

Background: Activated RhoA binds to the PH domain of its own GEF, PDZRhoGEF.
Results: Activated RhoA binds to all Lbc RhoGEFs and provides positive feedback regulation to these regulators.
Conclusion: Allosteric binding of activated RhoA to its own regulators facilitates other regulatory stimuli.
Significance: This autoregulatory mechanism could mediate robust localized signaling and may represent a general paradigm for regulation of other monomeric GTPases.
Keywords: Phospholipid Vesicle, Protein Translocation, RhoA, Signal Transduction, Small GTPases, PH Domain, RhoGEF, Feedback Regulation, Localization, p115RhoGEF
Abstract
The monomeric Rho GTPases are essential for cellular regulation including cell architecture and movement. A direct mechanism for hormonal regulation of the RhoA-type GTPases is their modulation by the G12 and G13 proteins via RH (RGS homology) containing RhoGEFs. In addition to the interaction of the G protein α subunits with the RH domain, activated RhoA also binds to the pleckstrin homology (PH) domain of PDZRhoGEF. The latter interaction is now extended to all seven members of the homologous Lbc family of RhoGEFs which includes the RH-RhoGEFs. This is evinced by direct measurements of binding or through effects on selected signaling pathways in cells. Overexpression of these PH domains alone can block RhoA-dependent signaling in cells to various extents. Whereas activated RhoA does not modulate the intrinsic activity of the RhoGEFs, activated RhoA associated with phospholipid vesicles can facilitate increased activity of soluble RhoGEFs on vesicle-delimited substrate (RhoA-GDP). This demonstrates feasibility of the hypothesis that binding of activated RhoA to the PH domains acts as a positive feedback mechanism. This is supported by cellular studies in which mutation of this binding site on PH strongly attenuates the stimulation of RhoA observed by overexpression of five of the RhoGEF DH-PH domains. This mutation is even more dramatic in the context of full-length p115RhoGEF. The utilization of this mechanism by multiple RhoGEFs suggests that this regulatory paradigm may be a common feature in the broader family of RhoGEFs.
Introduction
The Rho family of monomeric GTPases mediates regulation of a wide variety of cellular functions including the dynamics of the actin cytoskeleton, cell polarity, cell proliferation, and gene transcription programs. Disruptions in the regulation of these proteins can cause or contribute to several diseases (1–3). Signaling from a wide variety of extracellular and intracellular stimuli that regulate this subfamily of GTPases is facilitated and integrated by two families of proteins that either activate or deactivate the proteins. The Rho guanine nucleotide exchange factors (RhoGEFs)2 stimulate Rho proteins by facilitating exchange of GDP for GTP (4). RhoGAPs (Rho GTPase-activating proteins) inactivate Rho proteins by accelerating hydrolysis of bound GTP, thus returning Rho proteins to their inactive state with bound GDP (5).
The classic RhoGEFs constitute a family of approximately 70 proteins that are characterized by the presence of tandem DH (Dbl homology) and PH (pleckstrin homology) domains (4). The DH domain provides the active site for exchange of nucleotide on Rho proteins. The tandem PH domain subserves several functions in the various GEFs to include assisting in binding of the substrate for catalysis, stability of the DH domain, and localization of these regulatory factors. In addition, most of the RhoGEFs contain other domains or binding motifs for interaction with a wide variety of regulatory or structural molecules.
The RH-RhoGEFs are a subfamily of these exchange factors that act specifically on RhoA type GTPases and contain RH (RGS homology, regulator of G protein signaling) domains that interact exclusively with the activated α subunits of the heterotrimeric G12 and G13 proteins (6, 7). Whereas the RH domain of p115RhoGEF was first characterized as a GAP for the α subunits, it is also necessary for the RhoGEF to mediate direct regulation of RhoA by G protein-coupled receptors that activate G12 and G13 (8, 9). Recent studies indicate that the main function of the RH domain in activation is to bind and orient the GTPase so that the α-helical domain of Gα13 can interact effectively, albeit with low affinity, to the back side of the DH domain (10). In a mechanism not yet defined, this interaction stimulates the turnover rate of p115RhoGEF for activation of RhoA.
PRG (PDZRhoGEF) differs functionally from p115RhoGEF and LARG (leukemia-associated RhoGEF) in that its RH domain binds tightly to activated Gα13 but does not enhance the GTPase activity of the α subunit (11). This is due to retention of the anchoring sites for bivalent binding of the domain to Gα13 but a lack of key residues in the N-terminal acidic region that facilitate GAP activity (12). Furthermore, activated Gα13 does not stimulate the exchange activity of PRG (11). This suggests that regulation of PRG in cells may occur through regulated localization of the RhoGEF to the plasma membrane where it could functionally interact with its substrate, free RhoA. Hormones that regulate the G12 and G13 proteins would utilize binding of the RH domain to activated α subunits for localization of PRG.
A second form of regulation for PRG is indicated by the association of activated RhoA with its PH domain (13). This occurs at a hydrophobic surface of the PH domain that is distal to the substrate binding site. Formation of a trimeric complex between the PRG DH-PH domains with both its substrate, inactive RhoA, and activated RhoA, suggests that this interaction mediates feedback regulation. Because activated RhoA does not alter the catalytic activity of PRG, we hypothesized that binding of activated RhoA serves as a positive feedback mechanism to help maintain the RhoGEF at the plasma membrane where it could activate additional substrate (13). This idea is also supported by an observation that mutations in a homologous hydrophobic region of LARG reduced stimulation of RhoA-dependent pathways upon overexpression of the RhoGEF (14).
We now show that activated RhoA binds to the PH domains of all seven members of the Lbc-RhoGEF family, which includes the three RH-RhoGEFs and four RhoGEFs with high homology and similar specificity for activating RhoA (6). Mutations in the homologous hydrophobic regions of their PH domains attenuate this interaction. Expression of the PH domains by themselves inhibits a RhoA-dependent pathway stimulated by hormone. With five of the seven proteins, the constitutive activation of RhoA pathways by overexpression of the RhoGEFs is highly reduced by mutation of the binding site for activated RhoA. Finally, a phospholipid vesicle system is used to demonstrate that interaction of activated RhoA with the PH domain of PRG can support processive activation of membrane-delimited RhoA.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Coding regions for tandem DH-PH domains or PH domains of the indicated RhoGEFs (Fig. 1) and full-length RhoA were inserted into pGEX-KG-TEV either with or without a C-terminal His6 tag as described previously (10). Mutations were inserted by PCR sewing. Proteins were expressed in Escherichia coli strain BL21(DE3) and purified as described (13, 15). Briefly, expressed domains were purified by affinity and anion exchange chromatography either as the GST fusion protein or cleaved from GST with the tobacco etch virus (TEV) protease. RhoA with a C-terminal His6 tag was prepared by cleavage with TEV protease from the immobilized GST fusion protein and further purified by size-exclusion chromatography using Superdex 200/75 tandem gel filtration columns (Amersham Biosciences). YFP-RhoA utilized mCitrine (16) fused to the N terminus of full-length RhoA with a Gly-Ser linker. This was expressed as a GST fusion protein. Preparations of RhoA were activated with GTPγS as described previously (13).
FIGURE 1.

Lbc RhoGEFs and domains. The table lists the Lbc RhoGEFs; the domain constructs used in this work are indicated by their N- and C-terminal amino acids. Mutations in the PH domains that were used to disrupt binding to activated RhoA are indicated. The numbering of residues is based on reported sequences in GenBank (GB Accession numbers); all sequences are human except p114 (mouse). The lower panel compares the Lbc RhoGEFs. Numbers on the right indicate the total amino acids in each protein. The dendogram on the left shows the comparative homology among their DH-PH domains. Selected domains shown are: RH, DH, PH, PDZ, C (C1 homology domain), and PKA (binding site for protein kinase A).
Pulldown Assays
Immobilized GST-tagged RhoA was used to compare the relative ability of purified His6-tagged PH domains to bind activated RhoA. GST-RhoA (80 pmol), either basal (GDP) or preactivated with GTPγS, was mixed with 10 μl of glutathione-Sepharose 4B resin (GE Healthcare) in 100 μl of Buffer A (50 mm NaHEPES, pH 7.5, 50 mm NaCl, 1 mm DTT, 1 mm EDTA, 5 mm MgCl2, 0.3% (v/v) Triton X-100, and 0.01% (w/v) BSA) and incubated for 30 min at 4 °C. The resin was washed with Buffer A, and His6-tagged PH domains (40 pmol) were added to the immobilized GST-RhoA in Buffer A (100 μl) containing either 10 μm GDP or 10 μm GTPγS for basal or preactivated RhoA, respectively. The mixtures were incubated on a rotating platform for 15 min at room temperature and resin rapidly separated by spinning in a microcentrifuge. Supernatants containing free PH domains were removed and the resins rapidly washed four times with 600 μl of cold Buffer A. PH domains bound to the resins were released by incubating in 30 μl of 15 mm glutathione buffer for 5 min and visualized by SDS-PAGE and immunoblot analysis using an anti-His6 monoclonal antibody (R&D Systems).
Protein Blots
UltraBindTM US450 membranes (Pall Life Sciences) were pre-wet in Buffer A for 5 min. Purified GST-PH domains (20 μg) were manually spotted onto the membranes using a suction filter. Following application, the membranes were incubated with blocking buffer (phosphate-buffered saline containing 0.5% casein) for 30 min at room temperature. After washing three times with Buffer B (25 mm NaHEPES, pH 8.0, 100 mm NaCl, 5 mm MgCl2, and 1 mm DTT), membranes were incubated for 15 min at room temperature with either basal or activated YFP-RhoA (1 μm) in Buffer A containing 10 μm GDP or GTPγS, respectively. Membranes were then washed three times for 5 min with ice-cold Buffer B containing 0.1% Tween 20. YFP-RhoA bound to the membrane was visualized by scanning for fluorescence of the YFP with a Typhoon imager, model 9450. ImageQuant 5.2 software was used to quantify the fluorescence in individual spots.
Cell Culture
HeLa Tet-On cells (Clontech) were maintained in high glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Benchmark) and 100 μg/ml G418 (Invitrogen) in a 37 °C humidified 5% CO2 atmosphere.
Activity of Proteins and Domains in Mammalian Cells
Proteins and tandem DH-PH domains were expressed as myc-tagged proteins encoded in pcDNA3.1-myc. In this vector, the multicloning site of pcDNA3.1+ (Invitrogen) was replaced with a start site, sequence for expression of a myc tag, and cloning sites that matched those in pGEX-KG-TEV for direct insertion of domains and mutated domains in both vectors. PH domains for blocking experiments were encoded after the myc sequence in pCMV5-myc (17). Included amino acids differed from Fig. 1 as follows: p115 (Met609–Ala766); LARG (Ala981–Gln1137); Lbc (Tyr2195–Glu2339); p114 (Tyr302–Glu447); p190 (Glu1049–Lys1194); GEFH1 (Leu436–Pro574).
Activation of RhoA was assessed with the SRE.L luciferase reporter as described previously (17). Thus, the Renilla luciferase thymidine kinase (pRL-TK) reporter plasmid and the SRE.L firefly luciferase reporter plasmid (pSRE.L) are co-transfected, and the production of Renilla luciferase (RLuc) and firefly luciferase (FLuc) are assessed by use of the Dual Luciferase Assay kit (Promega) following the manufacturer's instructions.
The activities of RhoGEF DH-PH domains and full-length p115RhoGEF were examined in HeLa Tet-On cells grown on 48-well tissue culture plates with antibiotic-free medium. Cells were transfected at approximately 25% confluence with 160 ng of total DNA/well (100 ng of pSRE.L, 50 ng of pRL-TK, and 0–10 ng of mycDHPHpcDNA3.1 adjusted to 10 ng of DNA with mycpcDNA3.1) using FuGENE HD (Promega) according to the manufacturer's protocol. After 24 hours of normal culture, cells were starved in OptiMEM (Invitrogen) for 16 h. Cells were then washed in PBS, lysed in 65 μl of passive lysis buffer (Promega), and luciferase-assayed. Expression of protein was verified by application of 15 μl of cell lysate to SDS-PAGE and standard Western immunoblotting with anti-myc antibody (Santa Cruz sc-40).
The inhibitory activities of individual PH domains were assessed in Tet-On HeLa cells grown in 48 well plates. Cells were transfected at approximately 60% confluence with 500 ng of total DNA/well (100 ng of pSRE.L, 100 ng of pRL-TK, and 0–300 ng of pCMV5-myc-PH adjusted to 500 ng of total DNA with pCMV5-myc) using FuGENE HD (Promega). Four hours after transfection cells were switched to OptiMEM for overnight serum starvation (20 h). Cells were then stimulated with ligands and incubated for 5 h followed by lysis and transcriptional reporter assays as above. Lysates were also separated on 12% polyacrylamide SDS gels, transferred onto PDVF membranes and immunoblotted with anti-myc and anti-RhoGDI antibodies (Santa Cruz, sc-40 and sc-360, respectively). Blots were scanned and bands quantified using ImageJ software to assess the content of myc-PH domain in each sample.
Assays with Phospholipid Vesicles
Unilamellar phospholipid vesicles were prepared by extrusion through an Avanti Mini-Extruder using a 100-nm polycarbonate membrane (Avanti Polar Lipids). All lipids were obtained from Avanti. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (poPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (poPC), and 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (18:1 DGS-NTA(Ni)) were mixed in solvent at a mol ratio of 4.75:5:0.25, respectively. The mixture was dried under a stream of N2 gas for 30 min and placed under vacuum overnight. The lipids were then suspended to a concentration of 10 mm in 1 ml of buffer (20 mm NaHEPES, pH 7.5, 200 mm NaCl, 2 mm MgCl2, 5 mm β-mercaptoethanol). After five freeze-thaw cycles using an ethanol/dry ice bath, the mixture was passed through the extruder for 21 passages to form vesicles of ∼100-nm diameter.
RhoA-His6 was loaded with N-methylanthraniloyl-GDP (mant-GDP; Invitrogen) by incubation of 200 μm RhoA with 1 mm mant-GDP) in Buffer C (20 mm NaHEPES, pH 8.0, 1 mm EDTA, 1 mm DTT, 200 mm NaCl) plus 1 mm MgCl2 for 20 h at 25 °C. The RhoA with bound mant-GDP was exchanged into Buffer A plus 5 mm MgCl2 by dilution and concentration with an Amicon Ultra filtration device to remove excess nucleotide. A second incubation of 200 μm RhoA with 1 mm mant-GDP in Buffer A plus 5 mm MgCl2 for 20 h at 25 °C was performed to ensure complete loading. RhoA loaded with mant-GDP was separated from free nucleotide by gel filtration through tandem Superdex 200 and Superdex 75 10/100 GL columns with 20 mm NaHEPES, pH 8.0, 5 mm β-mercaptoethanol, 200 mm NaCl, and 5 mm MgCl2.
His-tagged RhoA-mant-GDP (1 μm) and 1 mm GDP, either with or without His-tagged RhoA-GTPγS, were incubated with vesicles (∼ 5 nm vesicles, 0.5 mm lipid) for 1 min at 25 °C to allow association of the proteins with the DGS-NTA-Ni on the vesicle surface. Other proteins were added as indicated and reactions started with the addition of RhoGEF proteins. The decrease in fluorescence due to dissociation of the mant-GDP was monitored with a Fluorolog-3 spectrofluorometer at 25 °C at λex = 356 nm, λem = 445 nm, and slits = 1/1 nm. Initial rates of exchange were assessed by fitting the change in fluorescence to a hyperbolic decay; maximal fluorescence change from this and other experiments and the known concentration of RhoA-His6-mant-GDP were used to convert fluorescence change into molar values.
RESULTS
Binding of Activated RhoA to the PH Domains of Lbc-RhoGEFs
We have shown previously that activated RhoA associates with a hydrophobic surface on the PH domain of PRG, and we speculated that this may represent a potential mechanism for positive feedback regulation of the RhoGEF (13). In contrast, p115RhoGEF did not bind to activated RhoA by a pulldown assay, even though it is highly homologous to PRG in this region. Because all of the PH domains in the Lbc family of RhoGEFs appear to have homologous sequences in this binding region (14), we tested all seven PH domains to define the potential spectrum of this interaction. The Lbc family of related RhoGEFs and the domains used in the following studies are shown in Fig. 1.
Fig. 2 shows that at least three (Lbc, p114, p190) of the other six PH domains in the Lbc family bind selectively to activated RhoA in pulldown assays with GST-RhoA. The LARG PH domain also appears to bind but with higher background. This association is confirmed using a dot blot assay in which the PH domains were spotted onto membranes and then incubated with either inactive (GDP) or activated (GTPγS) YFP-tagged RhoA (Fig. 3). In this format, six of the PH domains show binding to activated RhoA that is >4 S.D. greater than average binding to the GDP form of RhoA (10, σ = 2.5). Mutation of residues in the PH domain that interface with RhoA could abolish binding of the PRG PH domain to activated RhoA (13). Homologous mutations in two highly conserved hydrophobic residues were made in each of the PH domains. Whereas the mutated domains expressed as well as the wild-type domains, the mutations abolished binding to RhoA by all six of the positive domains as well as interactions with activated GST-RhoA by pulldowns (Fig. 2). This confirms specificity of binding and strongly suggests that the observed binding of RhoA is to equivalent surfaces on the PH domains.
FIGURE 2.
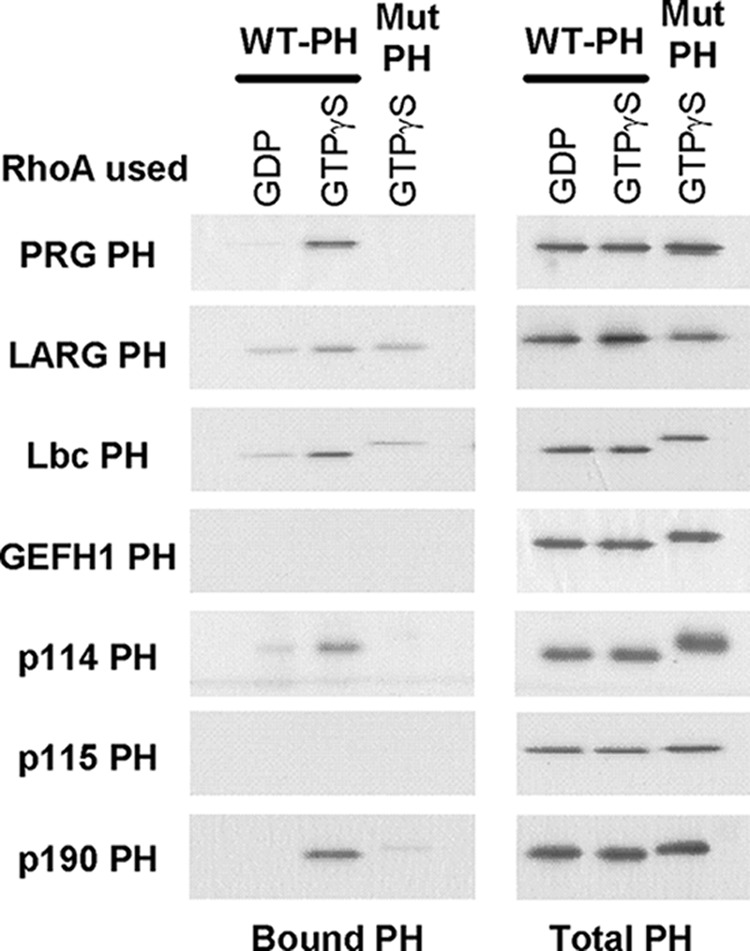
Interaction of PH domains from Lbc RhoGEFs with activated RhoA. Purified PH domains (40 pmol) were incubated with immobilized GST-RhoA (80 pmol) in the presence of either excess GDP or GTPγS, as indicated. For the GTPγS condition, the RhoA was preactivated as described. PH domains bound to RhoA were visualized by immunoblotting after separation though SDS-PAGE as described under “Experimental Procedures.” Mutant PH domains contained the mutations indicated in Fig. 1. The reason for slower migration of some of the mutated PH domains is unknown.
FIGURE 3.
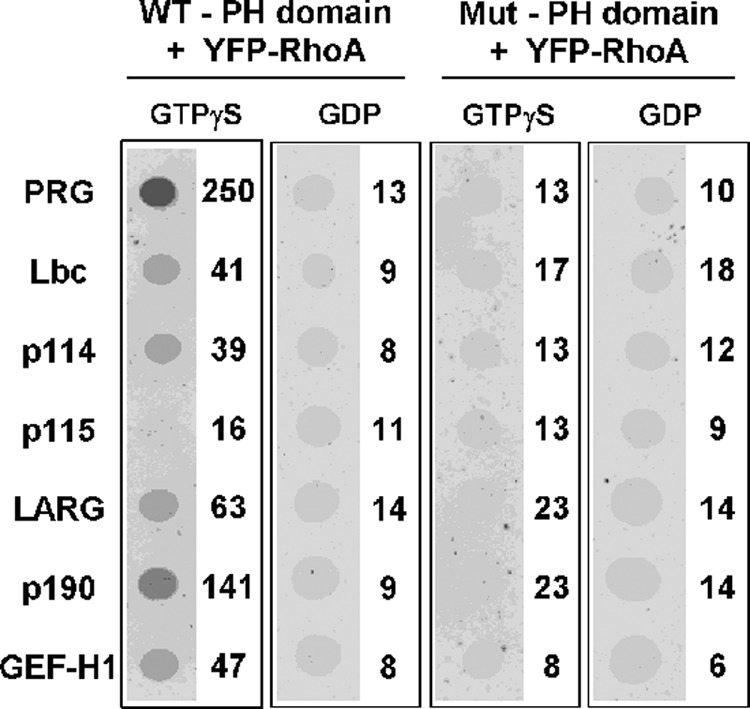
Binding of YFP-RhoA to immobilized PH domains. Purified GST-PH domains were applied to Ultrabind filters and exposed to either basal or activated YFP-RhoA, and bound RhoA was visualized as described under “Experimental Procedures.” Wild type (WT) or mutated (Mut) PH domains are described in Fig. 1. Quantified fluorescence (×10−5) for individual spots is shown to the right of the spot.
The scatter in signal strength suggests that the domains differ in their affinity for activated RhoA, although differences between the two assays disallow quantitative prediction of relative potencies at this time. Furthermore, the PH domain of GEFH1, which shows only weak binding in these assays, can form a stoichiometric complex with RhoA-GTPγS that is stable at high concentrations through gel chromatography (data not shown). Interestingly, the only PH domain that appears to be devoid of binding by these criteria is from p115RhoGEF.
The PH Domains of Lbc-RhoGEFs Block the RhoA Pathway in HeLa Cells
In HeLa cells, sphingosine 1-phosphate (S1P) causes activation of RhoA and its downstream pathways. This is mediated by S1P receptors and the heterotrimeric G12/13 proteins. If the Lbc PH domains bind activated RhoA, exogenous expression of the domains should attenuate downstream signaling by RhoA. To test this, we utilized the SRE.L transcriptional reporter assay to assess downstream function of RhoA. Increased expression of the PH domain from PRG blocked stimulation of luciferase expression by S1P in a concentration-dependent manner (Fig. 4A). To verify that this inhibition was dependent on binding of the PH domain to activated RhoA, we expressed the F1044A mutant (PRG-FA) which does not bind RhoA (13). The mutant PH domain had no effect on RhoA-dependent signaling by S1P at concentrations equivalent to expression of the wild-type domain (Fig. 4B). These data indicate that the free PH domain can interact with activated RhoA in the cellular environment and, at high concentrations, effectively sequester the GTPase from its downstream effectors. Indeed, this offers an alternative method for specifically blocking the RhoA pathway.
FIGURE 4.

PH domains of Lbc-RhoGEFs block activated RhoA in cells. A, concentration dependence. Tet-On HeLa cells were transfected with increasing amounts of plasmid encoding the PH domain of PRG along with the SRE.L reporter plasmids. After 20 h, cells were treated with either control (0.1 mg/ml BSA) or 1 μm S1P for 5 h, and expression of RLuc and FLuc was determined by activity assays. Stimulation of FLuc synthesis via the SRE.L promoter was normalized based on constitutive expression of RLuc. Stimulation by S1P is expressed as -fold over the signal from matched control-stimulated cells. PRG-FA is the mutated version of the PRG-PH domain (Fig. 1). B, comparison of PH domains from various Lbc-RhoGEFs. The ability of the indicated myc-tagged PH domains to block the RhoA-dependent S1P pathway was assessed at various concentrations of each PH domain as in A. Comparison is shown with samples representing the same concentration of each domain based on detection of the myc tag. Error bars represent the variance among four samples from two separate experiments for each domain. C, titration of the Lbc-PH domain. Experiments were performed as in A.
We next examined the PH domains from other members of the Lbc-RhoGEF family. Five of the domains attenuated stimulation of the SRE.L reporter by S1P to varying extents. Fig. 4B compares the efficacy of five PH domains expressed at similar concentrations. Whereas GEFH1 did not show strong signals in the binding assays (Figs. 2 and 3), it inhibits the RhoA pathway as well as the PRG-PH domain, an observation consistent with its ability to form a stoichiometric complex with activated RhoA. In contrast, the PH domains of p114RhoGEF and LARG were less efficacious. Surprisingly, the PH domain of p115RhoGEF blocked to a similar degree, although no binding was observed by the in vitro assessments. This suggests that the PH domain of p115RhoGEF does bind activated RhoA, albeit with low affinity. The ability of some domains to block RhoA in the cellular environment while showing variable or no detectable binding in vitro suggests that these domains may interact with other cellular membrane components that assist their localization and effective interaction with activated RhoA.
Two of the domains were harder to assess. The PH domain of p190-RhoGEF is a highly effective blocker but is not included in the comparison because it did not express to concentrations at which the other domains were compared. The PH domain from Lbc actually causes constitutive activation of the SRE.L reporter at very high levels of expression (Fig. 4C). Whereas inhibition of the S1P response by the Lbc-PH domain is clear at lower concentrations, the apparent complete attenuation of S1P response at the highest concentration may be due to nonspecific effects of the constitutive stimulation. Mutations in the Lbc-PH domain that prevent binding to activated RhoA eliminate the inhibitory impact of the domain on stimulation by S1P, but not the constitutive stimulation of SRE.L. The mechanism for this constitutive effect is not known.
Activated RhoA Facilitates Stimulation of RhoA in a Membrane-delimited System
The ability of PH domains to interact with activated RhoA in living cells supports the hypothesis that this interaction could mediate localization of the RhoGEFs to sites of RhoA activation. In effect, this would enable the RhoGEFs to remain at the membrane where they could successively activate more free RhoA, a mechanism for positive feedback regulation. To test this mechanism in vitro, we used phospholipid vesicles that contain the synthetic lipid, 18:1 DGS-NTA(Ni). This lipid contains a nickel-chelating head group for binding polyhistidine-tagged proteins. Thus, RhoA with a C-terminal His6 tag associates with the vesicles with the same orientation as free native RhoA on the plasma membrane. To assess nucleotide exchange, either dissociation of prebound mant-GDP from RhoA or binding of mant-GDP to RhoA is measured by the respective decrease or increase of intrinsic fluorescence of the mant-nucleotide.
The ability of activated membrane-delimited RhoA to drive nucleotide exchange on vesicle-associated RhoA is shown in Fig. 5. Basal exchange of nucleotide on RhoA is slow and only modestly stimulated by the soluble DH-PH domains of PRG added (filled circles). As increasing amounts of RhoA-His6-GTPγS are added to vesicles, the rates at which PRG affects exchange on basal RhoA are markedly increased. The dependence of this action on the concentration of RhoA is seen more clearly by plotting the initial rates of exchange observed upon addition of the RhoGEF (Fig. 5B). In this experiment, activated RhoA facilitated the activity of the PRG DH-PH domains approximately 40-fold. The EC50 for stimulation is approximately 50 nm, which is similar to the affinity measured previously for the interaction of activated RhoA with the PH domain of PRG (13).
FIGURE 5.

Membrane-delimited active RhoA facilitates activation of membrane localized RhoA by soluble RhoGEF. A, increasing amounts of RhoA-His6-GTPγS were reconstituted on the surface of phospholipid vesicles along with RhoA-His6-mant-GDP (1 μm, substrate) as described under “Experimental Procedures.” Facilitated dissociation of mant-GDP from the preloaded RhoA was initiated by addition of the PRG-DHPH domains (10 nm) and monitored by the decrease in fluorescence of dissociated mant-GDP. Concentrations of activated RhoA for representative curves are noted in the legend. B, initial rates for dissociation of mant-GDP were determined as described under “Experimental Procedures” and plotted against the concentration of RhoA-His6-GTPγS bound to vesicles.
The specific dependence for this facilitated RhoGEF activity on membrane-delimited RhoA-GTPγS and its interaction with the PH domain of the RhoGEF is further demonstrated in Fig. 6. The activity is not supported by membrane-delimited Rac1 and Cdc42, which do not bind to the PRG-PH domain; the facilitation is lost if activated RhoA is not localized to the vesicle, and mutation of the binding interface between the PH domain and RhoA abolishes the activity.
FIGURE 6.

Stimulation of guanine nucleotide exchange is dependent on membrane-associated RhoA-GTPγS and its interaction with the PH domain of PRG. A, stimulation is specific for activated RhoA. Substrate vesicles containing RhoA-His6-mant-GDP were incubated with the DH-PH domains of PRG (10 nm) alone or supplemented with the C-terminal His-tagged versions of RhoA, Rac1, or Cdc42, which had been preactivated with GTPγS: no GEF, filled circles; no activated GTPase, filled triangles; 100 nm RhoA, open circles; 30 nm RhoA, open triangles; 30 nm Cdc42, filled squares; 30 nm Rac1, open squares. B, soluble activated RhoA does not facilitate RhoGEF activity. Substrate vesicles containing RhoA-His6-mant-GDP were incubated with the DH-PH domains of PRG (10 nm) alone (open circles) or supplemented with activated RhoA lacking the His tag (100 nm, open triangles) or His-tagged RhoA (100 nm, filled triangles). C, binding of activated RhoA to the PRG PH domain is required for facilitated activity. Substrate vesicles containing 1 μm RhoA-His6-GDP were incubated alone (filled squares) or with either WT (circles) or mutant (triangles) DH-PH domains of PRG (10 nm) and 2.5 μm mant-GDP to measure nucleotide exchange. Vesicles were supplemented with 100 nm RhoA-His6-GTPγS (open symbols) to test for facilitated activity.
Mutations of the Binding Site for Active RhoA Reduce Stimulation of RhoA by Overexpression of the RhoGEFs
Overexpression of the RhoGEFs induces constitutive stimulation of Rho GTPases. One potential mechanism for this unregulated activity among the Lbc RhoGEFs is their association with activated RhoA in membranes and prolonged localization near additional substrate, as shown for vesicular RhoA in the previous experiments. In Fig. 7, this hypothesis is tested by exogenous expression of both WT and mutant DH-PH domains in HeLa cells. Mutant domains contain the dual mutations listed in Fig. 1 that attenuate interactions with activated RhoA; these domains express like the wild-type domains (Fig. 7B). The DH-PH domains from all seven of the Lbc RhoGEFs markedly increase expression of luciferase driven by the RhoA-responsive SRE.L promoter in a concentration-dependent manner (Fig. 7A). With the DH-PH domains from LARG, PRG, p115, Lbc, and p190, this stimulation is strongly attenuated by mutation of the binding surface for activated RhoA on their respective PH domains. This is most obvious for low levels of expression where the WT proteins show strong activity whereas the mutant proteins are largely ineffective. At more robust expression, the mutant proteins provide activation of the pathway.
FIGURE 7.

Binding of LbcRhoGEFs to activated RhoA in cells increases their tonic activity. A, HeLa Tet-On cells were co-transfected with SRE.L reporter plasmids and increasing amounts of pcDNA3.1-myc-DHPH (wild type or double mutant). Assays for luciferase activity are described under “Experimental Procedures” and Fig. 4; results are reported as -fold activation over transfection with empty pcDNA3.1-myc vector. Data shown are the mean of duplicate wells with average variance and are representative of three independent experiments. B, lysates (15 μl) from samples in A were resolved by SDS-PAGE and Western blotted for the myc epitope.
Two RhoGEFs do not show this behavior. Thus, mutation of the putative RhoA binding site on the DH-PH domains of p114 and GEFH1 shows little to no effect on their ability to stimulate the RhoA pathway. Although it is possible that these proteins have a different interface for binding active RhoA, it is more likely that these domains also have other mechanisms for localizing constitutively to the plasma membrane and their substrates.
Because constitutive activity by the RhoGEFs would be deleterious for survival and function of normal cells, there have to be mechanisms for keeping the RhoGEFs inactive in cells until needed. This could certainly be a function of other domains in the RhoGEFs. Therefore, we used p115RhoGEF and PRG to test for effects of mutating the RhoA binding site in the context of the full-length protein. In Fig. 8, we show that the wild-type and mutant proteins express to similar extents. As with the DH-PH domains alone, the WT proteins show strong stimulation of the RhoA reporter, whereas the mutant proteins are ineffective until much higher levels of expression. These results correspond with prior experiments with full-length LARG, where mutations in the hydrophobic region of its PH domain attenuated its ability to cause constitutive activation of RhoA pathways (14). We now know that these mutations should reduce binding to activated RhoA.
FIGURE 8.

Mutation of the RhoA-GTP binding interface in the PH domain of full-length p115RhoGEF and PRG reduces their tonic activity in cells. A, cells were transfected with reporter vectors and increasing amounts of vector expressing WT or mutant p115RhoGEF. Activation of the RhoA pathway was assessed as in Fig. 7 and shown as the ratio of FLuc to RLuc. Expression of p115RhoGEF was determined by Western analysis using detection of the myc tag. B, cells were transfected with increasing amounts of full-length WT or mutant PRG and analyzed as in A.
DISCUSSION
Previous studies showed that activated RhoA interacts with the PH domain of its own regulator, PRG. This interaction involves the switch regions in RhoA and a hydrophobic patch on the PRG-PH domain that is distal to the active site of the protein (13). PRG belongs to a subfamily of seven proteins known as the LbcRhoGEFs that are selective for catalyzing guanine nucleotide exchange on RhoA-like GTPases and have homologous DH-PH domains, especially residues corresponding to the hydrophobic patch of PRG which make contact with activated RhoA (13, 14). The hypothesis that this conserved region may mediate a common mechanism for regulation was confirmed in the demonstration that all seven of the RhoGEFs can interact with activated RhoA through this motif, either by direct biochemical binding in vitro (Figs. 1–3) or by selective measurements of intracellular activities (Figs. 4, 7, and 8). This also confirms and extends prior observations that full-length versions of p114 and p190 RhoGEF could interact with activated RhoA (18, 19).
The interaction of activated RhoA with the different PH domains shows an apparent range of affinities based on their detection by various assays with different sensitivities. The most obvious difference is the PH domain of p115RhoGEF, which shows essentially no interaction by in vitro assays but can inhibit a RhoA dependent pathway in cells (Fig. 4); further, mutations in this site highly attenuate activity of the overexpressed protein (Fig. 8). This indicates that the interaction has relatively low affinity but is still effective in vivo. A feature that may contribute to increased sensitivity of this interaction in living cells is the potential for other interactions with components of the plasma membrane that enhance the effective interaction with RhoA. We have hypothesized that such interactions may account for the relatively small effects of removing the RhoA binding site from the DH-PH domains of p114RhoGEF and GEFH1 (Fig. 7). A second feature that may influence biological activity is the capability of several of these proteins to form constitutive dimers, thus increasing potential binding energy through use of multiple concurrent interactions. Future studies to determine the influence of other interactive sites and the impact of these apparent differences in affinity should provide insight into specific biological roles for the different RhoGEFs.
The binding of activated RhoA to the PH domains of all seven Lbc-RhoGEFs suggests that this is a fundamental mechanism for the regulation of these proteins. The most obvious function for this interaction is feedback regulation that might directly impact the catalytic rate for these exchange factors. So far, attempts to demonstrate a direct effect of activated RhoA on catalytic activity of the DH-PH domains of the Lbc-RhoGEFs have been negative (Ref. 13 and data not shown). It is possible that such regulation requires other domains in the RhoGEFs, but this was not the case for PRG (13). In the studies with PRG, we speculated that regulation of the RhoGEF by RhoA used an indirect mechanism, that is, localization of the RhoGEF to the plasma membrane where it could use its basal, intrinsic activity to engage its localized substrate, free RhoA, more effectively. This hypothesis was tested directly through the use of phospholipid vesicles with associated RhoA to mimic the plasma membrane (Fig. 5). This experiment demonstrates that tethering of the RhoGEFs to the membrane surface by activated RhoA was sufficient to increase the effective turnover of basal RhoA. Whereas the activity of free RhoGEF is limited to its random collision frequency with substrate on vesicles, localized RhoGEF will be more efficiently directed to substrate by its diffusion in two dimensions on the membrane surface. Under conditions used here, this can result in an increased rate of exchange >40-fold. A key control shown in Fig. 6 is that activated RhoA that is not tethered to the vesicles has no effect on the activity of the RhoGEF. Although the tethered hypothesis and experimental result may seem obvious, this demonstration clearly shows that the orientation of the RhoGEF via binding to activated RhoA is consistent with, and perhaps even enhanced by, an orientation that allows effective interaction with substrate in the same membrane; this was predicted by prior studies (13).
Localization via activated RhoA constitutes an effective, positive feedback mechanism for robust signal generation by this family of RhoGEFs. The exact purpose for this is not yet established. However, this mechanism would enhance the sensitivity of the pathways to initial stimuli, such as activation of RH-RhoGEFs by hormonal signaling through G12/13 and may be able to facilitate very robust localized signaling that might be required for cellular behaviors like migration. The presence of a positive feedback mechanism also implies that there must be potent dampening mechanisms to prevent spontaneous stimulation as observed in the studies with overexpression of the RhoGEFs (e.g. Figs. 7 and 8). The ability to remove this positive mechanism by site-directed mutations should facilitate discovery of its importance and role in cellular physiology.
The PH domain is one of the most common domains in the human genome, and the classical RhoGEFs all contain this domain in conjunction with the catalytic DH domain. There is increasing evidence that these PH domains can facilitate activation of Rho GTPases through interaction with substrate, proteins, or phospholipids (4). A pertinent example is the interaction of activated Rac1 with the PH domain of Dbs (20), a RhoGEF with selectivity for RhoA and Cdc42. This invites the speculation that interaction of activated GTPases with the PH domains of RhoGEFs may be a common feature for the purpose of both feedback regulation and cross-talk between the signaling pathways.
This work was supported, in whole or in part, by National Institutes of Health Grant GM31954 (to P. C. S.). This work was also supported by Robert A. Welch Foundation Grant I-1262 (to P. C. S.) and the Alfred and Mabel Gilman Chair in Molecular Pharmacology (to P. C. S.).
- RhoGEF
- Rho guanine nucleotide exchange factor
- DH
- Dbl homology
- FLuc
- firefly luciferase
- GAP
- GTPase-activating protein
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- LARG
- leukemia-associated RhoGEF
- mant-GDP
- N-methylanthraniloyl-GDP
- NTA
- nitrilotriacetic acid
- PDZ
- postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (DlgA), and zonula occludens-1 protein (zo-1)
- PH
- pleckstrin homology
- PRG
- PDZ-RhoGEF
- RGS
- regulators for G protein signaling
- RH
- RGS homology
- RLuc
- Renilla luciferase
- S1P
- sphingosine 1-phosphate
- TEV
- tobacco etch virus.
REFERENCES
- 1. Etienne-Manneville S., Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Brown J. H., Del Re D. P., Sussman M. A. (2006) The Rac and Rho hall of fame. Circ. Res. 98, 730–742 [DOI] [PubMed] [Google Scholar]
- 3. Guilluy C., Garcia-Mata R., Burridge K. (2011) Rho protein cross-talk: another social network? Trends Cell Biol. 21, 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go: turning on Rho GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 5. Bos J. L., Rehmann H., Wittinghofer A. (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 [DOI] [PubMed] [Google Scholar]
- 6. Sternweis P. C., Carter A. M., Chen Z., Danesh S. M., Hsiung Y., Singer W. D. (2007) Regulation of Rho guanine nucleotide exchange factors by G proteins. In Advances in Protein Chemistry, (Sprang S. R. ed.) pp. 189–228, Academic Press, Orlando, FL: [DOI] [PubMed] [Google Scholar]
- 7. Aittaleb M., Boguth C. A., Tesmer J. J. (2010) Structure and function of heterotrimeric G protein-regulated Rho guanine nucleotide exchange factors. Mol. Pharmacol. 77, 111–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kozasa T., Jiang X., Hart M. J., Sternweis P. M., Singer W. D., Gilman A. G., Bollag G., Sternweis P. C. (1998) p115 RhoGEF, a GTPase-activating protein for Gα12 and Gα13. Science 280, 2109–2111 [DOI] [PubMed] [Google Scholar]
- 9. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 10. Chen Z., Guo L., Hadas J., Gutowski S., Sprang S. R., Sternweis P. C. (2012) Activation of p115-RhoGEF requires direct association of Gα13 and the Dbl homology domain. J. Biol. Chem. 287, 25490–25500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wells C. D., Liu M.Y., Jackson M., Gutowski S., Sternweis P. M., Rothstein J. D., Kozasa T., Sternweis P. C. (2002) Mechanisms for reversible regulation between G13 and Rho exchange factors. J. Biol. Chem. 277, 1174–1181 [DOI] [PubMed] [Google Scholar]
- 12. Chen Z., Singer W. D., Danesh S. M., Sternweis P. C., Sprang S. R. (2008) Recognition of the activated states of Gα13 by the rgRGS domain of PDZRhoGEF. Structure 16, 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen Z., Medina F., Liu M. Y., Thomas C., Sprang S. R., Sternweis P. C. (2010) Activated RhoA binds to the PH domain of PDZ-RhoGEF: a potential site for autoregulation. J. Biol. Chem. 285, 21070–21081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aittaleb M., Gao G., Evelyn C. R., Neubig R. R., Tesmer J. J. (2009) A conserved hydrophobic surface of the LARG pleckstrin homology domain is critical for RhoA activation in cells. Cell. Signal. 21, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Z., Guo L., Sprang S. R., Sternweis P. C. (2011) Modulation of a GEF switch: autoinhibition of the intrinsic guanine nucleotide exchange activity of p115-RhoGEF. Protein Sci. 20, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shaner N. C., Steinbach P. A., Tsien R. Y. (2005) A guide to choosing fluorescent proteins. Nat. Methods 2, 905–909 [DOI] [PubMed] [Google Scholar]
- 17. Wells C. D., Gutowski S., Bollag G., Sternweis P. C. (2001) Identification of potential mechanisms for regulation of p115 RhoGEF through analysis of endogenous and mutant forms of the exchange factor. J. Biol. Chem. 276, 28897–28905 [DOI] [PubMed] [Google Scholar]
- 18. Blomquist A., Schwörer G., Schablowski H., Psoma A., Lehnen M., Jakobs K. H., Rümenapp U. (2000) Identification and characterization of a novel Rho-specific guanine nucleotide exchange factor. Biochem. J. 352, 319–325 [PMC free article] [PubMed] [Google Scholar]
- 19. van Horck F. P., Ahmadian M. R., Haeusler L. C., Moolenaar W. H., Kranenburg O. (2001) Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 276, 4948–4956 [DOI] [PubMed] [Google Scholar]
- 20. Cheng L., Mahon G. M., Kostenko E. V., Whitehead I. P. (2004) Pleckstrin homology domain-mediated activation of the Rho-specific guanine nucleotide exchange factor Dbs by Rac1. J. Biol. Chem. 279, 12786–12793 [DOI] [PubMed] [Google Scholar]