

Background: Nitric oxide (NO) regulates class I and IIa histone deacetylase (HDAC) function. NO production is regulated by class III HDACs (sirtuins).
Results: NO functions as a bridging molecule between class I and sirtuins (SIRTs).
Conclusion: The SIRT-NO-class I HDAC axis provides key signals during wound repair.
Significance: Modulation of HDAC activity may play an important role in tissue regeneration.
Keywords: Epigenetics, Histone Deacetylase, Keratinocytes, Nitric Oxide, Sirtuins
Abstract
In a mouse model of skin repair we found that the class I-IIa histone deacetylase inhibitor trichostatin A accelerated tissue regeneration. Unexpectedly, this effect was suppressed by Sirtinol, a class III histone deacetylase (HDAC) (sirtuin)-selective inhibitor. The role of sirtuins (SIRTs) was then investigated by using resveratrol and a novel SIRT1-2-3 activator, the MC2562 compound we synthesized recently. Both resveratrol and MC2562 were effective in accelerating wound repair. The local administration of natural or synthetic SIRT activators, in fact, significantly accelerated skin regeneration by increasing keratinocyte proliferation. In vitro experiments revealed that the activation of SIRTs stimulated keratinocyte proliferation via endothelial NO synthase phosphorylation and NO production. In this condition, the class I member HDAC2 was found S-nitrosylated on cysteine, a post-transduction modification associated with loss of activity and DNA binding capacity. After deacetylase inhibitor or SIRT activator treatment, ChIP showed, in fact, a significant HDAC2 detachment from the promoter region of insulin growth factor I (IGF-I), fibroblast growth factor 10 (FGF-10), and Epithelial Growth Factor (EGF), which may be the final recipients and effectors of the SIRT-NO-HDAC signaling cascade. Consistently, the effect of SIRT activators was reduced in the presence of NG-nitro-l-arginine methyl ester (l-NAME), a general inhibitor of NO synthesis. In conclusion, the NO-dependent cross-talk among class III and I histone deacetylases suggests an unprecedented signaling pathway important for skin repair.
Introduction
NO has been reported recently as an important regulator of histone deacetylase (HDAC)4 activity (1). Experimental evidence has been provided about the effect of NO on the function of class I-IIa HDACs in different biological contexts (1, 2). Specifically, the NO-dependent S-nitrosylation of HDAC2, a member of class I HDACs, has been associated with enzyme inactivation and detachment from chromatin in neurons (3) and skeletal muscle precursors (4). In addition, NO regulates the nuclear shuttling of class IIa HDACs in endothelial cells and during mouse embryonic stem cell differentiation (5, 6). Conversely, class III HDACs, or sirtuins (SIRTs), among the multiplicity of their metabolic functions, act as positive regulators of NO production. Indeed, sirtuin1 (SIRT1) plays a fundamental role in the maintenance of vascular homeostasis in the presence of shear stress (7) by direct deacetylation and activation of endothelial NO synthase (eNOS), leading to a significant increase in the release of NO (8).
In wound healing (WH), NO exerts a pivotal role at all stages of regeneration, including inflammation, angiogenesis, re-epithelization, matrix deposition, and tissue remodeling (9–12). Although their role is still poorly characterized, SIRTs seem important in this process because SIRT1 regulates keratinocyte proliferation (13, 14). We recently synthesized novel SIRT activators with effects comparable with those of resveratrol (Resv) but on the basis of a different chemistry (15). Of note is that Resv is capable of promoting WH, activating SIRT1, and stimulating NO synthesis (16, 17). This work describes how, similarly to Resv, SIRT activation by the novel synthetic compound MC2562 (15) led to accelerated wound repair. Remarkably, the same effect was obtained with the class I pan-inhibitor trichostatin A (TSA), suggesting a concerted action for these molecules in wound repair. This process occurred via enhanced NO synthesis which, in turn, determined the functional inactivation of HDAC2. We propose here that, during skin repair and possibly other tissue regeneration processes (4), NO may act under the control of SIRTs as a hierarchically upstream negative regulator of class I HDACs.
EXPERIMENTAL PROCEDURES
Animal Skin Wound Model
Eight-week-old CD1 male mice were obtained from Charles River Laboratories, Inc. (Calco, Lecco, Italy) and were anesthetized with an intraperitoneal injection of 1 mg/kg medetomidine (Domitor, Vetem, Milan, Italy) and 75 mg/kg ketamine (Ketavet 100, Intervet Farmaceutici, Aprilia, Italy). The animal dorsum was clipped free of hair, and a full-thickness wound of 3.5-mm diameter was created using a biopsy punch. Drugs were applied daily at the indicated concentration in 20 μl of saline solution, directly in the wound area. Another group of mice was treated with diluting solvent in 20 μl of saline solution on the wound. The epigenetic drugs were administered in the wound area at the following concentrations: resveratrol (1 μm, Sigma), MC2562 (1 μm), Sirtinol (25 μm, Sigma), TSA (100 nm, Alexis), ITSA (100 nm, Sigma), MS275 (1 μm, Alexis) and MC1568 (1 μm). Knockdown of HDAC2 expression in the wound area was achieved with siRNA targeting HDAC2 (100 nm, Ambion). Specific siRNAs or scrambled sequence were applied directly on the wound using siPORT NeoFX according to the instructions of the manufacturer.
To compare the rate of wound closure between epigenic drug-treated and solvent-treated mice, animals were photographed at days 0, 3, 5, 6, 7, 10, and 14 after treatment. Pictures were processed digitally, and wound areas were calculated using the KS300 system (Zeiss, Jena, Germany). For each sample, the rate of the healing process was measured as a ratio of the area at each time point divided by the area at time 0 (that is, immediately after wounding). All experimental procedures complied with the Guidelines of the Italian National Institutes of Health and the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academy of Sciences, Bethesda, MD) and were approved by the Institutional Animal Care and Use Committee.
Histology and Wound Analysis
Mouse biopsies were fixed in formalin and embedded in paraffin (Bio-plast special, melting point 52–54 ºC). For immunohistochemical staining, 3-mm-thick sections were incubated with specific antibodies. The epithelial gap, which represents the distance between the leading edge of migrating keratinocytes, was measured by hematoxylin-eosin staining and reported in millimeters.
In Vivo Bioluminescence Imaging
Transgenic MITO-Luc mice were generated as described previously (18). Briefly, a transgene harboring the luciferase gene under control of an nuclear factor Y-dependent cyclin B2 promoter fragment was used as a sensor of nuclear factor Y activity in vivo. In these mice, bioluminescence imaging of nuclear factor Y activity visualizes areas of physiological cell proliferation and regeneration during response to injury (19). Light emission was monitored with an IVIS® Lumina imaging system. This system includes a highly sensitive charge-coupled device (CCD) camera, a light-tight imaging chamber with complete computer automation, and the Living Image® 2.2 software package for image acquisition and analysis (Caliper Life Sciences). Mice were anesthetized, and 150 mg/kg of D-luciferin (XenoLight D-luciferin, Keliper/PKI) was injected intraperitoneally. Ten minutes later, light emission was quantified in photons/second and visualized in a pseudocolor scaling. Time exposure ranged from 1–5 min depending on light intensity.
Cell Culture and Treatment
The transformed human keratinocyte cell line (HaCaT) (19) was cultured in DMEM (Lonza) supplemented with 10% FBS and antibiotics. Cells were treated with resveratrol (1 μm, Sigma), MC2562 (1 μm), Sirtinol (25 μm, Sigma), or TSA (100 nm) in the absence of serum. DMSO was used as solvent control.
Immunofluorescence and Immunohistochemistry
HaCaT cells were fixed in 4% paraformaldehyde solution for 10 min at room temperature, washed three times for 5 min with PBS, and blocked for 1 h in PBS containing 10% BSA. Cells were incubated with primary antibodies in PBS containing 1% BSA overnight, at 4 °C. Plates were washed twice for 5 min with PBS and incubated with α-rabbit-tetramethylrhodamine B isothiocyanate, α-mouse-fluorescein isothiocyanate, and α-goat-fluorescein isothiocyanate secondary antibodies for 1 h at room temperature in the dark. After three washes in PBS for 5 min, cells were incubated with 1 μg/ml Hoechst for 10 min at room temperature in the dark and then washed three times with PBS for 5 min. A total of 100 μl of DAKO cytomation-mounting fluorescent medium was used. Staining was visualized with an Apotome microscope. Pictures were obtained using an Axiocam and analyzed with KS 300 3.0 acquisition software (Zeiss).
For immunohistochemical staining, 3-μm-thick sections were deparaffinized, incubated at room temperature for 20 min with a methanol solution containing 0.03% H2O2, blocked for 1 h with 10% rabbit or goat serum in PBS, and incubated overnight at 4 °C with anti-FGF10 (Santa Cruz Biotechnology, Inc.), anti-EGF (Santa Cruz Biotechnology, Inc.), and anti-IGF1 (Santa Cruz Biotechnology, Inc.) antibodies, followed by biotinylated secondary antibody (7.5 μg/ml, Vector Laboratories, Peterborough, UK) and avidin-biotinylated peroxidase complex (ABC Elite kit, Vector Laboratories). The staining was visualized by treatment for 10 min in a 0.05% solution of 3-diaminobenzidine and 0.01% H2O2 in 0.1 m PBS. Sections were counterstained with hematoxylin to identify nuclei.
Western Blotting, Immunoprecipitation, and Antibodies
Cells were lysed in Laemmli buffer (WB) or radioimmune precipitation assay buffer (immunoprecipitation). WB was performed according to standard procedures, and results were quantified by ImageJ v1.43 software. Optical density values were internally normalized by α-tubulin and corrected further for the value of controls considered equal to 1. Data represent the means of at least three independent experiments ± S.E. SIRT1 and eNOS interaction and HDAC2 S-nitrosylation were determined by immunoprecipitation performed by using anti-SIRT1 (4 μg for 500 μg of total proteins, polyclonal, Abcam), anti-eNOS (4 μg for 500 μg total proteins, polyclonal, Abcam), anti-HDAC2 (4 μg for 500 μg total proteins, polyclonal, Santa Cruz Biotechnology, Inc.), or anti-S-nitrosylated cysteine (4 μg for 500 μg total proteins, polyclonal, Alpha Diagnostic). Paramagnetic beads (Ademtech's Bioadembeads) were used for specific protein separation. Negative controls were performed by using the same amount of protein extract from solvent samples immunoprecipitated with the corresponding purified normal IgG (Santa Cruz Biotechnology, Inc.) in the absence of primary antibody.
The following antibodies and dilutions were used for WB, immunofluorescence (IF), and immunohistochemistry analysis: anti-α-tubulin (WB, 1:4000, monoclonal, Cell Signaling Technology, Inc.), anti-luciferase (IF, 1:100, polyclonal, Santa Cruz Biotechnology, Inc.), anti-keratin 14 (IF, 1:100, monoclonal, Thermo Scientific), anti-acetylated tubulin (IF, 1:200, monoclonal, Sigma), anti-smooth muscle actin (IF, 1:30, monoclonal, Sigma), anti-peNOS(s1177) (IF, 1:150; WB, 1:1000; polyclonal; Cell Signaling Technology, Inc.), anti-eNOS (WB, 1:500, polyclonal, Abcam), anti-HDAC1 (WB, 1:500, polyclonal, Santa Cruz Biotechnology, Inc.), anti-HDAC2 (WB, 1:500, polyclonal, Santa Cruz Biotechnology, Inc.), anti-HDAC3 (WB, 1:1000, monoclonal, Cell Signaling Technology, Inc.), anti-acetylated histone H4 lysine 16 (WB, 1:1000, polyclonal, Abcam), anti-acetylated histone H3 lysine 14 (WB, 1:1000, polyclonal, Abcam), anti-S-nitrosylated cysteine (WB, 1:1000, polyclonal, Alpha Diagnostic), anti-SIRT1 (WB, 1:1000, polyclonal, Abcam), anti-FGF10 (immunohistochemistry and IF, 1:50, polyclonal, Santa Cruz Biotechnology, Inc.), anti-EGF (immunohistochemistry and IF, 1:50, monoclonal, Santa Cruz Biotechnology, Inc.), and anti-IGF-I (immunohistochemistry and IF, 1:50, polyclonal, Santa Cruz Biotechnology, Inc.).
HDAC Activity Assay
An HDAC colorimetric assay (Biovision) was performed according to the instructions of the manufacturer. HDAC2 specific activity was evaluated in HaCaT cells resuspended in radioimmune precipitation assay buffer after immunoprecipitation with specific antibodies as described previously (4).
FACS Analysis
Nitric oxide production was evaluated by adding 4,5-diaminofluorescein (DAF-2 DA) (Alexis) to the medium according to the instructions of the manufacturer. ChIP and quantitative real-time PCR were performed as described previously (20). Briefly, HaCaT cells treated with TSA, MC2562, or solvent were cross-linked, and ChIP was performed using specific antibodies to HDAC2 (Abcam). Negative controls were in absence of antibody (No Ab). DNA fragments were recovered and analyzed by quantitative real-time PCR in duplicate, and the data obtained were normalized to the corresponding DNA input control (used to generate standard curves). Data are represented as relative enrichment.
Primers for quantitative PCR were as follows (distance from transcription start site (TSS)): hEGFprom-4898 forward, 5′-CCACATTCGCATTTGCAAAC-3′ (-102 bp) and hEGFprom-4961 reverse, 5′-CAGGCTCCACCTCCTTTCC (-84 bp); hEGFprom-3574 forward, 5′-CACTGACACAGCGTGATGCTT-3′ (-1426bp) and hEGFprom-3708 reverse, 5′-AAATAACCTGCACTGACTCTTCGA-3′ (-1292bp); hFGF10prom forward, 5′-ACCCACGTCCACCATTTACC-3′ and hFGF10prom reverse, 5′-AACATCCATAACTCCTCGGAAAAG-3′; and hIGF1prom forward, 5′-GCCCTAAAGGGACCAATCCA-3′ and hIGF1prom reverse, 5′-TTGTCCCAGTTGCCAAGTGA-3′.
Wound Healing RT2 Profiler PCR Array
RNA was extracted using TRIzol (Invitrogen) according to the instructions of the manufacturer. Briefly, total mRNA was treated with column DNase treatment (Qiagen RNeasy mini kit) and converted to cDNA. The human wound healing PCR array (SABiosciences) was used to monitor the expression of 84 genes related to the WH process plus five housekeeping genes. Controls for genomic DNA contamination, RNA quality, and general PCR performance are included on each array. For data analysis, SABiosciences provides an integrated web-based software package for the PCR array system that performs all ΔΔCt-based fold change calculations of uploaded raw threshold cycle data as well as pairwise comparison between groups of experimental replicates defining fold changes and statistical significance thresholds or compares all of the groups side by side. cDNA template was mixed with the ready-to-use PCR master mix, and equal volumes were aliquoted to each well of the same plate to perform the real-time PCR cycling program.
Statistical Analysis
Data represent the mean of at least three independent experiments ± S.E. Variables were analyzed by two-side Student's t test and two-way analysis of variance (WH experiments).
RESULTS
Class I HDAC Inhibitors Accelerate Wound Healing
To investigate HDAC contribution to skin repair, excisional wounds were made on the back of mice by standard punch biopsy (3.5-mm diameter). Solvent (DMSO) or HDAC inhibitors (deacetylase inhibitor) were applied on the wound daily for 2 weeks. Digital pictures were taken at time 0 (t0) and at 3, 5, 7, 10, and 14 day post-wounding. The experiments revealed that the pan-inhibitor TSA accelerated the kinetics of wound repair (Fig. 1, A and B). This effect was abrogated by the selective TSA suppressor molecule ITSA in combination with TSA. The acetylation of tubulin was assessed by immunofluorescence analysis of the wound section. This assay was performed as a functional TSA readout (Fig. 1C). Fig. 1C shows the presence of a strong signal for acetylated tubulin in sections obtained from the TSA-treated wound. To evaluate the contribution of a different class of HDACs during skin repair, we used the class I selective inhibitor MS275 and the MC1568 compound, which represses class IIa HDAC function (21). Fig. 1D shows that MS275 accelerated the closure process, suggesting an active role for class I HDACs during WH, whereas the class II inhibitor MC1568 had no effect. Surprisingly, the positive effect of TSA on WH was abrogated by the class III HDAC inhibitor Sirtinol, initially used as additional control, thus indicating a potential molecular cross-talk between HDAC classes I and III (Fig. 1E).
FIGURE 1.
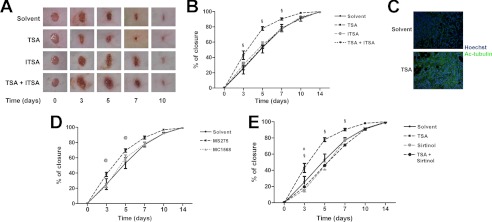
Class I HDAC inhibitors accelerate wound healing. A, representative pictures of mouse wound healing experiments. Animals were treated topically with TSA (n = 10), ITSA (n = 7), and TSA and ITSA (n = 7) in combination each day. DMSO was used as a solvent control (n = 12). B, kinetics of skin wound healing in CD1 mice treated with TSA, ITSA, and TSA and ITSA in combination. DMSO was used as a control solvent (n = 12). §, p < 0.05 versus solvent. C, immunofluorescence analysis showing the presence of acetylated tubulin in the wound after local TSA administration compared with the solvent. Green, acetylated tubulin; blue, nuclei counterstained with Hoechst (magnification ×20). D, kinetics of skin wound healing in CD1 mice treated with the deacetylase inhibitor MS275 (n = 8) and MC1568 (n = 8). DMSO was used as a control solvent (n = 12). @, p < 0.05 versus solvent. E, kinetics of skin wound healing in CD1 mice treated with TSA (n = 10), Sirtinol (n = 12), and TSA and Sirtinol (n = 7) in combination. DMSO was used as a control solvent (n = 12). § and #, p < 0.05 versus solvent.
Natural and Synthetic SIRT Activators Enhance Cell Motility and Wound Healing via Keratinocyte Proliferation
In vivo experiments were performed to evaluate the direct effect of SIRT modulators during the closing of experimental wounds. Fig. 2A shows that Resv and MC2562 accelerated wound repair compared with solvent and Sirtinol. The latter retarded wound closure significantly at the day 3 time point. Consistently, histology revealed that SIRT activators decreased the epithelial gap at day 5, whereas Sirtinol had no effect or was detrimental (Fig. 2B). To identify the cellular population responsive to treatments, a series of experiments was performed with the transgenic MITO-Luc mice (18). In this transgenic in vivo reporter system, luciferase expression occurs under control of a portion of the cyclin B2 promoter cassette encompassing two “CAAT” boxes specifically recognized by members of the nuclear factor Y family. In this model, only proliferating cells can be visualized non-invasively by bioluminescence imaging (19). In this context, SIRT activators enhanced cell proliferation significantly during the early stage of the repair process (days 2 and 3 after wounding) (Fig. 2, C and D). Immunofluorescence analyses performed on MITO-Luc tissue samples revealed that keratinocytes, recognized by keratin 14 expression, had the brightest luciferase signal (Fig. 2E). To further investigate the role of SIRTs during keratinocyte activation, the HaCaT cell line derived from transformed human keratinocytes was used (19). The acetylation level of lysine 16 of histone H4 (H4K16Ac) was utilized as a readout of sirtuin activity (22). After overnight starvation, HaCaT cells were treated with the SIRT activator MC2562 (1 μm) (15). Resv (1 μm) and DMSO were used as positive and negative controls, respectively. Fig. 3A shows that both MC2562 and Resv induced significant H4K16 deacetylation compared with controls, whereas the acetylation of other lysine residues, including histone 3 lysine 14 (H3K14Ac), was unchanged. SIRT1 activity was evaluated further by an enzymatic assay performed with HaCaT nuclear extracts in the presence of MC2562 (1 μm), Resv (1 μm), Sirtinol (25 μm), and DMSO. As shown in Fig. 3B, MC2562 enhanced SIRT1 activity at levels comparable with or higher than Resv. The effect of SIRT activators on cell motility was evaluated in vitro by a scratch assay and revealed a significant increase after Resv and MC2562 treatments compared with the solvent control (Fig. 3, C and D). As expected, Sirtinol inhibited cell motility, confirming the involvement of SIRTs in this process.
FIGURE 2.

Sirtuin modulators regulate cell motility, proliferation, and wound healing. A, kinetics of skin wound healing in CD1 mice treated with SIRT modulators. Animals were treated topically with Resv (n = 13), MC2562 (n = 12), or Sirtinol (n = 12) each day. DMSO was used as a solvent control (n = 12). *, °, and #, p < 0.05 versus solvent. B, epithelial gap histological evaluation at day 5. Resv or MC2562 treatment reduces the epithelial gap significantly at day 5 after punching. * and °, p < 0.05 versus solvent. C, bioluminescence imaging of representative MITO-Luc mice experiments. The images were collected on five animals for each treatment, and one representative animal is shown. D, quantification of emitted light from MITO-Luc mouse would healing. * and °, p < 0.05 versus solvent. E, immunofluorescence panels showing keratinocyte activation after wound healing in MITO-Luc mice. Green, keratin 14; red, luciferase. Magnification ×40).
FIGURE 3.

Sirtuin modulators regulate keratinocytes motility. A, Western blot analysis of histone H4 acetylated lysine 16 (H4K16Ac) and histone H3 acetylated lysine 14 (H3K14Ac) in the presence of Resv, MC2562, and solvent. Relative densitometry is shown in the right panel. B, SIRT1 activity assay performed in HaCaT cells treated with Resv, MC2562, or Sirtinol. DMSO was used as a control solvent. *, p < 0.05 versus solvent. C, phase contrast panels showing an in vitro scratch assay after 24-h treatment with SIRT modulators in HaCaT cells. 10% FCS condition represents a positive control. D, percentage of closure quantification in the in vitro scratch assay. *, °, and #, p < 0.05 versus solvent.
Nitric Oxide Mediates a Functional Cross-talk between Sirtuins and Class I HDACs during Wound Healing
To evaluate the effect of SIRT activators on NO production, experiments were performed in which NO levels were monitored by 4,5-diaminofluorescein diacetate fluorescence in HaCaT cells treated with Resv or MC2562. Both compounds enhanced NO production as early as 1 h after application (Fig. 4A, upper panel), whereas Sirtinol abrogated this effect (lower panel). In this condition, coimmunoprecipitation and immunofluorescence experiments (Fig. 4) were performed to analyze eNOS and SIRT1 association. The results show that SIRT activators stabilized the SIRT1-eNOS complex (Fig. 4B) and induced eNOS phosphorylation in serine 1177 (Ser-1177), suggesting a possible involvement of upstream serine/threonine kinases in this process (C and D). These experiments also revealed SIRT-dependent eNOS activation and NO production in keratinocytes. Prompted by this observation, we explored whether the SIRT-dependent NO production could affect class I HDAC (Fig. 4E). The results show a higher level of HDAC2 S-nitrosylation induced by SIRT activation that correlated well with the loss of specific activity (Fig. 4F). In consideration of the emerging role of HDAC2 in wound closure and scar formation (23), interference experiments were performed by local administration of siRNA oligos to down-modulate HDAC2 expression at the wound site in the presence or absence of SIRT modulators. Fig. 5A shows that HDAC2 inhibition significantly enhanced wound repair compared with the scrambled control. Sirtinol abrogated the positive closure effect of HDAC2 siRNAs, whereas the SIRT activator MC2562 still enhanced skin repair. Immunofluorescence analysis (Fig. 5B) confirmed the down-modulation of HDAC2. The inhibition of NO synthesis by L-NAME, applied directly to the wound alone or in combination with SIRT activators (Fig. 5C), reduced healing efficiency significantly. In vitro L-NAME prevented NO production, counteracting the SIRT activator-dependent HDAC2 nitrosylation in HaCaT cells (Fig. 5D), thus indicating that the functional cross-talk between class I and III HDACs is dependent on NO production.
FIGURE 4.

Nitric Oxide mediates a functional cross-talk between class I and III HDACs. A, SIRT activation induces NO release. Upper panel, representative FACS analysis of 4,5-diaminofluorescein diacetate (DAF) positivity. HaCaT cells exposed to SIRT activators exhibited an increase in NO production. The right graph shows the quantification of 4,5-diaminofluorescein diacetate-positive cells. * and °, p < 0.05 versus solvent. Lower panel, representative FACS analysis of DAF positivity. Sirtinol inhibits Resv-dependent NO release. *, p < 0.05 versus solvent. B, immunoprecipitation (IP) analysis showing the association of eNOS with SIRT1 after 1 h of Resv or MC2562 treatment in HaCaT cells. Relative densitometry is shown in the lower panel. *, p < 0.05 versus solvent. C, Western blotting analysis of eNOS (Ser-1177) phosphorylation after 1 h of Resv or MC2562 treatment in HaCaT cells. Relative densitometry is shown in the lower panel. *, p < 0.05 versus solvent. D, immunofluorescence showing the eNOS (Ser-1177) phosphorylation level (green). Nuclei were counterstained with Hoechst (blue) (magnification ×20). E, immunoprecipitation analysis of HDAC2 cysteine (SNO-Cys) nitrosylation via SIRT activation. Relative densitometries in respect to input are shown in the right panel. * and °, p < 0.05 versus solvent. F, bar graph showing HDAC2-specific activity in HaCaT cells treated with SIRT activators for 1 h. * and °, p < 0.05 versus solvent.
FIGURE 5.
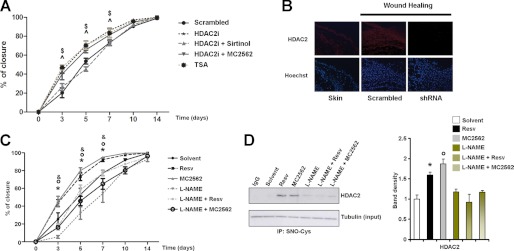
In vivo HDAC2 knockdown by siRNAs and the inhibition of NO synthesis by L-NAME show opposite effects on skin repair. A, graph showing kinetics of skin wound healing in CD1 mice treated topically with siRNA specific for HDAC2 alone (n = 8) or in combination with Sirtinol (n = 7) or MC2652 (n = 7). Scrambled RNA oligos (n = 8) and TSA (n = 7) were used as controls. B, immunofluorescence showing HDAC2 expression (red). Nuclei were counterstained with Hoechst (blue) (magnification ×20). $ and ^, p < 0.05 versus scrambled. C, L-NAME prevents SIRT-dependent skin regeneration in the kinetics of wound healing. CD1 mice were treated with Resv (n = 13), MC2562 (n = 12), L-NAME (n = 8), and L-NAME in combination with Resv (n = 8) or MC2562 (n = 7). DMSO was used as a control solvent (n = 10). *, °, and &, p < 0.05 versus solvent. D, immunoprecipitation analysis of HDAC2 cysteine nitrosylation (SNO-Cys) in presence of the general inhibitor of NO synthesis l-NAME alone or in combination with SIRT activators. Relative densitometries respect input are shown in right. *, °, and &, p < 0.05 versus solvent.
Growth Factors Are Common Targets of Class I HDAC Inhibitors and SIRT Activators
A series of experiments was performed to evaluate the effect of TSA treatment on keratinocyte secretome. The expression of 84 key genes central to WH response was profiled by human WH PCR array (see “Experimental Procedures” and supplemental Table 1). In this condition, three different TSA-dependent growth factors were found to be up-regulated significantly: EGF, keratinocyte growth factor 2/fibroblast growth factor 10 (FGF10), and insulin-like growth factor 1 (IGF-1) (supplemental Table 2). The in vivo expression of these factors was validated in wounded skin samples treated topically with TSA and taken at the day 5 time point after wounding. Immunohistochemistry confirmed the TSA-dependent expression of EGF, FGF10, and IGF-1 during the repair processes (Fig. 6A). Mechanistically, the role of HDAC2 in the regulation of these genes was explored by a series of ChIP experiments performed in HaCaT cells. Remarkably, a significant HDAC2 detachment from the selected GF promoters was found with both TSA and MC2562 (Fig. 6B), whereas the HDAC2 expression levels did not change (C). Immunofluorescence analysis confirmed the expression of EGF, FGF10, and IGF-1 in HaCaT cells treated with TSA and MC2562 (Fig. 6D). These results further support the evidence of a negative regulatory role of class I HDAC and indicated important GFs, including the Keratinocyte Growth Factor 2 (FGF10) (24), as final targets of the SIRT-NO-HDAC2 signaling cascade.
FIGURE 6.

Growth factors are common targets of Class I HDAC inhibitor and sirtuin activators during wound healing. A, representative immunohistochemical staining for EGF, FGF-10, and IGF1 in mouse skin treated with solvent or TSA 5 days after wounding. B, chromatin immunoprecipitations. Graphs show the relative HDAC2 enrichment on growth factor gene promoters (EGF, FGF-10, and IGF1) after 1 h of treatment with TSA, MC2562, or control solvent in HaCaT cells. C, Western blot analysis of HDAC2 expression in HaCaT cells after TSA or MC2562 treatment versus solvent. D, immunofluorescence analysis of EGF, FGF-10, and IGF1 expression (green) in HaCaT cells after TSA or MC2562 treatments versus solvent. Nuclei were counterstained with Hoechst (blue) (magnification ×40).
DISCUSSION
Important epigenetic processes occur during the healing of wounds. Specifically, the silencing of polycomb or the activation of HDACs and SIRTs have been reported during the various phases of skin repair but their role in the regulation of these processes is yet poorly characterized (25, 26). Intriguingly, NO has been found functionally relevant in all phases of WH and recent observations have shown that it modulates gene expression by regulating the function of epigenetic enzymes such as histone acetylases and deacetylases (4, 27). Noteworthy, members of the class-III HDACs, the sirtuins, have been described to modulate eNOS function eliciting a significant increase in NO production by a lysine deacetylation-dependent process (8). To date, the biological relevance of this mechanism, beyond the regulation of endothelial function, remains unclear.
In this study we explored the role of SIRTs, NO and that of class I HDACs in an experimental mouse model of WH in which angiogenesis, cell proliferation and tissue regeneration all take coordinately place. We explored the effect of a new generation SIRT1, 2 and 3 direct activator, MC2526 (15), compared with that of Resv which acts through the inhibition of phosphodiesterase 4 and the consequent metabolic switch that makes more NAD+ available to sirtuins (28). Not surprisingly all SIRT activators accelerated the healing process promoting keratinocytes proliferation, as indicated by the MITO-Luc assay (18). Unexpectedly, the pan-class I-IIa HDAC inhibitor TSA or the class I selective inhibitor MS275, also had a positive effect on skin repair and significantly accelerated healing in a fashion similar to that of SIRT activators. This finding prompted us to evaluate the involvement of class I HDACs in this process and its relationship with SIRTs. To date, HDAC2 is the only member of this class known to be S-nitrosylated, a post-translational modification that depends on the direct action of NO (3, 4). For this reason we first investigated whether HDAC2 could be S-nitrosylated in the presence of SIRT activators and found that this was the case. In vivo HDAC2 knockdown experiments confirmed the positive effect of HDAC2 inactivation on the WH process. These results suggest that class I HDACs, specifically HDAC2, which expression is increased during scar formation (23), could be one of the downstream effectors of the SIRT-dependent NO signaling. The observation that Sirtinol, a pan-sirtuin inhibitor, negatively regulates healing in the presence of a reduced HDAC2 tissue content raises, however, the possibility that additional SIRTs-dependent pathways could be relevant in this process. The evidence that the expression of some of the growth factors up-regulated during WH is further enhanced by TSA (see Fig. 6 and supplemental Tables 1 and 2) reinforces the role of class I HDACs as negative regulators of healing. Their inhibition, however, although sufficient to trigger the process, seems under the hierarchical control of SIRTs, as indicated by the Sirtinol experiment. The positive consequence of SIRT activation on the production of NO may help to explain these findings. In our model system, in fact, it is the SIRT-dependent production of NO that plays the most important role, as indicated by the retarded wound closure determined by Sirtinol as well as the NO synthase inhibitor L-NAME (Fig. 7).
FIGURE 7.

Schematic representation of the NO-dependent functional cross-talk between SIRTs and Class I HDACs during skin repair. The schematic shows the class I and III HDAC functional cross-talk important during wound healing. Specifically, SIRT induction by Resv or MC2562 enhances NO production via eNOS activation. Among its pleiotropic functions, NO acts as an inhibitor of HDAC2 via S-nitrosylation. This modification determines the detachment of HDAC2 from the promoter regions of growth factors, including EGF, FGF-10, and IGF-I, relevant to keratinocyte activation. The class I HDAC inhibitors TSA and MS275 or the selective siRNA oligos aimed at HDAC2 knockdown enhanced skin repair. Notably, the Sirtinol inhibition of SIRT activity or the reduction in NO synthesis, determined by L-NAME, retarded wound healing regardless of HDAC2 interference. This evidence suggests that SIRT-dependent NO synthesis plays a dominant role in skin repair.
In conclusion, our study reveals an unprecedented mechanism by which SIRT and class I HDACs cross-talk in an NO-dependent manner. This observation shows a novel mechanism through which NO regulates keratinocyte proliferation and skin repair. Modulating this molecular machinery may offer new therapeutic perspectives in the case of chronic ulcers, where regulatory molecules, active on different HDAC families, should be considered as potential therapeutic tools.
Supplementary Material
This work was supported by Italian Ministry of Education, University, and Research Grants PRIN2010TYCL9B_006 and FIRB RBFR087JMZ_002 (to A. F.). This work was also supported by the LOEWE Center for Cell and Gene Therapy, Goethe University Frankfurt, Germany (Funding Number III L 4-518/17.004 (2010) (to C. G. and F. S.), by the Duchenne Parent Project, The Netherlands (to C. C.), by grants from AIRC (IG-13234) and the CARIPLO Foundation (grant no. 2009–2439) (to G.P.), and by the Ministry of Health, Ricerca Corrente (RC) 4.3 (to the Istituto Dermopatico dell'Immacolata-IRCCS).

This article contains supplemental Tables 1 and 2.
- HDAC
- histone deacetylase
- SIRT
- sirtuin
- eNOS
- endothelial NO synthase
- WH
- wound healing
- Resv
- resveratrol
- TSA
- trichostatin A
- DMSO
- dimethyl sulfoxide
- WB
- Western blotting
- IF
- immunofluorescence
- ITSA
- Inhibitor of Thrichostatin A.
REFERENCES
- 1. Illi B., Colussi C., Grasselli A., Farsetti A., Capogrossi M. C., Gaetano C. (2009) NO sparks off chromatin. Tales of a multifaceted epigenetic regulator. Pharmacol. Ther. 123, 344–352 [DOI] [PubMed] [Google Scholar]
- 2. Watson P. M., Riccio A. (2009) Nitric oxide and histone deacetylases. A new relationship between old molecules. Commun. Integr. Biol. 2, 11–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nott A., Watson P. M., Robinson J. D., Crepaldi L., Riccio A. (2008) S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455, 411–415 [DOI] [PubMed] [Google Scholar]
- 4. Colussi C., Mozzetta C., Gurtner A., Illi B., Rosati J., Straino S., Ragone G., Pescatori M., Zaccagnini G., Antonini A., Minetti G., Martelli F., Piaggio G., Gallinari P., Steinkuhler C., Steinkulher C., Clementi E., Dell'Aversana C., Altucci L., Mai A., Capogrossi M. C., Puri P. L., Gaetano C. (2008) HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. U.S.A. 105, 19183–19187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Illi B., Dello Russo C., Colussi C., Rosati J., Pallaoro M., Spallotta F., Rotili D., Valente S., Ragone G., Martelli F., Biglioli P., Steinkuhler C., Gallinari P., Mai A., Capogrossi M. C., Gaetano C. (2008) Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 102, 51–58 [DOI] [PubMed] [Google Scholar]
- 6. Spallotta F., Rosati J., Straino S., Nanni S., Grasselli A., Ambrosino V., Rotili D., Valente S., Farsetti A., Mai A., Capogrossi M. C., Gaetano C., Illi B. (2010) Nitric oxide determines mesodermic differentiation of mouse embryonic stem cells by activating class IIa histone deacetylases. Potential therapeutic implications in a mouse model of hindlimb ischemia. Stem Cells 28, 431–442 [DOI] [PubMed] [Google Scholar]
- 7. Chen Z., Peng I. C., Cui X., Li Y. S., Chien S., Shyy J. Y. (2010) Shear stress, SIRT1, and vascular homeostasis. Proc. Natl. Acad. Sci. U.S.A. 107, 10268–10273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mattagajasingh I., Kim C. S., Naqvi A., Yamamori T., Hoffman T. A., Jung S. B., DeRicco J., Kasuno K., Irani K. (2007) SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 104, 14855–14860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broughton G., 2nd, Janis J. E., Attinger C. E. (2006) The basic science of wound healing. Plast. Reconstr. Surg. 117, 12S-34S [DOI] [PubMed] [Google Scholar]
- 10. Soneja A., Drews M., Malinski T. (2005) Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol. Rep. 57, 108–119 [PubMed] [Google Scholar]
- 11. Isenberg J. S., Ridnour L. A., Espey M. G., Wink D. A., Roberts D. D. (2005) Nitric oxide in wound-healing. Microsurgery 25, 442–451 [DOI] [PubMed] [Google Scholar]
- 12. Luo J. D., Chen A. F. (2005) Nitric oxide. A newly discovered function on wound healing. Acta Pharmacol. Sin. 26, 259–264 [DOI] [PubMed] [Google Scholar]
- 13. Blander G., Bhimavarapu A., Mammone T., Maes D., Elliston K., Reich C., Matsui M. S., Guarente L., Loureiro J. J. (2009) SIRT1 promotes differentiation of normal human keratinocytes. J. Invest. Dermatol. 129, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dal Farra C., Domloge N. (2006) SIRT1, the human homologue to SIR2, is expressed in human skin and in cultured keratinocytes fibroblasts and HaCaT cells; and its levels is closely related to stress and aging. J. Cosmet. Sci. 57, 187–188 [PubMed] [Google Scholar]
- 15. Mai A., Valente S., Meade S., Carafa V., Tardugno M., Nebbioso A., Galmozzi A., Mitro N., De Fabiani E., Altucci L., Kazantsev A. (2009) Study of 1,4-dihydropyridine structural scaffold. Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 52, 5496–5504 [DOI] [PubMed] [Google Scholar]
- 16. Bashmakov Y. K., Assaad-Khalil S., Petyaev I. M. (2011) Resveratrol may be beneficial in treatment of diabetic foot syndrome. Med. Hypotheses 77, 364–367 [DOI] [PubMed] [Google Scholar]
- 17. Khanna S., Venojarvi M., Roy S., Sharma N., Trikha P., Bagchi D., Bagchi M., Sen C. K. (2002) Dermal wound healing properties of redox-active grape seed proanthocyanidins. Free Radic. Biol. Med. 33, 1089–1096 [DOI] [PubMed] [Google Scholar]
- 18. Goeman F., Manni I., Artuso S., Ramachandran B., Toietta G., Bossi G., Rando G., Cencioni C., Germoni S., Straino S., Capogrossi M. C., Bacchetti S., Maggi A., Sacchi A., Ciana P., Piaggio G. (2012) Molecular imaging of nuclear factor-Y transcriptional activity maps proliferation sites in live animals. Mol. Biol. Cell 23, 1467–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schoop V. M., Mirancea N., Fusenig N. E. (1999) Epidermal organization and differentiation of HaCaT keratinocytes in organotypic coculture with human dermal fibroblasts. J. Invest. Dermatol. 112, 343–353 [DOI] [PubMed] [Google Scholar]
- 20. Nanni S., Benvenuti V., Grasselli A., Priolo C., Aiello A., Mattiussi S., Colussi C., Lirangi V., Illi B., D'Eletto M., Cianciulli A. M., Gallucci M., De Carli P., Sentinelli S., Mottolese M., Carlini P., Strigari L., Finn S., Mueller E., Arcangeli G., Gaetano C., Capogrossi M. C., Donnorso R. P., Bacchetti S., Sacchi A., Pontecorvi A., Loda M., Farsetti A. (2009) Endothelial NOS, estrogen receptor β, and HIFs cooperate in the activation of a prognostic transcriptional pattern in aggressive human prostate cancer. J. Clin. Invest. 119, 1093–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mai A., Massa S., Pezzi R., Simeoni S., Rotili D., Nebbioso A., Scognamiglio A., Altucci L., Loidl P., Brosch G. (2005) Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J. Med. Chem. 48, 3344–3353 [DOI] [PubMed] [Google Scholar]
- 22. Vaquero A., Sternglanz R., Reinberg D. (2007) NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 26, 5505–5520 [DOI] [PubMed] [Google Scholar]
- 23. Fitzgerald O'Connor E. J., Badshah I. I., Addae L. Y., Kundasamy P., Thanabalasingam S., Abioye D., Soldin M., Shaw T. J. (2012) Histone deacetylase 2 is upregulated in normal and keloid scars. J. Invest. Dermatol. 132, 1293–1296 [DOI] [PubMed] [Google Scholar]
- 24. Marchese C., Felici A., Visco V., Lucania G., Igarashi M., Picardo M., Frati L., Torrisi M. R. (2001) Fibroblast growth factor 10 induces proliferation and differentiation of human primary cultured keratinocytes. J. Invest. Dermatol. 116, 623–628 [DOI] [PubMed] [Google Scholar]
- 25. Rafehi H., El-Osta A., Karagiannis T. C. (2011) Genetic and epigenetic events in diabetic wound healing. Int. Wound J. 8, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shaw T., Martin P. (2009) Epigenetic reprogramming during wound healing. Loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 10, 881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Colussi C., Scopece A., Vitale S., Spallotta F., Mattiussi S., Rosati J., Illi B., Mai A., Castellano S., Sbardella G., Farsetti A., Capogrossi M. C., Gaetano C. (2012) P300/CBP associated factor regulates nitroglycerin-dependent arterial relaxation by N(ε)-lysine acetylation of contractile proteins. Arterioscler. Thromb. Vasc. Biol. 32, 2435–2443 [DOI] [PubMed] [Google Scholar]
- 28. Park S. J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A. L., Kim M. K., Beaven M. A., Burgin A. B., Manganiello V., Chung J. H. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.