for 30 min prior to PDGF-BB stimulation. Cells were harvested at 0 min (untreated), 30 min (to represent transient phosphorylation), and 120 min (to represent sustained phosphorylation), and total cellular protein was subjected to Western blot analysis. Blots were immunostained for pAktSer-473 and GAPDH (loading control), followed by peroxidase-conjugated secondary antibodies. Results are mean ± S.E. from three (A) or four (B) independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To establish whether CaM is involved in the sustained calcium-mediated component of Akt Ser-473 phosphorylation, we pre-incubated ST88-14 cells for 30 min with the CaM antagonist W7 (10 μm) before PDGF-BB treatment. Similar to BAPTA-AM-treated cells, only the sustained component (120 min) of Akt Ser-473 phosphorylation was inhibited in the presence of W7 (Fig. 3B). For a control, we used the less potent CaM inhibitor W5, which appeared to have less of an effect (Fig. 3B). These combined data indicate that the sustained phosphorylation of Akt at Ser-473 in PDGF-BB-stimulated ST88-14 cells is calcium/CaM-dependent, whereas transient Akt phosphorylation is not.
PI3K Inhibitors Block PDGF-BB-induced Akt Ser-473 Phosphorylation
The dependence of PDGF-BB-induced transient (calcium/CaM-independent) and sustained (calcium/CaM-dependent) Akt Ser-473 phosphorylation on PI3K activity was examined by preincubating cells for 30 min with 50 μm LY294002 prior to stimulation with PDGF-BB. In the presence of the PI3K inhibitor, there was almost a complete inhibition of Akt Ser-473 phosphorylation over the 120-min time period (Fig. 4). A similar effect was obtained with 0.2 μm wortmannin (data not shown). These results indicate that both the calcium/CaM-independent and -dependent components of Akt phosphorylation require PI3K activity.
FIGURE 4.
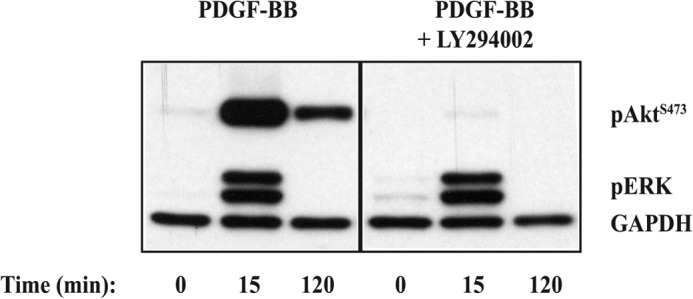
Effect of PI3K inhibition with LY294002 on PDGF-BB-stimulated transient and sustained Akt Ser-473 phosphorylation. ST88-14 cells were serum-starved for 4 h and then pretreated with vehicle or 50 μm LY294002 for 30 min prior to PDGF-BB (20 ng/ml) stimulation. Cells were harvested at 0 min (untreated), 15 min (to represent transient phosphorylation), and 120 min (to represent sustained phosphorylation), and total cellular protein was subjected to Western blot analysis. Blots were immunostained for pAktSer-473, pERK, and GAPDH (loading control), followed by peroxidase-conjugated secondary antibodies. Results are representative of three independent experiments.
Co-immunoprecipitation of CaM with Akt
Because both the intracellular calcium chelator BAPTA-AM and the CaM antagonist W7 inhibited only the sustained (120 min) component of PDGF-BB-induced Akt phosphorylation, we considered the possibility that CaM interacts directly with Akt to form a functional complex that could be involved in maintaining Akt in a phosphorylated state. To establish whether direct interactions between CaM and Akt occur in a PDGF-BB-dependent manner, we carried out co-immunoprecipitation experiments in which CaM was immunoprecipitated from ST88-14 cells treated with PDGF-BB for either 30 or 120 min. We consistently found that Akt was associated with CaM in unstimulated cells and that PDGF-BB stimulation increased the amount of this association over the 120-min time period (Fig. 5). These results indicate that PDGF-BB induces a direct and sustained association of CaM with Akt, which coincides with the sustained phosphorylation of Akt.
FIGURE 5.
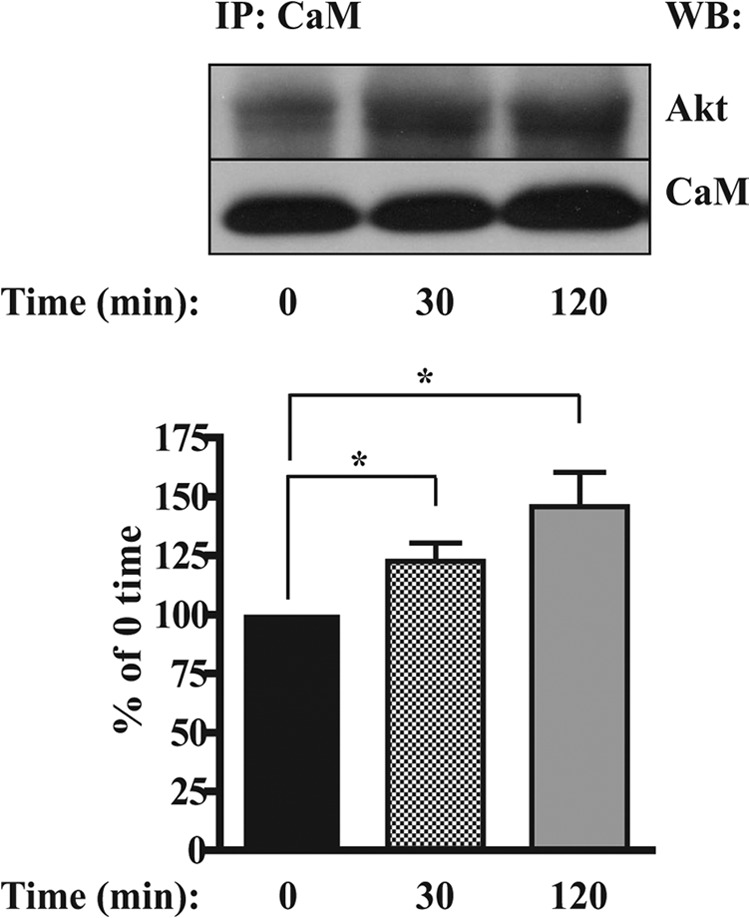
PDGF-BB-dependent co-immunoprecipitation of Akt with CaM. ST88-14 cells (1.5 × 106/time point) were serum-starved for 4 h and then treated with 20 ng/ml PDGF-BB for 0 (untreated), 30, and 120 min. Cells were solubilized in lysis buffer, and the lysates were incubated overnight with anti-CaM antibody and precipitated with protein A/G-agarose. Immunoprecipitated Akt was detected by Western blotting (WB) using an antibody to total Akt. Results are mean ± S.E. from five independent experiments. *, p < 0.05. IP, immunoprecipitation.
CaM Antagonist W7 Blocks the Anti-apoptotic Effect of PDGF-BB in ST88-14 Cells
Given that Akt activation promotes cell survival, we next investigated whether PDGF-BB stimulation could enhance ST88-14 cell survival under conditions that would be expected to drive the cells toward apoptosis and, if so, whether this survival stimulus is dependent on CaM. To induce apoptosis, cells were cultured in low glucose MEM under conditions of serum deprivation. Treatment with PDGF-BB alone or after a 30-min pretreatment with W7 or the control analog W5 was used to determine the dependence of PDGF-BB-induced cell survival on CaM. Cellular morphology (Fig. 6, A–F) and cleavage of nuclear PARP (Fig. 6, G and H) were used to evaluate the apoptotic status of the cells after 24 h. Relative to cells grown in medium containing serum (Fig. 6A), the cells maintained in serum-free/low glucose MEM for 24 h showed cell contraction and rounding (Fig. 6B, arrowheads), characteristic of cells undergoing cell death, with significant cleavage of PARP. PDGF-BB added to serum-free/low glucose MEM helped maintained normal cellular morphology (Fig. 6C) and prevented PARP cleavage, indicating that PDGF-BB promotes survival under these conditions. The anti-apoptotic effect of PDGF-BB was blocked by the CaM antagonist W7, which caused cell contraction and rounding (Fig. 6D, arrowheads) and produced PARP cleavage beyond that of the serum-free/low glucose medium alone. This result suggests that CaM may have an additional role in promoting survival that is independent of PDGF-BB stimulation. In contrast, cells pretreated with the control analog W5 (Fig. 6E) showed less cell contraction and rounding and PARP cleavage than the W7-treated cells. SCF, which produced only a transient phosphorylation of Akt at Ser-473 (Fig. 2A), showed minimum effectiveness in reversing the outcomes of serum deprivation and low glucose, i.e. morphology associated with dying cells (Fig. 6F), and preventing PARP cleavage. Considered together with our evidence that W7 inhibits only the sustained portion of Akt Ser-473 phosphorylation (Fig. 3B), these results suggest that PDGF-BB-induced activation of CaM plays an important role in promoting survival of ST88-14 cells, most likely through sustained Akt activation.
FIGURE 6.

Effect of CaM antagonist W7 on PDGF-BB-induced cell survival. Shown is the morphology of ST88-14 cells incubated for 24 h in normal medium (DMEM and 5% FBS) (A), serum-free/low glucose MEM (B), MEM + 20 ng/ml PDGF-BB (C), MEM + 20 ng/ml PDGF-BB + 10 μm W7 (D), MEM + 20 ng/ml PDGF-BB + 10 μm W5 (E), or MEM + 20 ng/ml SCF (F). W7 or W5 was added to the medium 30 min prior to PDGF-BB treatment. Arrowheads in B and D indicate cells with morphologies consistent with dying cells. Scale bar = 100 μm. G and H, after incubating ST88-14 cells for 24 h under the conditions indicated in A–F, cells were harvested, and total cellular protein was subjected to Western blot analysis. Blots were immunostained for cleaved PARP, PARP, and GAPDH (loading control), followed by peroxidase-conjugated secondary antibodies. Results are mean ± S.E. from four independent experiments. *, p < 0.05; **, p < 0.01.
DISCUSSION
Neurofibromin-deficient cells generated from NF1 tumors characteristically hyperproliferate as a consequence of their constitutively elevated Ras activity and subsequent activation of signaling pathways involved in the regulation of cell growth (1, 17–19). NF1-derived Schwann cells also have been shown to overexpress growth factor receptors (3–5), which, when coupled with aberrant intracellular signaling, can cause these cells to express phenotypic traits characteristic of tumors, such as unregulated growth and survival. In this study, we focused on the atypical intracellular calcium signaling coupled to the activation of overexpressed PDGFR-β (6), and we have demonstrated that calcium/CaM plays a necessary role in a sustained but not transient Akt phosphorylation resulting from PDGF-BB stimulation. We also present evidence that PDGF-BB-induced activation of CaM has a prosurvival influence on ST88-14 cells placed under conditions that promote cell death, suggesting that PDGF-BB-induced sustained Akt activation may be a mechanism by which NF1-derived MPNST cells evade apoptosis.
In our initial characterization, we found that the extent of Akt expression and phosphorylation in unstimulated ST88-14 cells was essentially the same as in nhSc, indicating that constitutively elevated Ras-GTP was not affecting the basal (unstimulated) levels of pAkt and that there was no amplification or overexpression of Akt by the ST88-14 cells. In contrast and consistent with elevated Ras-GTP as an upstream activator of the MAPK pathway, pERK was significantly increased in the unstimulated ST88-14 cells relative to nhSc, which is in agreement with previous characterizations of the ST88-14 cell line (16, 18, 20) and other neurofibromin-deficient cell types (18, 20). Interestingly, even though ST88-14 cells had levels of pERK that were constitutively elevated, the amount of total ERK protein was reduced compared with that expressed by nhSc. Thus, relative to nhSc, the levels of pERK were higher in unstimulated ST88-14 cells even though the amount of total ERK protein was lower. This result might suggest that chronic elevation of pERK in ST88-14 cells results in a negative feedback mechanism that reduces the expression of ERK protein in an effort to limit ERK activation.
Although nhSc typically do not express receptors for SCF, PDGFR-β is expressed by both nhSc and ST88-14 cells (6), which allowed us to compare PDGF-BB activation of the PI3K/Akt signaling pathway between the two cell types. Our results show that the duration of PDGF-BB-stimulated Akt Ser-473 phosphorylation in ST88-14 cells extended beyond 120 min, whereas in PDGF-BB-stimulated nhSc, pAktSer-473 had returned to nearly unstimulated levels by 60 min. The longer duration of Akt phosphorylation was also specific to PDGF-BB-mediated signaling in the ST88-14 cells because stimulation with SCF resulted in a time course for pAkt formation that was transient, similar to that for PDGF-stimulated nhSc. A previous study from our laboratory showed that there is an increase in intracellular calcium in response to PDGF-BB stimulation of NF1-derived Schwann cell lines but not nhSc and that this calcium response is associated with CaM activation as indicated by CaM kinase II phosphorylation (6). On the basis of this earlier work, we hypothesized that a calcium/CaM-dependent mechanism was responsible for sustained Akt phosphorylation.
Our studies with the intracellular calcium chelator BAPTA-AM and the CaM antagonist W7 demonstrate that the PDGF-BB-induced calcium activation of CaM is responsible for the sustained portion of Akt Ser-473 phosphorylation and that the transient phosphorylation of Akt at Ser-473 is a separate event independent of either calcium or CaM. This is a notable observation because it shows that Akt phosphorylation can be regulated concurrently by more than one mechanism, in this case, calcium/CaM-independent and -dependent mechanisms. Previous reports in which CaM was shown to be involved in Akt activation include CaM association with the SH2 (Src homology 2) domain of the 85-kDa regulatory subunit of PI3K (21) and CaM activation of CaM kinase kinase, which then proceeds to phosphorylate Akt directly (14). However, in our hands, experiments to co-immunoprecipitate CaM with the 85-kDa regulatory subunit of PI3K or to block sustained Akt phosphorylation with the specific CaM kinase kinase inhibitor STO609 provided no evidence for either of these two mechanisms occurring in PDGF-BB-stimulated ST88-14 cells (data not shown).
More recent studies with mouse mammary carcinoma and human breast cancer cell lines have provided evidence for an EGF-induced direct association between CaM and Akt that may serve to translocate Akt to the plasma membrane, where it becomes activated (12, 13). Our finding that PDGF-BB increases the formation of a CaM and Akt immunoprecipitable complex in ST88-14 cells is consistent with this type of functional complex being formed between the two molecules, although it remains to be determined whether the PDGF-BB-induced formation of this complex serves to translocate Akt to the plasma membrane or other sites within the cell. Nonetheless, our co-immunoprecipitation results, together with the inhibition of sustained Akt phosphorylation by W7 and, to a lesser extent, by the weaker antagonist W5, strongly suggest that this PDGF-BB-induced physical association between CaM and Akt is responsible for its sustained activation.
A major difference between our results and those of Deb et al. (12) and Coticchia et al. (13) is that, in mouse mammary and human breast cancer cell lines, treatment with the CaM antagonist W7 blocked all of Akt phosphorylation, whereas in our studies with ST88-14 cells, W7 inhibited only sustained Akt phosphorylation without a discernible effect on the transient portion. Thus, their results indicate that all of the EGF-induced Akt activation occurred through one CaM-dependent mechanism, whereas our results clearly indicate that two separate mechanisms are involved concurrently in the activation of Akt.
Both the calcium/CaM-dependent and -independent mechanisms were sensitive to the PI3K inhibitors LY294002 and wortmannin, indicating that each is PI3K-dependent and requires Akt to associate with phosphatidylinositol 3,4,5-trisphosphate at the plasma membrane prior to activation by phosphorylation. It is worth considering in this context that two binding sites for calcium/CaM have been identified within Akt (11). One of these sites is at the N terminus of the kinase catalytic domain, and the other is within the first half of the pleckstrin homology domain, which also is the domain necessary for Akt to associate with phosphatidylinositol 3,4,5-trisphosphate at the plasma membrane. It is possible that calcium/CaM binds the pleckstrin homology domain of activated Akt, thereby releasing it from the membrane and preventing or delaying its inactivation by an Akt phosphatase (22).
Although we could not ascertain definitively that Akt plays a direct role in the PDGF-BB-induced survival of serum-deprived ST88-14 cells, our results do show that the PDGF-BB-induced calcium/CaM response is necessary for this survival. By blocking the CaM-dependent sustained Akt phosphorylation with W7 or by stimulating the cells with SCF, which produced only a transient Akt response, we showed that transient activation of Akt is not sufficient to counteract the apoptotic response (PARP cleavage) induced by serum and glucose deprivation. Given the suggested role of CaM in directing Akt to specific intracellular sites (12, 13), together with recent evidence indicating that distinct subcellular pools of Akt are important for its specific functions (23), it is possible that, in ST88-14 cells, CaM may serve to target activated Akt to a particular intracellular site(s) where anti-apoptotic substrates for Akt reside.
It is not known whether there is an Akt isoform dependence for NF1, and information on this topic for other Ras-induced tumors is limited. One Ras-induced model in which Akt isoform specificity has been studied is mutant K-Ras-mediated lung tumorigenesis (24), in which Akt1 was identified as the only Akt isoform required for tumor initiation and progression. This study is consistent with Akt1 knock-out mice being growth-impaired (25). However, other studies have linked Akt-dependent cell survival to Akt3 (26) or, in some tumor cell lines, all three Akt isoforms (27), suggesting that the survival function of an Akt isoform may be cell line-specific. Indeed, with respect to potential therapeutic targets, it will be important to ascertain whether a particular Akt isoform is responsible for its sustained phosphorylation mediated by calcium/CaM in NF1 tumor cells.
To date, emphasis has been placed primarily on how growth factors affect proliferation of NF1 tumor cells. Along this line, EGF has been shown to be the most potent growth factor for stimulating proliferation of NF1-derived Schwann cells (3), whereas PDGF-BB is only slightly stimulatory (5), and there is little if any stimulation of proliferation by SCF (4). Thus, PDGF-BB, which does not appear to have a major role in proliferation of MPNST cells, may enhance their survival by stimulating a calcium/CaM-dependent mechanism involved in maintaining Akt in an active state. We propose that PDGF signaling allows NF1 Schwann cells to evade normal programmed cell death and that this aberrant survival is a component of tumor formation.
This work was supported by the Department of Veterans Affairs, Illinois Neurofibromatosis, Inc., and United States Army Medical Research and Materiel Command Contract Grant DAMD17-98-8607.
- MPNST
- malignant peripheral nerve sheath tumor
- NF1
- neurofibromatosis type 1
- PDGFR-β
- PDGF receptor-β
- CaM
- calmodulin
- SCF
- stem cell factor
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl) ester
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- pERK
- phospho-ERK
- pAkt
- phospho-Akt
- PARP
- poly(ADP-ribose) polymerase
- MEM
- minimum essential medium
- nhSc
- normal human Schwann cells
- pAktSer-473
- Akt phosphorylated at Ser-473.
REFERENCES
- 1. Carroll S. L., Stonecypher M. S. (2005) Tumor suppressor mutations and growth factor signaling in the pathogenesis of NF1-associated peripheral nerve sheath tumors. II. The role of dysregulated growth factor signaling. J. Neuropathol. Exp. Neurol. 64, 1–9 [DOI] [PubMed] [Google Scholar]
- 2. Thomas S. L., De Vries G. H. (2009) Neurofibromatosis type I: from genetic mutation to tumor formation. in Handbook of Neurochemistry and Molecular Neurobiology (Lajtha A., ed) 3rd Ed., pp. 107–130, Springer, New York [Google Scholar]
- 3. DeClue J. E., Heffelfinger S., Benvenuto G., Ling B., Li S., Rui W., Vass W. C., Viskochil D., Ratner N. (2000) Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J. Clin. Invest. 105, 1233–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Badache A., Muja N., De Vries G. H. (1998) Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene 17, 795–800 [DOI] [PubMed] [Google Scholar]
- 5. Badache A., De Vries G. H. (1998) Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J. Cell. Physiol. 177, 334–342 [DOI] [PubMed] [Google Scholar]
- 6. Dang I., De Vries G. H. (2005) Schwann cell lines derived from malignant peripheral nerve sheath tumors respond abnormally to platelet-derived growth factor-BB. J. Neurosci. Res. 79, 318–328 [DOI] [PubMed] [Google Scholar]
- 7. Franke T. F. (2008) PI3K/Akt: getting it right matters. Oncogene 27, 6473–6488 [DOI] [PubMed] [Google Scholar]
- 8. Gonzalez E., McGraw T. E. (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8, 2502–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarbassov D. D., Guertin D. A., Ali S. M., Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 10. Yue Y., Lypowy J., Hedhli N., Abdellatif M. (2004) Ras GTPase-activating protein binds to Akt and is required for its activation. J. Biol. Chem. 279, 12883–12889 [DOI] [PubMed] [Google Scholar]
- 11. Dong B., Valencia C. A., Liu R. (2007) Ca2+/calmodulin directly interacts with the pleckstrin homology domain of AKT1. J. Biol. Chem. 282, 25131–25140 [DOI] [PubMed] [Google Scholar]
- 12. Deb T. B., Coticchia C. M., Dickson R. B. (2004) Calmodulin-mediated activation of Akt regulates survival of c-Myc-overexpressing mouse mammary carcinoma cells. J. Biol. Chem. 279, 38903–38911 [DOI] [PubMed] [Google Scholar]
- 13. Coticchia C. M., Revankar C. M., Deb T. B., Dickson R. B., Johnson M. D. (2009) Calmodulin modulates Akt activity in human breast cancer cell lines. Breast Cancer Res. Treat. 115, 545–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yano S., Tokumitsu H., Soderling T. R. (1998) Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396, 584–587 [DOI] [PubMed] [Google Scholar]
- 15. Casella G. T., Bunge R. P., Wood P. M. (1996) Improved method for harvesting human Schwann cells from mature peripheral nerve and expansion in vitro. Glia 17, 327–338 [DOI] [PubMed] [Google Scholar]
- 16. DeClue J. E., Papageorge A. G., Fletcher J. A., Diehl S. R., Ratner N., Vass W. C., Lowy D. R. (1992) Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69, 265–273 [DOI] [PubMed] [Google Scholar]
- 17. Donovan S., See W., Bonifas J., Stokoe D., Shannon K. M. (2002) Hyperactivation of protein kinase B and ERK have discrete effects on survival, proliferation, and cytokine expression in Nf1-deficient myeloid cells. Cancer Cell 2, 507–514 [DOI] [PubMed] [Google Scholar]
- 18. Zhang Y. Y., Vik T. A., Ryder J. W., Srour E. F., Jacks T., Shannon K., Clapp D. W. (1998) Nf1 regulates hematopoietic progenitor cell growth and Ras signaling in response to multiple cytokines. J. Exp. Med. 187, 1893–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ingram D. A., Hiatt K., King A. J., Fisher L., Shivakumar R., Derstine C., Wenning M. J., Diaz B., Travers J. B., Hood A., Marshall M., Williams D. A., Clapp D. W. (2001) Hyperactivation of p21ras and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J. Exp. Med. 194, 57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Basu T. N., Gutmann D. H., Fletcher J. A., Glover T. W., Collins F. S., Downward J. (1992) Aberrant regulation of Ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356, 713–715 [DOI] [PubMed] [Google Scholar]
- 21. Joyal J. L., Burks D. J., Pons S., Matter W. F., Vlahos C. J., White M. F., Sacks D. B. (1997) Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 272, 28183–28186 [DOI] [PubMed] [Google Scholar]
- 22. Gao T., Furnari F., Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 23. Maiuri T., Ho J., Stambolic V. (2010) Regulation of adipocyte differentiation by distinct subcellular pools of protein kinase B (PKB/Akt). J. Biol. Chem. 285, 15038–15047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hollander M. C., Maier C. R., Hobbs E. A., Ashmore A. R., Linnoila R. I., Dennis P. A. (2011) Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene 30, 1812–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho H., Thorvaldsen J. L., Chu Q., Feng F., Birnbaum M. J. (2001) Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 276, 38349–38352 [DOI] [PubMed] [Google Scholar]
- 26. Liu X., Shi Y., Birnbaum M. J., Ye K., De Jong R., Oltersdorf T., Giranda V. L., Luo Y. (2006) Quantitative analysis of anti-apoptotic function of Akt in Akt1 and Akt2 double knock-out mouse embryonic fibroblast cells under normal and stressed conditions. J. Biol. Chem. 281, 31380–31388 [DOI] [PubMed] [Google Scholar]
- 27. Koseoglu S., Lu Z., Kumar C., Kirschmeier P., Zou J. (2007) AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol. Ther. 6, 755–762 [DOI] [PubMed] [Google Scholar]