

Background: Tau fibrillization inhibitors hold promise as potential therapeutic agents for neurodegenerative disease.
Results: The Tau fibrillization inhibitors, aminothienopyridazines and methylene blue, promote disulfide bond formation in Tau.
Conclusion: These compounds affect Tau fibrillization by a relatively nonspecific oxidative mechanism.
Significance: Understanding the mechanism by which these compounds affect Tau fibril formation provides insights into their potential as therapeutic agents.
Keywords: Alzheimer Disease, Disulfide, Drug Action, Oxidation-Reduction, Tau, Fibrils, Methylene Blue
Abstract
Alzheimer disease and several other neurodegenerative disorders are characterized by the accumulation of intraneuronal fibrils comprised of the protein Tau. Tau is normally a soluble protein that stabilizes microtubules, with splice isoforms that contain either three (3-R) or four (4-R) microtubule binding repeats. The formation of Tau fibrils is thought to result in neuronal damage, and inhibitors of Tau fibrillization may hold promise as therapeutic agents. The process of Tau fibrillization can be replicated in vitro, and a number of small molecules have been identified that inhibit Tau fibril formation. However, little is known about how these molecules affect Tau fibrillization. Here, we examined the mechanism by which the previously described aminothieno pyridazine (ATPZ) series of compounds inhibit Tau fibrillization. Active ATPZs were found to promote the oxidation of the two cysteine residues within 4-R Tau by a redox cycling mechanism, resulting in the formation of a disulfide-containing compact monomer that was refractory to fibrillization. Moreover, the ATPZs facilitated intermolecular disulfide formation between 3-R Tau monomers, leading to dimers that were capable of fibrillization. The ATPZs also caused cysteine oxidation in molecules unrelated to Tau. Interestingly, methylene blue, an inhibitor of Tau fibrillization under evaluation in Alzheimer disease clinical trials, caused a similar oxidation of cysteines in Tau and other molecules. These findings reveal that the ATPZs and methylene blue act by a mechanism that may affect their viability as potential therapeutic agents.
Introduction
A number of neurodegenerative diseases are characterized by the presence within brain neurons of deposits of the protein Tau (1, 2). The most prevalent of these “tauopathies” is Alzheimer disease (AD),3 but this group of disorders also includes various forms of frontotemporal lobar degeneration, including progressive supranuclear palsy, corticobasal degeneration, Pick's disease, and a rare familial disorder known as frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (3–6). Tau is normally found in axons, where it stabilizes microtubules (MTs) through an interaction mediated by the three (3-R) or four (4-R) repeated MT binding domains found in Tau isoforms that are generated by differential splicing (7, 8). However, there is a redistribution of Tau in tauopathies such that inclusions form within the cell soma and processes, where they are referred to as neurofibrillary tangles and neuropil threads, respectively (4). The Tau aggregates in these diseases are made up of well defined fibrils comprised of an ordered array of misfolded Tau monomers, which are recognized by amyloid binding dyes such as thioflavin S and Congo Red.
Tau inclusions are thought to contribute to the neuronal dysfunction and death that occurs in the tauopathies. For example, there is a correlation between neurofibrillary tangle density and cognitive decline in AD (9–11). Moreover, Tau mutations have been shown to cause FTDP-17 (5, 6). The neurotoxicity associated with Tau may result from a direct effect of misfolded Tau oligomers (12) or fibrils and/or from a loss of Tau-mediated MT stabilization due to its disengagement from MTs and sequestration into aggregates (3, 13, 14). Thus, a potential therapeutic strategy to reduce neurotoxicity in tauopathies would be to prevent the formation of Tau oligomers and fibrils (15, 16).
The formation of insoluble Tau fibrils is believed to depend on a nucleation-elongation process whereby misfolded Tau monomers assemble relatively slowly into ill-defined nucleating cores followed by rapid elongation from the core structures to yield fibrils (17, 18). The process of Tau fibrillization can be replicated in vitro, where the addition of anionic co-factors such as heparin or arachidonic acid to recombinant Tau results in the formation of fibrils that resemble those observed in the brains of tauopathy patients (19–22). This has led several laboratories to identify a relatively diverse set of molecules that inhibit Tau fibrillization (15, 23, 24). Among these, one compound, methylene blue (MB), has progressed to testing in AD patients (25), where potentially promising Phase II clinical data have been reported that await confirmation in larger Phase III studies. However, very little is known about how MB or other reported compounds prevent the assembly of Tau into fibrils. We previously described an aminothienopyridazine (ATPZ) series of Tau fibrillization inhibitors with drug-like physical-chemical properties (26–28) and identified a brain-penetrant example that is well tolerated upon repeated dosing in mice (27, 28). Here, we have investigated the molecular mechanism by which active ATPZ molecules prevent Tau fibril formation and have identified a mode of action that involves the oxidation of cysteine residues within Tau. This results in the formation of an intramolecular disulfide linkage in 4-R Tau that renders the monomer fibrillization-incompetent as well as the generation of an intermolecular disulfide bond in 3-R Tau that promotes dimerization and fibrillization. Interestingly, this mechanism is not unique to the ATPZ series but is also observed with the AD clinical candidate, MB.
EXPERIMENTAL PROCEDURES
Compounds
All ATPZs and oleocanthal were synthesized as previously described (27–29), with purities of >95% as determined by NMR and LC-MS/MS. MB was purchased from Sigma. A 10-mer peptide (NRCSQGSCWN) comprised of a sequence unrelated to Tau was purchased from Sigma.
Tau Constructs and Preparation
Full-length Tau constructs containing the two alternatively spliced N-terminal exons and either four (T40) or three (T39) semi-conserved MT binding regions as well as the truncated K18 and K19 constructs containing only the MT binding regions from 4-R and 3-R Tau, respectively (30), were cloned into the pRK172 expression vector, expressed, and purified as previously described (6). K18PL, which is the K18 construct containing the P301L mutation found in inherited FTDP-17 (5) that confers an enhanced fibrillization propensity to Tau (31), was prepared in a similar manner. Mutated versions of K18PL were prepared by site-directed mutagenesis to yield alanine substitutions of cysteine 291 (C291A), cysteine 322 (C322A), or both (2XCA), with the resulting proteins purified as with the other Tau preparations.
Tau Pre-reduction and Pre-oxidation
Recombinant Tau was reduced by incubating Tau preparations in 1 mm DTT for 30 min at 60 °C. DTT-reduced Tau protein underwent centrifugation through a Zebra spin desalting column (Thermo Scientific 89882) that had been pre-equilibrated in 100 mm sodium acetate buffer, pH 7.0, to remove DTT. Tau protein was subsequently quantified by BCA assay (Pierce) to account for protein losses on the column. Tau oxidation can be facilitated by treatment with DMSO as described (32) or can be allowed to occur spontaneously when 20 μm Tau in 100 mm sodium acetate buffer, pH 7.0, is incubated overnight at room temperature.
Tau Compound Incubation
Tau proteins were typically incubated at 20 μm concentration with 50 μm ATPZs or MB for 1 h at 37 °C in 0.1 m sodium acetate, pH 7.0, unless otherwise noted under “Experimental Procedures”.
Tau Fibrillization
Heparin-induced Tau fibrillization was performed as previously described (26, 33). Briefly, 15 μl of 33.3 μm recombinant Tau in fibrillization buffer (100 mm sodium acetate buffer, pH 7.0) was dispensed into wells of black non-treated polystyrene 384-well plates (NUNC) followed by a pin tool dispense of 250-fold concentrated test compound that was dissolved in 100% DMSO. This was immediately followed by the addition of 10 μl of 100 μm heparin (Sigma; 5 kDa mean molecular mass) in fibrillization buffer, such that the final Tau and heparin concentrations were 20 and 40 μm, respectively. Compound concentration-response analyses were performed starting at 80 μm, with 2-fold serial dilutions down to 0.16 μm. In some studies, oleocanthal (50 μm) was included as a positive control inhibitor of Tau fibrillization (29), and vehicle alone was used as a non-fibrillizing control. Plates were incubated at 37 °C overnight followed by the addition of 25 μl of 25 μm thioflavin T (ThT; Sigma) in 100 mm glycine, pH 8.5. After incubation for 1 h at room temperature, ThT fluorescence was read at an excitation of 450 nm and an emission of 510 nm with a cutoff of 475 nm on a Spectramax M5 spectrophotometer (Molecular Devices). Z′ scores (34) for each dose-response experiment were greater than 0.5. In some cases ThT was omitted, and the samples were subjected to sedimentation analysis as described below.
Tau Sedimentation Assay
The contents of the wells from fibrillization reactions as described above were removed and centrifuged at 100,000 × g for 30 min (Beckman Coulter TL-100 centrifuge). Supernatants were removed, and one part 5× SDS-PAGE buffer (62.5 mm Tris, pH 6.8, 10% SDS, 25% glycerol, 10 mm DTT, 0.01% bromphenol blue) was added to four parts supernatant. The pellet was dissolved in a volume of 1× SDS-PAGE buffer equal to the final supernatant volume followed by SDS-PAGE of equal volumes of the supernatant and pellet fraction (typically 10 μl for K18 or K19 Tau species or 2 μl for full-length Tau), with subsequent Coomassie Blue (R250) staining. Gel band intensities were quantified using ImageQuant software (Molecular Dynamics), and fibrillization inhibition was determined by comparing the percent of Tau remaining in the supernatant fraction of compound-treated samples relative to the full fibrillization (vehicle only) controls.
Native PAGE
The native gel electrophoresis protocol utilized buffer systems as previously described (35). Gels were prepared from 7.5 or 15% acrylamide (37.5:1 acrylamide/bisacrylamide; Bio-Rad) for full-length Tau or truncated Tau proteins, respectively, in a low conductivity acidic buffer (30 mm β-alanine (Sigma) and 20 mm lactic acid (Sigma), pH 3.8). Tau samples were prepared by mixing with 2.5× sample buffer (75 mm β-alanine and 50 mm lactic acid, pH 3.8, 0.01% methyl green, and 25% glycerol) to achieve 1× final sample buffer followed by loading into the wells of the gel. Gels were run at 4 °C on a Bio-Rad Mini Protean III system at 180 V for 2 h with the polarity reversed, then stained with Coomassie Blue. Fully reduced Tau, fully oxidized Tau, and vehicle-treated Tau were typically included on each gel in lieu of molecular weight markers.
Size-exclusion Chromatography (SEC)
K18PL, K19, K18PL-C291A, K18PL-C322A, or K18PL2xCA (20 μm) were incubated with ATPZ or MB (50 μm) in 100 mm sodium acetate, pH 7.0, for 1 h at 37 °C. SEC was performed using an Acquity UPLC system equipped with a photodiode array detector (Waters Corp., Milford, MA). Injections of 15 μl were separated with an Acquity BEH200 SEC 1.7 μm (4.6 × 300 mm including a 4.6 × 30 guard column) using 100 mm sodium acetate, pH 5, with 300 mm NaCl at 0.3 ml/min over 30 min. Sample peaks were detected and analyzed using absorbance at 220 nm.
Reversed-phase Chromatography
The 10-mer peptide (NRCSQGSCWN) at 20 μm concentration was incubated with 50 μm ATPZ or MB in 100 mm sodium acetate, pH 5, for 30 min at 37 °C. Reversed-phase HPLC was performed using an Acquity UPLC system equipped with an Acquity BEH C18 1.7 μm (2.1 × 50 mm) column at 35 °C with detection using a photodiode array detector and a TQ mass spectrometer. The MS electrospray source was operated in positive ion mode. A water/acetonitrile gradient containing 0.1% formic acid from 5 to 35% acetonitrile over 1.5 min at a flow rate of 0.6 ml/min was used to separate peptide after 5-μl sample injections. Sample peaks were detected and analyzed using absorbance at 280 nm. DTT at 20 μm was incubated with 50 μm CNDR-51348 in 100 mm sodium acetate, pH 5, for 30 min at 37 °C. Reversed-phase chromatography was performed using an Acquity UPLC system equipped with an Acquity HSS T3 1.8 μm (2.1 × 100 mm) column at 35 °C, with detection using a photodiode array detector and a TQ mass spectrometer. The MS electrospray source was operated in negative ion mode. Isocratic elution conditions using 2% acetonitrile with 0.1% formic acid at 0.6 ml/min were used to separate components after a 10-μl sample injection. Sample peaks were detected and analyzed using absorbance at 210 nm and compared to reduced or oxidized DTT standards.
Oregon Green-Iodoacetamide Labeling of Tau
Iodoacetamide labeled with Oregon Green 488 (IAA-OG, Invitrogen) was dissolved in N,N-dimethylformamide to achieve a 10 mm stock solution. Tau (20 μm) in fibrillization buffer with or without 50 μm test compound at a final volume of 0.1 ml was incubated for 1 h at 37 °C. A 20-μl aliquot of this solution was removed and added to a tube containing 1 μl of IAA-OG reagent (25-fold molar excess of IAA-OG to Tau) and incubated 30 min at room temperature followed by analysis by SDS-PAGE. Upon completion of electrophoresis, the unbound IAA-OG reagent at the gel dye front was cut away, and the gel was imaged on a Fuji LAS 3000 imager using the existing green fluorescent protein setting. The fluorescent bands were quantified using ImageQuant 5.0 software for Windows (Molecular Dynamics), and intensity was compared with vehicle only and pre-reduced controls, the latter of which defines the maximal possible IAA-OG signal. After fluorescent imaging, the gel was stained with Coomassie Blue to confirm equal protein loading between the samples.
CNDR-51348 HPLC-MS
CNDR-51348 (50 μm) was incubated with 20 μm K18PL in 100 mm sodium acetate, pH 7, for 1 h at 37 °C. LC-MS analyses were performed using an Acquity UPLC system equipped with a TQ mass spectrometer. Samples were separated with an Acquity BEH C18 1.7 μm (2.1 × 50 mm) column at 35 °C. A water/acetonitrile gradient containing 0.1% formic acid from 5 to 95% acetonitrile over 2 min at a flow rate of 0.6 ml/min was used to separate CNDR-51348 after a 5-μl sample injection. The MS electrospray source was operated in negative ion mode. Source and analyzer voltages were optimized using the MassLynx auto tune utility. MS scans from 100 to 600 m/z with 0.5-s scan times were acquired. Mass spectra were then analyzed for the loss of ATPZ or the appearance of chemically reduced products.
Peroxide Quantification and Tau Treatment with Peroxide
Compound-mediated peroxide generation was measured using the PeroXOquantTM assay (Pierce 23280) per the manufacturer's kit instructions. Tau (20 μm) or 1 mm DTT was incubated with 50 μm compound in water at a final volume of 0.1 ml for periods of time ranging from 1 to 18 h. Aliquots (10 μl) were removed and transferred into a clear non-treated polystyrene 384-well assay plate (NUNC) to which 90 μl of kit reagent was added and allowed to incubate 20 min at room temperature. The absorbance at 595 nm was subsequently read on a Spectramax M5 spectrophotometer (Molecular Devices). Hydrogen peroxide controls were prepared by dilution from a 30% (8.8 m) hydrogen peroxide stock (Fisher) into water. A linear relationship between peroxide concentration and absorbance was observed between 62.5 and 2 μm peroxide and was used as a standard curve for quantification of peroxide in the compound samples. K18PL protein (20 μm) was treated with either 20 μm or 1 mm hydrogen peroxide in 0.1 m sodium acetate, pH 7.0, for 1 h at 37 °C in the absence or presence of 24 milliunits of catalase (Sigma), as described below, followed by native gel electrophoresis.
Catalase Treatment of Tau
Catalase (Sigma) was added to Tau samples incubated with either peroxide-generating compounds or with varying concentrations of hydrogen peroxide. In addition, an identical amount of catalase was added to Tau fibrillization reactions that were conducted as described above in the presence of peroxide-generating compounds. The catalase was added to the Tau solutions at a 50,000-fold dilution to yield a 24 milliunits/ml final concentration before compound or hydrogen peroxide was added. Fibrillization was assayed by ThT fluorescence as above. Catalase activity was confirmed to be identical to the manufacturer's specification when used in fibrillization buffer.
RESULTS
ATPZ Inhibitors of Tau Fibrillization Promote Formation of an Intramolecular Disulfide Bond in 4-R Tau Monomers
The ATPZ inhibitors of Tau fibril formation were initially identified after high-throughput screening of ∼300,000 compounds (26), and subsequent medicinal chemistry efforts resulted in the development of many analogues and the elucidation of structure-activity relationships (27, 28). An initial SEC characterization of the Tau species found within an ATPZ-treated fibrillization reaction revealed a large increase in Tau monomer concentration relative to an uninhibited fibrillization mixture (26). These data combined with the observation that the IC50 values of ATPZ inhibitors were generally near the stoichiometric equivalence of Tau concentration suggested that the ATPZs might prevent fibril formation through interaction with Tau monomers, although association with multimeric Tau species could not be excluded. To further investigate the possible interaction of ATPZs with monomeric Tau, additional SEC studies were conducted after incubation of an active ATPZ (CNDR-51348; see structure in Table 1) with a well described truncated Tau protein (K18PL) comprising the four MT binding repeats found in full-length 4-R Tau. This protein fibrillizes rapidly in the presence of a co-factor such as heparin (26, 33). SEC analyses were also performed with K18PL Tau that was incubated in the absence of an ATPZ or with an ATPZ analog (CNDR-51449; see the structure in Table 1) that does not inhibit Tau fibrillization (26). Interestingly, co-incubation of the Tau with CNDR-51348, but not CNDR-51449, resulted in the formation of a later-eluting peak that was present only as a minor peak when K18PL was incubated in the absence of compound (Fig. 1).
TABLE 1.
The relative activity of various ATPZ compounds in a 4-R Tau fibrillization reaction correlates with their ability to induce a disulfide-containing tau compact monomer
K18PL Tau fibrillization was monitored in the absence and presence of ATPZs using ThT fluorescence as previously described (26, 27). Compound activity is expressed both as the inhibitory log EC50 and as the maximum percent inhibition of Tau fibrillization. The compounds were also evaluated for their ability to induce the faster migrating, disulfide-containing compacted form of K18PL observed by native gel electrophoresis, as in Fig. 2B.
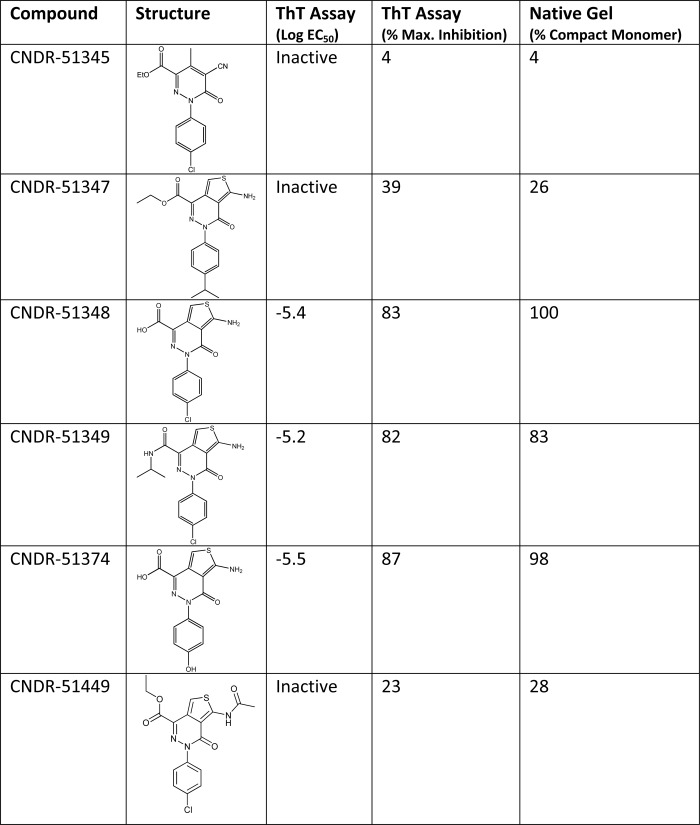
FIGURE 1.

Active ATPZs cause the formation of a compact 4-R Tau monomer. 4-R Tau K18PL protein was incubated for 1 h at 37 °C in the absence (K18PL) or presence of the active ATPZ, CNDR-51348 (K18PL:51348). The Tau protein was also either treated with an inactive ATPZ, CNDR-51449 (K18PL:51449) or was pre-reduced (K18PL Red) or pre-oxidized (K18PL Ox). The samples were subsequently analyzed by SEC.
A compacted form of 4-R Tau has been described previously in which the single cysteine residues found within both the second and third MT binding repeats are oxidized to form an intramolecular disulfide bridge (30). To investigate whether K18PL that was incubated with CNDR-51348 might correspond to a cysteine-oxidized form of the peptide, K18PL was treated with DTT to ensure complete reduction of cysteine residues. The DTT-reduced Tau (Fig. 1, K18PL Red) had a retention time that corresponded to the major peak in the untreated Tau sample. In contrast, K18PL samples that were incubated with DMSO (32) or incubated overnight in oxygenated buffer to facilitate cysteine oxidation (Fig. 1; K18PL Ox) had an elution time that was identical to the Tau peptide treated with CNDR-51438. These data are consistent with the ATPZ-treated Tau undergoing oxidization to generate a more compacted structure.
To further test this hypothesis, K18PL Tau was treated with CNDR-51348 or CNDR-51449 and subsequently reacted with IAA-OG, which preferentially forms a covalent adduct with the sulfhydryl moiety of reduced cysteine residues (36). K18PL was also pre-reduced or pre-oxidized before incubation with IAA-OG. As depicted in Fig. 2A, SDS-PAGE of the IAA-OG-reacted samples followed by fluorographic detection of the covalently linked Oregon Green fluorophore showed that the reduced Tau sample displayed significant IAA-OG labeling that was greater than that observed with untreated protein, suggesting that the latter contained a mixture of reduced and oxidized K18PL. However, very little IAA-OG modification was detected in the pre-oxidized Tau or the Tau treated with CNDR-51348.
FIGURE 2.

ATPZs promote intramolecular cysteine disulfide formation in 4-R Tau. A, K18PL was incubated for 1 h at 37 °C in the absence (−) or presence of CNDR-51348 (51348) or CNDR-51449 (51449) or was pre-reduced (R) or pre-oxidized (O). The samples were then analyzed for IAA-OG reactivity. Both the IAA-OG fluorescence and corresponding Coomassie blue staining are shown for each IAA-OG-treated sample after SDS-PAGE. B, K18PL, K18, or full-length 4-R Tau (T40) were incubated for 1 h at 37 °C in the absence (−) or presence of CNDR-51348 (51348) or CNDR-51449 (51449) or were pre-reduced (R) or pre-oxidized (O) followed by native gel electrophoresis. C, K18PL in which both cysteines were mutated to alanine (2xCA) was incubated for 1 h at 37 °C in the absence (2xCA) or presence of CNDR-51348 (2xCA:51348) followed by SEC analysis.
To confirm that ATPZ treatment of K18PL resulted in the formation of a compacted structure, the Tau was incubated in the absence or presence of CNDR-51348 or CNDR-51449, or pretreated to generate fully reduced and oxidized protein and subsequently analyzed by acidic native gel electrophoresis (Fig. 2B). The K18PL treated with CNDR-51348 had a greater electrophoretic mobility than the reduced Tau and migrated with the same apparent size as pre-oxidized Tau, whereas the untreated or CNDR-51449-treated Tau contained a mixture of the reduced and oxidized species. A small amount of the protein in all samples was found to migrate with greater apparent size, and this material likely corresponds to dimers. A comparable result was obtained with K18 Tau lacking the P301L mutation or with full-length 4-R Tau (T40), as incubation of these Tau species with CNDR-51348 resulted in conversion of the Tau to a compacted form that migrated identically to pre-oxidized Tau (Fig. 2B). Thus, all 4-R Tau species seem to be equally affected by active ATPZs to yield compact structures resulting from an intramolecular disulfide bond between the two available cysteine residues, and therefore, truncated and full-length Tau preparations were used interchangeably in subsequent studies.
To provide further evidence that the cysteine residues of 4-R Tau were critical to the formation of the compact monomer, CNDR-51348 was incubated with K18PL in which both of the cysteine residues were replaced with alanine (K18PL2xCA). As expected, this modified peptide failed to yield the compacted structure, as evidenced by an absence of retention time shift upon SEC analysis (Fig. 2C). These and the previous data demonstrate that the ATPZs, which prevent 4-R Tau fibrillization, promote the formation of an intramolecular disulfide within the Tau monomer.
It has previously been reported that a compact species of 4-R Tau harboring an intramolecular disulfide bridge does not readily assemble into fibrils in vitro (30, 37, 38), thereby providing a potential explanation as to why the active ATPZs impede 4-R Tau fibrillization. To confirm that oxidized 4-R Tau does not readily form fibrils, the K18 peptide was first pre-reduced, pre-oxidized, or treated with CNDR-51348 and then added to a standard fibrillization reaction mixture that contains heparin as a co-factor. Whereas the reduced Tau showed rapid fibrillization, as determined by increased ThT fluorescence, essentially no change in ThT signal was observed in the pre-oxidized Tau mixture or in that treated with CNDR-51348 (supplemental Fig. S1). Thus, oxidation of the cysteines within 4-R Tau and consequent formation of a compact structure renders the Tau fibrillization-incompetent. In fact there was excellent correlation between the relative activity of ATPZ molecules in preventing K18PL Tau fibrillization and in their ability to induce the compacted Tau structure (Table 1).
ATPZs Promote the Formation of Intermolecular Disulfide Bonds in 3-R Tau
As 3-R Tau contains only a single cysteine residue due to the absence of the second MT binding repeat found in 4-R Tau, it is incapable of forming an intramolecular disulfide bond. However, it has been reported that 3-R Tau monomers can form an intermolecular disulfide, with the resulting dimer having an enhanced propensity to fibrillize (30, 37, 38). To investigate the consequences of ATPZ incubation with 3-R Tau, we utilized a previously described truncated Tau protein (K19) comprising the three MT binding repeats found in full-length 3-R Tau (30). As with the K18PL studies, the K19 Tau was incubated in the presence or absence of CNDR-51348 or CNDR-51449 or was pre-reduced or pre-oxidized. Subsequent SEC analyses revealed that the untreated K19 eluted primarily as a single peak, with a small amount of earlier eluting material (Fig. 3A). The peak eluting at 10.5 min upon SEC was determined to be Tau monomer, as this corresponds with the elution time of the pre-reduced K19. Conversely, pre-oxidized K19 eluted with an earlier retention time of 9.3 min, consistent with the formation of a dimer containing an intermolecular disulfide bond. The CNDR-51348-treated K19 had a similar retention time to that of pre-oxidized K19, whereas the K19 treated with the inactive ATPZ, CNDR-51449, had an elution profile resembling the vehicle-treated control sample (Fig. 3A). Thus, CNDR-51348 appeared to facilitate the formation of disulfide-linked 3-R Tau dimers. This was further confirmed by native gel electrophoresis using K19 or full-length 3-R Tau (T39). As revealed in Fig. 3B, the vehicle-treated K19 and T39 ran primarily as a monomer, with a small amount of dimer, and pre-reduction resulted in both K19 and T39 migrating as a monomer. Conversely, pre-oxidation of these Tau preparations led to the majority of the protein running as a dimer, and treatment with CNDR-51348 led to enhanced formation of both K19 and T39 dimers (Fig. 3B). Thus, these data reveal that the active ATPZ facilitated intermolecular disulfide formation in 3-R Tau.
FIGURE 3.

ATPZ-treated 3-R Tau undergoes intermolecular disulfide formation. A, the 3-R Tau K19 protein was incubated for 1 h at 37 °C in the absence (K19) or presence of CNDR-51348 (K19:51348) or CNDR-51449 (K19:51449) or was pre-reduced (K19 red) or pre-oxidized (K19 ox) before SEC analysis. B, K19 or full-length 3-R Tau (T39) were incubated for 1 h in the absence (−) or presence of CNDR-51348 (51348) or CNDR-51449 (51449) or were pre-reduced (R) or pre-oxidized (O) followed by native gel electrophoretic analysis. The bands corresponding to monomer (M) and dimer (D) are indicated.
3-R Tau dimers have been suggested to facilitate fibrillization (30), and we confirmed that K19 that has been dimerized by pre-oxidization or ATPZ treatment shows more rapid fibrillization than pre-reduced K19 (supplemental Fig. S2A). Thus, these data provide an explanation as to why the ATPZs, which inhibit 4-R Tau fibril formation, are ineffective inhibitors of 3-R Tau fibrillization. As the Tau inclusions in AD are comprised of a mixture of 3-R and 4-R Tau, a study was undertaken to determine the extent of inhibition of Tau fibrillization in an equimolar mixture of K18 (4-R) Tau and K19 (3-R) Tau that was allowed to incubate overnight under standard fibrillization conditions in the presence of CNDR-51348. As a control, the mixed 4-R/3-R Tau fibrillization reaction was conducted in the absence of ATPZ, and reactions were also performed with K18 or K19 alone in the absence or presence of CNDR-51348. As expected, the reactions of K18 or K19 conducted in the absence of the ATPZ resulted in full Tau fibrillization, with recovery of nearly all of the Tau in the pellet fraction after centrifugation, as previously described (27, 33), with very little soluble Tau (supplemental Fig. S2B). The majority of the K18 and K19 proteins from the mixed fibrillization reaction performed without ATPZ were also found in the pellet fraction after centrifugation, although there was a greater percentage of each Tau protein in the soluble fraction than was observed when the individual Tau proteins were incubated alone. This suggests that 3-R and 4-R Tau might slightly inhibit the fibrillization of each other. As expected, the addition of CNDR-51348 to the pure K18 fibrillization reaction resulted in a substantial increase in the amount of soluble Tau, whereas there was essentially no soluble K19 after treatment of the K19-only reaction with ATPZ. A similar specificity of fibrillization inhibition was observed in the reaction comprised of the mixture of K18 and K19 as only an increase of soluble K18, and not K19, was observed upon CNDR-51348 treatment (supplemental Fig. S2B).
To explore whether the ability of ATPZs to promote intermolecular disulfide formation is unique to 3-R Tau, we added CNDR-51348 or CNDR-51449 to K18PL proteins in which each of the two cysteine residues was singly mutated to alanine (K18PL-C291A or K18PL-C322A). Interestingly, pre-oxidation of each of these proteins resulted in the formation of dimers that eluted earlier than the untreated or reduced proteins (Fig. 4). Similarly, the mutant K18PL proteins treated with CNDR-51348 showed the presence of dimers, with the extent of ATPZ-mediated dimerization of K18PL-C322A being somewhat less than that observed with K18PL-C291A. As expected, the inactive ATPZ, CNDR-51449, did not promote formation of the earlier eluting dimer with either of the mutated proteins. Thus, it appears that active ATPZs can facilitate intermolecular cysteine disulfide formation in both K19 Tau and K18PL Tau mutants containing a single cysteine residue.
FIGURE 4.

4-R Tau proteins harboring single cysteine mutations form intermolecular disulfide bonds after ATPZ treatment. A, K18PL Tau protein containing the C291A mutation was incubated for 1 h at 37 °C in the absence (C291A) or presence of CNDR-51348 (C291A:51348) or CNDR-51449 (C291A:51449) or was pre-reduced (Reduced) or pre-oxidized (Oxidized) before SEC analysis. B, K18PL Tau protein containing the C322A mutation was incubated for 1 h at 37 °C in the absence (C322A) or presence of CNDR-51348 (C322A:51348) or CNDR-51449 (C322A:51449) or was pre-reduced (Reduced) or pre-oxidized (Oxidized) before SEC analysis.
ATPZs Oxidize Tau and Other Thiols via a Redox-cycling Mechanism
The ATPZ-facilitated cysteine oxidation of Tau was unexpected, as we previously demonstrated that ATPZs, which inhibited 4-R Tau fibrillization, were inactive in a caspase enzymatic assay that is sensitive to cysteine-oxidizing compounds (26). Moreover, an analysis of the relative target selectivity of the initial ATPZ leads identified from the NIH Chemical Genomics Center revealed very low promiscuity (26), which would be inconsistent with this series having a nonspecific oxidative mechanism-of-action. To further explore the selectivity of ATPZ-mediated cysteine oxidation, we incubated CNDR-51348 or the inactive analog, CNDR-51449, with a 10-mer peptide composed of a sequence unrelated to Tau but which contains two cysteine residues separated by 4 amino acids (39). Pre-oxidation of this peptide resulted in the formation of a species with a molecular mass consistent with an intramolecular disulfide, which can be readily separated from the reduced peptide by reversed-phase HPLC-MS (Fig. 5A; reduced peptide had an m/z = 1154 and eluted at 1.25 min, and oxidized peptide had an m/z = 1152 and eluted at 1.15 min). CNDR-51348 greatly enhanced the rate of formation of the intramolecular disulfide-containing form of the peptide which was not observed upon incubation with the inactive ATPZ, CNDR-51449 (Fig. 5A). To further investigate the specificity of sulfhydryl oxidation by APTZs, CNDR-51348 was incubated with DTT. CNDR-51348 addition led to the oxidation of DTT, such that the ATPZ-treated sample showed the same molecular mass and retention time upon HPLC-MS analysis as pre-oxidized DTT (Fig. 5B). Thus, these data reveal that the active ATPZs are not Tau-specific in their action and can promote the formation of disulfide bonds in several unrelated molecules.
FIGURE 5.

ATPZs facilitate disulfide formation in thiol-containing molecules other than Tau. A, a 10-mer peptide containing two cysteine residues was incubated for 0.5 h at 37 °C in the presence or absence of CNDR-51348 or CNDR-51449, and the mixtures were subsequently analyzed by reversed-phase HPLC-MS. In addition, a pre-oxidized sample of the 10-mer was subjected to HPLC-MS analysis. Peaks with molecular masses corresponding to a peptide with oxidized cysteines and reduced cysteines eluted at 1.15 and 1.25 min, respectively. B, DTT was incubated in the absence (DTT) or presence of CNDR-51348 (DTT:51348) or was pre-oxidized (DTTox) before HPLC-MS analysis. The peaks eluting at 2.91 and 4.44–4.48 min have molecular masses corresponding to the reduced and oxidized forms of DTT, respectively, whose structures are depicted.
ATPZ-mediated cysteine oxidation does not appear to coincide with a reduction of the ATPZ molecule, as MS analysis revealed that CNDR-51348 that was incubated with K18PL had an unchanged molecular mass relative to the parent compound (see Fig. 7A). This raised the possibility that the active ATPZs facilitated cysteine oxidation through a catalytic redox cycling mechanism that might involve the generation of reactive superoxide or hydrogen peroxide. Such redox cycling between heterocyclic compounds and thiols has been previously noted (40). To determine whether this was the case, the level of peroxide was compared after CNDR-51348 was incubated in the absence or presence of K18PL for 6 h at a concentration (20 μm) typically used in Tau fibrillization reactions. As demonstrated in Fig. 6B, the combination of the ATPZ and Tau resulted in the formation of a low (5–10 μm) but significant amount of peroxide. To investigate whether peroxides might be involved in the ATPZ-facilitated oxidation of Tau, we co-incubated reaction mixtures of CNDR-51348 and K18PL with catalase to remove peroxides and also examined whether the addition of hydrogen peroxide directly to K18PL would result in Tau cysteine oxidation. As demonstrated by native gel electrophoresis and IAA-OG analysis, 6 h of treatment of K18PL with 20 μm peroxide (PLo in Fig. 6C) did not lead to an appreciable conversion of K18PL to the oxidized compact monomer, although a much higher concentration of peroxide (1 mm; PHi in Fig. 6C) caused nearly complete oxidation of the Tau cysteines and formation of the compacted structure. This peroxide-mediated formation of disulfides was largely inhibited by co-incubation with catalase (Fig. 6C), but the addition of catalase did not prevent the oxidation of K18PL cysteines by CNDR-51348 (Fig. 6C) nor did it affect CNDR-51348 inhibition of Tau fibrillization (Fig. 6D). These results coupled with the observation that CNDR-51348 is not consumed during the oxidation of Tau leads us to hypothesize that hydrogen peroxide is a byproduct of a catalytic redox cycle reaction whereby ATPZs facilitate cysteine oxidation by molecular oxygen, as depicted in Fig. 6E.
FIGURE 7.

MB also facilitates formation of disulfide-stabilized 4-R Tau compact monomer as well as intermolecular disulfides in 3-R Tau and 4-R Tau with a mutated cysteine. A, K18PL was incubated for 1 h at 37 °C in the absence (K18PL) or presence of CNDR-51348 (K18PL:51348) or MB (K18PL:MB). The samples were subsequently analyzed by SEC. B, 3-R Tau K19 was incubated for 1 h in the absence (K19) or presence of CNDR-51348 (K19:51348) or MB (K19:MB) followed by SEC analyses. C, K18PL containing the C291A mutation was incubated for 1 h at 37 °C in the absence (C291A) or presence of CNDR-51348 (C291A:51348) or MB (C291A:MB).
FIGURE 6.

ATPZs facilitate molecular oxygen-mediated cysteine oxidation. A, HPLC-MS analysis of CNDR-51348 that was incubated for 1 h at 37 °C in the absence (51348) or presence of K18PL (51348:K18PL). The molecular mass of the compound was unchanged after oxidation of Tau. The two major m/z values (320 and 322) correspond to CNDR-51348 containing the naturally occurring 35Cl and 37Cl isotopes. B, CNDR-51348 or vehicle (water) were incubated for 1 h in the absence or presence of K18PL followed by the determination of peroxide concentrations in the incubation mixtures. C, K18PL was left untreated (−) or was pre-reduced (R). In addition, K18PL was treated with CNDR-51348 or with 20 μm (PLo) or 1 mm (PHi) hydrogen peroxide for 1 h at 37 °C in the presence or absence of catalase. The samples were subsequently analyzed by native gel electrophoresis or underwent reaction with IAA-OG followed by SDS-PAGE to evaluate the extent of cysteine oxidation. Both the IAA-OG fluorescence and corresponding Coomassie blue staining are shown for each IAA-OG-treated sample. D, fibrillization reactions were conducted with K18PL in the presence of CNDR-51348 (51348) or the presence of both CNDR-51348 and catalase (51348 + cat). E, shown is the proposed reaction scheme of ATPZ-mediated oxidation of Tau.
MB Also Inhibits 4-R Tau Fibrillization via Cysteine Oxidation
The discovery that ATPZs inhibit 4-R Tau fibrillization through the formation of a compact monomeric structure that is stabilized by an intramolecular disulfide bond raises the question of whether other reported Tau fibrillization inhibitors might act by a similar mechanism. Arguably, the most well known Tau fibrillization inhibitor is MB, which is undergoing evaluation in clinical trials as a therapy for AD (25). MB has been shown to inhibit 4-R Tau fibrillization (41–43) and is also known to be capable of redox cycling (44, 45). We examined whether MB might behave like the ATPZs and induce the formation of a compact monomer of K18PL. As shown in Fig. 7A, MB treatment resulted in a delayed retention time for K18PL upon SEC that is identical to that observed after incubation with CNDR-51348. In addition, a small earlier-eluting peak (8.9 min) was observed after incubation with MB that may represent a K18PL dimer. Similarly, incubation of MB with K19 led to the appearance of an earlier eluting peak that coincided with the K19 dimer peak seen after CNDR-51348 treatment (Fig. 7B). Finally, we found that MB treatment of the K18PL-C291A protein also resulted in the formation of a dimeric species (Fig. 7C), as was observed with CNDR-51348 (Figs. 4A and 7C). These results thus suggest that MB, like the ATPZs, oxidizes cysteine residues within the MT binding repeats of Tau peptides.
To further confirm this, native gel electrophoresis studies were conducted in which K18PL and full-length T40 were incubated in the absence or presence of MB. As observed with active ATPZs, MB caused the formation of a K18PL compact monomer on native gels that migrated with the same electrophoretic mobility as the pre-oxidized protein (Fig. 8A). Moreover, MB treatment led to a time-dependent shift in the electrophoretic mobility of T40 to a faster migrating species as was also seen upon treatment of T40 with CNDR-51348 (compare Fig. 8A and Fig. 2B). Finally, treatment of K19 3-R Tau with MB led to a decrease in the amount of monomer and increased formation of the intermolecular disulfide-containing dimer (Fig. 9A). These data establish that MB treatment results in the oxidation of cysteine residues in the Tau peptides. To examine whether this effect is specific to Tau, the 10-mer peptide containing two cysteine residues was incubated with MB, and this resulted in the formation of a disulfide-stabilized conformer that eluted upon HPLC analysis with the same retention time (1.09 min) as pre-oxidized peptide (Fig. 8B). Note that additional peaks were observed on reversed-phase HPLC separation of the MB-treated 10-mer peptide, which corresponded to reduced MB (1.30 min) and demethylated MB (azure B; shoulder on 1.09 min peak). The oxidation of the 10-mer peptide reveals that MB acts as a nonspecific oxidizer of cysteine sulfhydryls.
FIGURE 8.

MB induces disulfide formation in molecules other than Tau and does not inhibit 3-R Tau fibrillization. A, K18PL or K19 were incubated for 1 h at 37 °C in the absence (−) of presence of MB (MB) or were pre-reduced (Red) or pre-oxidized (Ox). The samples were then analyzed by native gel electrophoresis. In addition, T40 was incubated in the absence (−) or presence of MB (+MB) for 15 and 30 min followed by analysis by native gel electrophoresis. B, a 10-mer peptide containing two cysteine residues was incubated for 1 h at 37 °C in the absence (10-mer) or presence of MB (10-mer:MB) or was pre-oxidized (10-mer:Ox), and the mixtures were subsequently analyzed by reversed-phase HPLC-MS. Peaks with masses corresponding to a peptide with oxidized cysteines (m/z = 1152) and reduced cysteines (m/z = 1154) eluted at 1.08–1.09 and 1.20–1.21 min, respectively. The peak eluting at 1.30 min in the MB-treated sample is reduced methylene blue, and the shoulder on the peak eluting at 1.09 min is azure B, which is a demethylated form of MB that is found as a minor contaminant in MB preparations. C, K19 and K18 fibrillization reactions were incubated with increasing concentrations (in μm) of MB or oleocanthal (Oleo). After completion of the reactions, the samples were subjected to centrifugation to separate fibrillar Tau (pellet fraction (P)) from non-fibrillar Tau (supernatant fraction (S)). Both the pellet and supernatant fractions were analyzed by SDS-PAGE and Coomassie Blue staining. D, K18PL was left untreated (−) or was pre-reduced (R) or pre-oxidized (O). In addition, K18PL was treated with MB or with 20 μm (PLo) or 1 mm (PHi) hydrogen peroxide both in the presence and absence of catalase. The samples subsequently underwent reaction with IAA-OG followed by SDS-PAGE to evaluate the extent of cysteine oxidation. Both the IAA-OG fluorescence and corresponding Coomassie blue staining are shown for each IAA-OG-treated sample. In A and D, the vertical lines designate sites where non-pertinent gel lanes were removed and figures were subsequently spliced.
FIGURE 9.

Tau cysteine oxidation by ATPZ or MB is inhibited by cellular concentrations of GSH. K18 (A) or K19 (B) were incubated for 1 h at 37 °C in the absence (−) or presence of CNDR-51348 (51348), CNDR-51449 (51449) or MB or was pre-reduced (R) or pre-oxidized (O). These reactions were conducted either in the absence (−GSH) or presence (+GSH) of 5 mm GSH. The samples were then analyzed by native gel electrophoresis.
Although our data indicate that MB can oxidize sulfhydryl groups, we have also observed that Tau that has been incubated for prolonged periods with MB shows multiple higher molecular weight species upon electrophoretic analysis. To determine whether the reported ability of MB to inhibit Tau fibrillization depends primarily on its ability to oxidize cysteine residues and/or perhaps an alternative mechanism such as covalent cross-linking, we examined the effect of MB on 3-R Tau (K19) fibrillization. Because MB can promote the formation of a K19 dimer that is stabilized by an intermolecular cysteine bond and as K19 dimerization promotes fibrillization, one would expect little or no inhibition of K19 fibrillization if the primary mechanism of MB action on Tau is cysteine oxidation. Conversely, if MB inhibits Tau fibrillization by an alternative mechanism, then the compound may still affect 3-R Tau fibrillization. As MB interferes with ThT fluorescence, we monitored the effect of MB on both K19 and K18 fibrillization using the previously described sedimentation assay (see supplemental Fig. S2) that separates Tau fibrils from soluble Tau (26, 28). Parallel fibrillization reactions were conducted in the presence of oleocanthal, which has been demonstrated previously to inhibit Tau fibril formation via covalent modification of lysines within Tau (29). As demonstrated in Fig. 8C, oleocanthal caused inhibition of both K19 and K18 Tau fibrillization, as evidenced by decreasing amounts of Tau in the insoluble pellet fraction. Note that oleocanthal can cause Tau cross-linking, resulting in the formation of soluble multimers that migrate more slowly on SDS-PAGE than monomeric Tau (29). This leads to a reduction in the amount of monomer band observed in the soluble fraction at higher oleocanthal concentrations, as observed in Fig. 8C. In contrast, whereas MB showed a concentration-dependent inhibition of 4-R Tau fibrillization as evidenced by an increasing percentage of soluble K18, no effect on K19 fibrillization was observed. This outcome is consistent with cysteine oxidation being the primary mechanism by which MB affects both 3-R and 4-R Tau.
As with the ATPZs, the oxidation of Tau by MB does not appear to depend on peroxides. This is demonstrated in Fig. 8D, where IAA-OG labeling reveals that the addition of catalase in an amount that reversed K18PL cysteine oxidation by 1 mm peroxide had no effect on MB-mediated oxidation of cysteines in K18PL. Thus, these data reveal substantial similarities in the mechanism of action of MB and the ATPZ class of Tau fibrillization inhibitors.
Glutathione (GSH) Prevents Tau Oxidation by ATPZs and MB
The generation of a disulfide-stabilized compact 4-R monomer by ATPZs and MB provides a mechanistic understanding of how these compounds act to inhibit Tau fibrillization in vitro. However, it is possible that Tau oxidation by these compounds may be attenuated by the reducing environment inside of cells, wherein GSH is found at low mm concentrations (46). To investigate this possibility, both K18 and K19 Tau proteins were treated with APTZs and MB in the presence or absence of 5 mm GSH. As previously demonstrated, in the absence of GSH both CNDR-51348 and MB facilitated the production of a K18 compact monomer and a K19 dimer, whereas CNDR-51349 was inactive (Fig. 9, A and B). However, these disulfide-stabilized Tau species were not observed after CNDR-51348 and MB treatment in the presence of GSH (Fig. 9, A and B). These data thus suggest that the cysteine-oxidizing activity of these compounds would largely be inhibited in the cytoplasm of cells, where Tau aggregation would occur. This conclusion is supported by our observation that CNDR-51348 and MB no longer effectively inhibits 4-R Tau fibrillization in the presence of 5 mm GSH (supplemental Fig. S3).
DISCUSSION
The ATPZ series of Tau fibrillization inhibitors was first identified from a large quantitative high-throughput screening campaign in which nearly 300,000 compounds were interrogated at multiple doses (26). This screen was conducted with a heparin-facilitated 4-R Tau fibrillization assay that did not contain a reducing agent such as DTT, as prior experience had revealed that many false positives are obtained with compounds that are capable of redox cycling in the presence of a reducing agent (33, 40). Among the secondary analyses conducted on the APTZs identified from this screen was a caspase-8 enzyme assay that is sensitive to oxidizing agents (26, 47). The ATPZs identified from the screen were inactive in the caspase assay and showed a low level of target promiscuity as evidenced by a low hit rate in the many high-throughput screens conducted at the NIH Chemical Genomics Center (26). These features as well as the absence of an inhibitory effect on Aβ peptide fibrillization or Tau-mediated microtubule assembly (26) led to the belief that these molecules were relatively specific inhibitors of 4-R Tau fibril assembly.
As very little is known about how Tau fibrillization inhibitors act in vitro, we undertook studies to gain a better understanding of ATPZ-mediated inhibition of Tau fibril formation. The finding that active ATPZs promote the oxidation of Tau cysteine residues was unexpected given the aforementioned assay results with these compounds. The observation that active ATPZs can promote the generation of dimers of 3-R, which contain an intermolecular cysteine bond, or dimers of 4-R Tau in which one cysteine residue has been mutated to an alanine, suggests that both forms of Tau first associate to form non-covalent dimers, with the active ATPZ molecules then catalyzing the formation of the intermolecular disulfide bond.
The cysteine-oxidizing mechanism of ATPZ action is shared by MB, including the ability to oxidize cysteines within 4-R and 3-R Tau as well as a 10-mer peptide with a sequence that is unrelated to Tau. These observations have potential ramifications with regard to the utility of these molecules as candidates for the treatment of AD and related tauopathies. First, although the active ATPZs and MB inhibit 4-R Tau fibrillization, they do not inhibit 3-R Tau fibril assembly. Compounds of this type would presumably be ineffectual in the treatment of Pick's disease, which is primarily a 3-R tauopathy (4, 48). Moreover, Tau inclusions found in the brains of patients with AD, the most prevalent tauopathy, are comprised of both 3-R Tau and 4-R Tau. Although there is uncertainty as to whether these AD Tau aggregates are composed of heteropolymeric and/or homopolymeric 3-R and 4-R fibrils, it is possible that molecules that only inhibit 4-R Tau fibrillization would allow for the continued deposition of 3-R Tau fibrils in AD.
A second important point related to the therapeutic potential of MB or the ATPZs concerns their apparent nonspecific cysteine oxidation, which might affect multiple proteins upon systemic administration and lead to the appearance of dose-limiting side effects. However, there is a long history of MB utilization in humans, and MB is still administered for several conditions, including methemoglobinemia and urinary tract infections (45, 49). Thus, MB appears to be fairly well tolerated. Although the ATPZs have not undergone the same degree of in vivo characterization as MB, an orally absorbed and brain-penetrant example has been administered at 50 mg/kg/day to mice for 1 month without any notable adverse effects (27). The apparent safety of these molecules is somewhat surprising given their potential for nonspecific oxidations, and it is thus possible that cysteine oxidization is tolerated in animals or, alternatively, that the extent of oxidation observed in vitro is not recapitulated in vivo. In this regard, our data demonstrating that Tau cysteine oxidation by the ATPZs and MB is significantly blunted in the presence of cellular concentrations of GSH suggests that this latter hypothesis may be correct. If so, then the ATPZs and MB would be predicted to be relatively ineffective inhibitors of Tau fibrillization in vivo, although it is possible that a pleiotropic drug like MB (45, 49) may inhibit Tau inclusion formation by mechanisms that do not depend on the inhibition of Tau fibrillization. For example, there are studies which suggest that MB enhances proteasomeal (50, 51) and autophagic degradation (52) of Tau.
In conclusion, our studies provide new mechanistic insights into how the structurally unrelated compounds, ATPZs and MB, affect Tau fibril assembly in vitro. Moreover, these studies suggest that compounds that affect Tau structure by a similar cysteine-oxidizing mechanism may have limitations that will negatively affect their in vivo efficacy and/or safety. Finally, our experience with the discovery and subsequent characterization of the ATPZ class of molecules provides lessons to be applied to future searches for drug-like Tau fibrillization inhibitors, including careful consideration of the redox properties of candidate compounds.
Supplementary Material
Acknowledgment
We thank Julia Durante for technical assistance in these studies.
This work was supported, in whole or in part, by National Institutes of Health Grant AG17586 (NIA; V. M.-Y. L.). This work was also supported by grants from the Alzheimer Disease Research program of the American Health Assistance Foundation (to K. R. B.) and the Coins for Alzheimer's Research Trust (to K. R. B.), a pilot grant from the Comprehensive Neuroscience Center at the University of Pennsylvania (to C. B.), and by the Marian S. Ware Alzheimer's Program.

This article contains supplemental Figs. S1—S3.
- AD
- Alzheimer disease
- ATPZ
- aminothienopyridazine
- FTDP-17
- frontotemporal dementia with parkinsonism linked to chromosome 17
- IAA-OG
- iodoacetamide-Oregon Green
- MB
- methylene blue
- MT
- microtubule
- SEC
- size-exclusion chromatography
- ThT
- thioflavin T
- 3-R
- three microtubule-binding repeats
- 4-R
- four microtubule-binding repeats
- AU
- absorbance units.
REFERENCES
- 1. Kidd M. (1963) Paired helical filaments in electron microscopy of Alzheimers disease. Nature 197, 192–193 [DOI] [PubMed] [Google Scholar]
- 2. Lee V. M., Balin B. J., Otvos L., Jr., Trojanowski J. Q. (1991) A68, a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678 [DOI] [PubMed] [Google Scholar]
- 3. Ballatore C., Lee V. M., Trojanowski J. Q. (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663–672 [DOI] [PubMed] [Google Scholar]
- 4. Lee V. M., Goedert M., Trojanowski J. Q. (2001) Neurodegenerative tauopathies. Annu. Rev. Neurosci. 24, 1121–1159 [DOI] [PubMed] [Google Scholar]
- 5. Hutton M., Lendon C. L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A., Hackett J., Adamson J., Lincoln S., Dickson D., Davies P., Petersen R. C., Stevens M., de Graaff E., Wauters E., van Baren J., Hillebrand M., Joosse M., Kwon J. M., Nowotny P., Che L. K., Norton J., Morris J. C., Reed L. A., Trojanowski J., Basun H., Lannfelt L., Neystat M., Fahn S., Dark F., Tannenberg T., Dodd P. R., Hayward N., Kwok J. B., Schofield P. R., Andreadis A., Snowden J., Craufurd D., Neary D., Owen F., Oostra B. A., Hardy J., Goate A., van Swieten J., Mann D., Lynch T., Heutink P. (1998) Association of missense and 5′-splice-site mutations in Tau with the inherited dementia FTDP-17. Nature 393, 702–705 [DOI] [PubMed] [Google Scholar]
- 6. Hong M., Zhukareva V., Vogelsberg-Ragaglia V., Wszolek Z., Reed L., Miller B. I., Geschwind D. H., Bird T. D., McKeel D., Goate A., Morris J. C., Wilhelmsen K. C., Schellenberg G. D., Trojanowski J. Q., Lee V. M. (1998) Mutation-specific functional impairments in distinct Tau isoforms of hereditary FTDP-17. Science 282, 1914–1917 [DOI] [PubMed] [Google Scholar]
- 7. Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977) Physical and chemical properties of purified Tau factor and role of Tau in microtubule assembly. J. Mol. Biol. 116, 227–247 [DOI] [PubMed] [Google Scholar]
- 8. Cleveland D. W., Hwo S. Y., Kirschner M. W. (1977) Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225 [DOI] [PubMed] [Google Scholar]
- 9. Wilcock G. K., Esiri M. M. (1982) Plaques, tangles, and dementia. A quantitative study. J. Neurol. Sci. 56, 343–356 [DOI] [PubMed] [Google Scholar]
- 10. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimers disease. Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 11. Braak H., Braak E. (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 [DOI] [PubMed] [Google Scholar]
- 12. Brunden K. R., Trojanowski J. Q., Lee V. M. (2008) Evidence that non-fibrillar Tau causes pathology linked to neurodegeneration and behavioral impairments. J. Alzheimers Dis. 14, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee V. M., Daughenbaugh R., Trojanowski J. Q. (1994) Microtubule stabilizing drugs for the treatment of Alzheimers disease. Neurobiol. Aging 15, S87–S89 [DOI] [PubMed] [Google Scholar]
- 14. Avila J. (2006) Tau phosphorylation and aggregation in Alzheimer's disease pathology. FEBS Lett. 580, 2922–2927 [DOI] [PubMed] [Google Scholar]
- 15. Brunden K. R., Ballatore C., Crowe A., Smith A. B., 3rd, Lee V. M., Trojanowski J. Q. (2010) Tau-directed drug discovery for Alzheimer's disease and related tauopathies. A focus on Tau assembly inhibitors. Exp. Neurol. 223, 304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brunden K. R., Trojanowski J. Q., Lee V. M. (2009) Advances in tau-focused drug discovery for Alzheimer's disease and related tauopathies. Nat. Rev. Drug Discov. 8, 783–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Margittai M., Langen R. (2004) Template-assisted filament growth by parallel stacking of Tau. Proc. Natl. Acad. Sci. U.S.A. 101, 10278–10283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Congdon E. E., Kim S., Bonchak J., Songrug T., Matzavinos A., Kuret J. (2008) Nucleation-dependent Tau filament formation. The importance of dimerization and an estimation of elementary rate constants. J. Biol. Chem. 283, 13806–13816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chirita C. N., Necula M., Kuret J. (2003) Anionic micelles and vesicles induce Tau fibrillization in vitro. J. Biol. Chem. 278, 25644–25650 [DOI] [PubMed] [Google Scholar]
- 20. Wilson D. M., Binder L. I. (1997) Free fatty acids stimulate the polymerization of Tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer's disease. Am. J. Pathol. 150, 2181–2195 [PMC free article] [PubMed] [Google Scholar]
- 21. Goedert M., Jakes R., Spillantini M. G., Hasegawa M., Smith M. J., Crowther R. A. (1996) Assembly of microtubule-associated protein Tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553 [DOI] [PubMed] [Google Scholar]
- 22. Wille H., Drewes G., Biernat J., Mandelkow E. M., Mandelkow E. (1992) Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein Tau in vitro. J. Cell Biol. 118, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ballatore C., Brunden K. R., Trojanowski J. Q., Lee V. M., Smith A. B., 3rd, Huryn D. (2011) Modulation of protein-protein interactions as a therpeutic strategy for the treatment of neurodegenerative tauopathies. Curr. Top. Med. Chem. 11, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bulic B., Pickhardt M., Mandelkow E. M., Mandelkow E. (2010) Tau protein and Tau aggregation inhibitors. Neuropharmacology 59, 276–289 [DOI] [PubMed] [Google Scholar]
- 25. Staff R. T., Ahearn T. S., Murray A. D., Benthan P., Seng K. M., Wischik C. (2008) Tau aggregation inhibitor (TAI) therapy with rember arrests the trajectory of rCBF decline in brain regions affected by Tau pathology in mild to moderate Alzheimer's disease. Alzheimer Dement. 4, T775 [Google Scholar]
- 26. Crowe A., Huang W., Ballatore C., Johnson R. L., Hogan A. M., Huang R., Wichterman J., McCoy J., Huryn D., Auld D. S., Smith A. B., 3rd, Inglese J., Trojanowski J. Q., Austin C. P., Brunden K. R., Lee V. M. (2009) The identification of aminothienopyridazine inhibitors of Tau assembly by quantitative high-throughput screening. Biochemistry 48, 7732–7745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ballatore C., Crowe A., Piscitelli F., James M., Lou K., Rossidivito G., Yao Y., Trojanowski J. Q., Lee V. M., Brunden K. R., Smith A. B., 3rd (2012) Aminothienopyridazine inhibitors of Tau aggregation. Evaluation of structure-activity relationship leads to selection of candidates with desirable in vivo properties. Bioorg. Med. Chem. 20, 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ballatore C., Brunden K. R., Piscitelli F., James M. J., Crowe A., Yao Y., Hyde E., Trojanowski J. Q., Lee V. M., Smith A. B., 3rd (2010) Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of Tau aggregation. J. Med. Chem. 53, 3739–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li W., Sperry J. B., Crowe A., Trojanowski J. Q., Smith A. B., 3rd, Lee V. M. (2009) Inhibition of Tau fibrillization by oleocanthal via reaction with the amino groups of Tau. J Neurochem. 110, 1339–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barghorn S., Mandelkow E. (2002) Toward a unified scheme for the aggregation of Tau into Alzheimer paired helical filaments. Biochemistry 41, 14885–14896 [DOI] [PubMed] [Google Scholar]
- 31. Barghorn S., Zheng-Fischhöfer Q., Ackmann M., Biernat J., von Bergen M., Mandelkow E. M., Mandelkow E. (2000) Structure, microtubule interactions, and paired helical filament aggregation by Tau mutants of frontotemporal dementias. Biochemistry 39, 11714–11721 [DOI] [PubMed] [Google Scholar]
- 32. Tam J. P., Wu C. R., Liu W., Zhang J. W. (1991) Disulfide bond formation in peptides by dimethyl sulfoxide. Scope and applications. J. Am. Chem. Soc. 113, 6657–6662 [Google Scholar]
- 33. Crowe A., Ballatore C., Hyde E., Trojanowski J. Q., Lee V. M. (2007) High throughput screening for small molecule inhibitors of heparin-induced Tau fibril formation. Biochem. Biophys. Res. Commun. 358, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 35. McLellan T. (1982) Electrophoresis buffers for polyacrylamide gels at various pH. Anal. Biochem. 126, 94–99 [DOI] [PubMed] [Google Scholar]
- 36. Gorman J. J. (1987) Fluorescent labeling of cysteinyl residues to facilitate electrophoretic isolation of proteins suitable for amino-terminal sequence analysis. Anal. Biochem. 160, 376–387 [DOI] [PubMed] [Google Scholar]
- 37. Walker S., Ullman O., Stultz C. M. (2012) Using intramolecular disulfide bonds in Tau protein to deduce structural features of aggregation-resistant conformations. J. Biol. Chem. 287, 9591–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schweers O., Mandelkow E. M., Biernat J., Mandelkow E. (1995) Oxidation of cysteine 322 in the repeat domain of microtubule-associated protein Tau controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. U.S.A. 92, 8463–8467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruddock L. W., Hirst T. R., Freedman R. B. (1996) pH dependence of the dithiol-oxidizing activity of DsbA (a periplasmic protein thiol:disulphide oxidoreductase) and protein disulphide-isomerase. Studies with a novel simple peptide substrate. Biochem. J. 315, 1001–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soares K. M., Blackmon N., Shun T. Y., Shinde S. N., Takyi H. K., Wipf P., Lazo J. S., Johnston P. A. (2010) Profiling the NIH small molecule repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev. Technol. 8, 152–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T., Goedert M., Hasegawa M. (2005) Inhibition of heparin-induced Tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614–7623 [DOI] [PubMed] [Google Scholar]
- 42. Chang E., Congdon E. E., Honson N. S., Duff K. E., Kuret J. (2009) Structure-activity relationship of cyanine Tau aggregation inhibitors. J. Med. Chem. 52, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wischik C. M., Edwards P. C., Lai R. Y., Roth M., Harrington C. R. (1996) Selective inhibition of Alzheimer disease-like Tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. U.S.A. 93, 11213–11218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oz M., Lorke D. E., Hasan M., Petroianu G. A. (2011) Cellular and molecular actions of methylene blue in the nervous system. Med. Res. Rev. 31, 93–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wainwright M., Crossley K. B. (2002) Methylene Blue. A therapeutic dye for all seasons? J. Chemother. 14, 431–443 [DOI] [PubMed] [Google Scholar]
- 46. Zhang H., Forman H. J. (2012) Glutathione synthesis and its role in redox signaling. Semin. Cell Dev. Biol. 23, 722–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith G. K., Barrett D. G., Blackburn K., Cory M., Dallas W. S., Davis R., Hassler D., McConnell R., Moyer M., Weaver K. (2002) Expression, preparation, and high-throughput screening of caspase-8. Discovery of redox-based and steroid diacid inhibition. Arch. Biochem. Biophys. 399, 195–205 [DOI] [PubMed] [Google Scholar]
- 48. Hasegawa M. (2006) Biochemistry and molecular biology of tauopathies. Neuropathology 26, 484–490 [DOI] [PubMed] [Google Scholar]
- 49. Schirmer R. H., Adler H., Pickhardt M., Mandelkow E. (2011) “Lest we forget you “methylene blue. ” Neurobiol. Aging 32, 2325. [DOI] [PubMed] [Google Scholar]
- 50. Jinwal U. K., Miyata Y., Koren J., 3rd, Jones J. R., Trotter J. H., Chang L., O'Leary J., Morgan D., Lee D. C., Shults C. L., Rousaki A., Weeber E. J., Zuiderweg E. R., Gestwicki J. E., Dickey C. A. (2009) Chemical manipulation of Hsp70 ATPase activity regulates Tau stability. J. Neurosci. 29, 12079–12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Leary J. C., 3rd, Li Q., Marinec P., Blair L. J., Congdon E. E., Johnson A. G., Jinwal U. K., Koren J., 3rd, Jones J. R., Kraft C., Peters M., Abisambra J. F., Duff K. E., Weeber E. J., Gestwicki J. E., Dickey C. A. (2010) Phenothiazine-mediated rescue of cognition in Tau transgenic mice requires neuroprotection and reduced soluble Tau burden. Mol. Neurodegener. 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Congdon E. E., Wu J. W., Myeku N., Figueroa Y. H., Herman M., Marinec P. S., Gestwicki J. E., Dickey C. A., Yu W. H., Duff K. E. (2012) Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 8, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.